Journal of Modern Physics
Vol.10 No.09(2019), Article ID:94157,10 pages
10.4236/jmp.2019.109073
Imaginary Time Density Functional Calculation of Ground States of Atoms Using CWDVR Approach
Naranchimeg Davgiikhorol1, Munkhsaikhan Gonchigsuren1*, Khenmedekh Lochin1, Sukh Ochir1, Tsogbadrakh Namsrai2
1Department of Physics, School of Applied Sciences, Mongolian University of Science and Technology, Ulaanbaatar, Mongolia
2Department of Physics, School of Arts and Sciences, National University of Mongolia, Ulaanbaatar, Mongolia

Copyright © 2019 by author(s) and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: June 17, 2019; Accepted: August 3, 2019; Published: August 6, 2019
ABSTRACT
The self-consistent Kohn-Sham equations for many-electron atoms are solved using the Coulomb wave function Discrete Variable Method (CWDVR). Wigner type functional is used to incorporate correlation functional. The discrete variable method is used for the uniform and optimal spatial grid discretization and solution of the Kohn-Sham equation. The equation is numerically solved using the CWDVR method. First time we have reported the solution of the Kohn-Sham equation on the ground state problem for the many-electronic atoms by the CWDVR method. Our results suggest CWDVR approach shown to be an efficient and precise solution of ground-state energies of atoms. We illustrate that the calculated electronic energies for He, Li, Be, B, C, N and O atoms are in good agreement with other best available values.
Keywords:
Density Functional Theory, Effective Potential, Electronic Energies, Kohn-Sham Equation

1. Introduction
Numerical approach of many-electron systems is extremely difficult computation. Density functional theory (DFT) [1] [2] is a computational quantum mechanical modeling method used to investigate many-electron systems, in particular atoms, molecules, and the condensed phases [3] [4] . It provides a powerful alternative technique to ab-initio wave function approach, since the electron density possesses only three spatial dimensions no matter how large the system is. The DFT proves accurate and computationally much less expensive than usual ab-initio wave function methods, such as a Hartree Fock method. However, the majority of the applications are restricted to the ground states. The exchange-correlation energy functional, which is a functional of the total electron density, is not known exactly, and thus approximate exchange-correlation energy functional must be used. Moreover, while the highest-occupied orbital energy corresponds to the negative of the ionization potential for well-behaved potentials vanishing at infinity, energies of other occupied and unoccupied orbitals have no rigorous correspondence to the excitation energies. Nevertheless, it has been shown recently [5] that the unoccupied true Kohn-Sham eigenvalues can also provide good excitation energies which the commonly used approximate density functionals usually do not satisfy. Nevertheless, numerous attempts [3] [6] have been made in this direction over the years; the ensemble density functional method [7] [8] [9] [10] [11] , the method based on partitioning of the wave function, the time-dependent density functional theory (TDDFT) approach [12] [13] [14] [15] . The DFT based upon the Hohenberg-Kohn (HK) energy functional [3] [4] focuses on the solution of exchange-correlation energy and it had been used in many calculations of ground state properties an atomic system. An efficient and accurate grid method for solving the time-dependent Schrödinger equation for an atomic system interacting with an intense laser pulse has been shown by Liang-Y.Peng and A.F. Starace [16] . Instead of the usual finite difference (FD) method, the radial coordinate is discretized using the discrete variable representation (DVR) constructed from Coulomb wave functions. The DVR method has its origin in the transformation method devised by Harris et al. [17] , where it was further developed by Dickinson and Certain [18] . Light et al. [19] first explicitly used the DVR method as a basis representation for quantum problems, where after different types of DVR methods have found wide applications in different fields of physical and chemical problems. The DVR method gives an idea; associated basis functions are localized about discrete values of the coordinate under consideration. The DVR simplifies the evaluation of Hamiltonian matrix elements. The matrix elements of kinetic energy can also be calculated very simply and analytically in most cases [20] . Since the CWDVR method is able to treat the Coulomb singularity naturally, it is suitable for atomic systems [16] . The Kohn-Sham equation is shown to be solved by the Coulomb wave function discrete variable representation method.
To the best of our knowledge, no one reported the solution of the Kohn-Sham equation on the ground state problem for the many-electronic atoms by the CWDVR method. Furthermore, we accurately present the ground state energies of the many-electronic atoms by the CWDVR method.
This paper consists of methodology and results obtained within the CWDVR method. We show that ground state energy values calculated by the present method are in good agreement with other precise theoretical calculations. Finally, we present conclusions. Here, atomic units (a.u.) are used throughout this paper unless otherwise specified.
2. CWDVR Method
In this section, we first give a brief introduction to the DVR constructed from orthogonal polynomials and Coulomb wave functions, which will be used to solve the Kohn-Sham equation for many-electron atomic systems. The DVR approach basis functions can be constructed from any complete set of orthogonal polynomials, defined in the domain with the corresponding weight function [21] .
It is known that a Gaussian quadrature can also be constructed using nonclassical polynomials. The DVR derived from the Legendre polynomials has been shown by Machtoub and Zhang [22] to provide very precise results for the metastable states of the exotic helium atom. An appropriate quadrature rule for the Coulomb wave function was given by Dunseath et al. [23] with explicit expressions for the weights.
It is known that a Gaussian quadrature can also be constructed using nonclassical polynomials. The DVR derived from the Legendre polynomials has been shown by Machtoub and Zhang [22] to provide very precise results for the metastable states of the exotic helium atom. An appropriate quadrature rule for the Coulomb wave function was given by Dunseath et al. [23] with explicit expressions for the weights.
The time dependent single particle Kohn-Sham equation has the form
(1)
here, is the single particle Kohn-Sham orbit of N electron atom, —atomic Hamiltonian, is the time dependent effective potential, and charge density depends on the coordinates and time and is given by
(2)
However, one can write Equation (1) in imaginary time and substitute , t being the real time, to obtain a diffusion-type equations:
(3)
The Kohn-Sham effective local potential contains both classical and quantum potentials and can be written as:
. (4)
here; the first term is inter-electronic Coulomb repulsion, the second is the electron-nuclear attraction term, the third is exchange-correlation term, and last term comes from interaction with the external field (in the present case, this interaction is zero). A simple local energy functional form has been applied for the atoms, and the exchange part can be found to be [24] ,
(5)
. (6)
The simple local parameterized Wigner-type correlation energy functional [25] used for ground states:
(7)
(8)
where: , , are respectively.
The solution of Equation (1) is used split time method, for split time . It can be written
(9)
One of the main features of the DVR is that a function can be approximated by interpolation through the given grid points:
(10)
here: is the interpolation function, is the cardinal function.
The Coulomb wave function is defined by radial grid points. Interpolation function is obtained by using the radial function that is derived from the cardinal functions.
By noting that is the Coulomb function, is the first derivative from at the position , is found to be .
The propagation in the energy space (step first in equation) can now be achieved through
(11)
The cardinal functions in Equation (10) are given by the following expression
(12)
where the points
. (13)
Since the Coulomb wave functions was expressed in quadrature rule with expressions for the weight , then DVR basis function satisfies the eigenvalue for the radial Kohn-Sham type equation:
(14)
and
(15)
The DVR greatly simplifies the evaluation of Hamiltonian matrix elements. The potential matrix elements involve merely the evaluation of the interaction potential at the DVR grid points, where no integration is needed.
The DVR basis function is constructed from the cardinal function as follows
, (16)
here the weight is given by [23] :
. (17)
(18)
The second derivative of the cardinal function is given,
(19)
where is given by Equation (18) and . Here kinetic energy matrix elements calculated using:
, (20)
. (21)
In the Equation (15), to expand in the eigenvectors of the Hamiltonian , we first solve the eigenvalue problem for after discretization of coordinate, the differential equation for this problem can be written as:
(22)
here denotes the symmetrized second derivative of the cardinal function that is given as,
(23)
(24)
Equation (2) is then numerically solved to achieve a self-consistent set of orbitals, using the DVR method. These orbitals are used to construct various Slater determinants arising out of that particular electronic configuration and its energies computed in the usual manner.
A key step in the time propagation of Equation (9) is to construct the evolution operator through an accurate and efficient representation of . Here we extend the DVR method to achieve optimal grid discretization and an accurate solution of the eigenvalue problem of .
In the present work, we are particularly interested in the exploration of the improvement of the Kohn Sham type equation in electron structure calculation. Thus we choose the Slater wave function as our initial state at . Note that, the differential equation for time propagation is normalized at each time step. Here the 152 grid points are used for the DVR discretization of the radial coordinates and , with 500 iterations is used in the time propagation to achieve convergence.
3. Results and Discussions
In this section we present results from nonrelativistic electronic structure calculation of the ground states of He, Li, Be, B, C, N and O atoms. Wolfram Mathematica Software has used for the calculations. Here, parameters of the Coulomb wave function such as wave number and effective charges are chosen to be and . Table 1 summarizes the main results for the mentioned atoms. The first row shows the present results. The results from the Amlan

Table 1. Calculated ground-state properties of He, Li, Be, B, C, N and O atoms (in au) along with literature data for comparison.
K Roy [26] for energies for the ground states for He, Li, Be, B, C, N and O atoms are shown below the present results. The corresponding Hartree-Fock values from the literature are listed for comparison. For all atoms considered, we found the present results of the total electronic energies considerably match the Hartree-Fock values and are significantly better than the results from Amlan K Roy [26] .
It is satisfying that the CWDVR approach can be used to perform a high precision calculation of the Kohn-Sham type equation with the use of only a few grid points.
Analyses of the results for exchange and correlation energies are given in the same table separately. The results from exchange energies (Ex) calculations of the present calculations show a good agreement with the Hartree-Fock results [3] . For the Be, B, C and O atoms, the calculated exchange energy is nearly exact, while for He, Li and N, there is an underestimation by 1.05% - 1.63%. The calculated exchange energy for He and N gives close results to Amlan J Roy, underestimated about 0.68% - 1.05%, the rest atoms are being the worst case. This indicates that the simple local exchange functional in Equation (5) is well accurate, compared to those of Amlan J Roy [26] , which show a closer agreement with Hartree-Fock exchange energies.
The “exact” correlation energies are considered for the Li, Be, B, C, N and O atoms in Table 1 due to the comparison with other results. The Wigner-type correlation energy functional likely seems to be sufficiently enough for the systems considered. Compared with Hartree-Fock results for He, Be, C and O atoms, it is nearly exact, otherwise underestimated by about 0.24% - 4.94%; the Li, B and N atoms are being the worst case. The calculated correlation energy for He atom is nearly exact to Amlan J Roy, but worse results for other atoms.
We note that the primary purpose of this work is to explore the feasibility of extending the CWDVR to the solution of the Kohn-Sham type differential equation with imaginary time propagation. LDA-type xc energy functional can be easily adopted in the present CWDVR approach.
Table 1 shows that the virial theorem is nearly satisfied for all atoms. The calculated kinetic energy term for the O atom is reasonably exact to Hartree-Fock, while for rest atoms there is an underestimation by 1.03% - 3.44%.
The results for total energies are given in the same table separately. The results from total energies calculations of the present calculations show a good agreement with the Hartree-Fock results [3] . For the Be, and O atoms, the calculated exchange energy is nearly exact, while for rest atoms there is an underestimation by 1.02% - 2.25%. This indicates that the total energy functional is well accurate, compared to those of Amlan J Roy [26] , which show a closer agreement with Hartree-Fock total energies.
Results from the calculations of radial densities of atoms were created images of correspondent plots. Examples of radial density plots are shown in Figure 1 and Figure 2. In Figure 1, we report the radial density plots for beryllium. The inset (a) reports the result from the present calculation; the inset (b) shows the
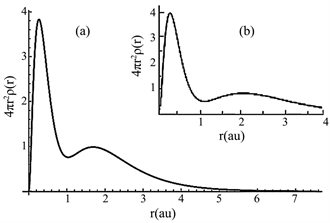
Figure 1. Radial density plot of Be (in au). The inset (a) reports the result from the present calculation; the inset (b) shows the Hartree-Fock plot for comparison.

Figure 2. Radial density plot of C (in au) obtained from the present calculation.
Hartree-Fock plot for comparison. Here, the radial density plot shape from our calculation is in good agreement with Hartree-Fock plot [3] .
Figure 2 shows the radial density plot for carbon. We note that the radial density of carbon calculated maintain the expected shell structure and closely resemble the Hartree-Fock density, where Hartree-Fock plot is not shown.
4. Conclusion
In this paper, we present that nonrelativistic ground-state properties of He, Li, Be, B, C, N and O atoms can be calculated by means of time-dependent Kohn-Sham equations and an imaginary time evolution methods. The CWDVR approach is shown to be an efficient and precise solution of ground-state energies of atoms. The calculated electronic energies are in good agreement with the Hartree-Fock values and are significantly better than the results in the literature [26] . The approach likely opens a road to the solution of ionization and excitation states of many electron atoms.
Acknowledgements
This work was supported by the Science Technology Foundation Project (Code: ShUS-2019/08) of Mongolia.
Conflicts of Interest
The authors declare no conflicts of interest regarding the publication of this paper.
Cite this paper
Davgiikhorol, N., Gonchigsuren, M., Lochin, K., Ochir, S. and Namsrai, T. (2019) Imaginary Time Density Functional Calculation of Ground States of Atoms Using CWDVR Approach. Journal of Modern Physics, 10, 1134-1143. https://doi.org/10.4236/jmp.2019.109073
References
- 1. Hohenberg, P. and Kohn, W. (1964) Physical Review, 136, B864. https://doi.org/10.1103/PhysRev.136.B864
- 2. Kohn, W. and Sham, L.J. (1965) Physical Review, 140, A1133. https://doi.org/10.1103/PhysRev.140.A1133
- 3. Parr, R.G. and Yang, W. (1989) Density-Functional Theory of Atoms and Molecules. Oxford Univ. Press, New York.
- 4. Gross, E.K.U. and Dreizler, R.M. (1995) Density Functional Theory. Plenum, New York.
- 5. Savin, A., Umrigar, C.J. and Gonze, X. (1998) Chemical Physics Letters, 288, 391-395. https://doi.org/10.1016/S0009-2614(98)00316-9
- 6. Singh, R. and Deb, B.M. (1999) Physics Reports, 311, 47. https://doi.org/10.1016/S0370-1573(98)00081-7
- 7. von Barth, U. (1979) Physical Review A, 20, 1693. https://doi.org/10.1103/PhysRevA.20.1693
- 8. Ziegler, T., Rauk, A. and Baerends, E.J. (1977) Theoretica Chimica Acta, 43, 261. https://doi.org/10.1007/BF00551551
- 9. Ziegler, T. (1991) Chemical Reviews, 91, 651. https://doi.org/10.1021/cr00005a001
- 10. Daul, C. (1994) International Journal of Quantum Chemistry, 52, 867. https://doi.org/10.1002/qua.560520414
- 11. Nagy, A. (1991) Journal of Physics B, 24, 4691.https://doi.org/10.1088/0953-4075/29/3/007
- 12. Petersilka, M., Gossmann, U.J. and Gross, E.K.U. (1996) Physical Review Letters, 76, 1212. https://doi.org/10.1103/PhysRevLett.76.1212
- 13. Petersilka, M. and Gross, E.K.U. (1996) International Journal of Quantum Chemistry, 60, 181. https://doi.org/10.1002/(SICI)1097-461X(1996)60:7<1393::AID-QUA21>3.0.CO;2-4
- 14. Casida, M.E., Jamorski, C., Casida, K.C. and Salahub, D.R. (1998) The Journal of Chemical Physics, 108, 4439. https://doi.org/10.1063/1.475855
- 15. Grabo, T., Petersilka, M. and Gross, E.K.U. (2000) Journal of Molecular Structure: THEOCHEM, 501, 353. https://doi.org/10.1016/S0166-1280(99)00445-5
- 16. Peng, L.-Y. and Starace, A.F. (2006) The Journal of Chemical Physics, 125, Article ID: 154311. https://doi.org/10.1063/1.2358351
- 17. Harris, D.O., Engerholm, G.G. and Gwinn, W.D. (1965) The Journal of Chemical Physics, 43, 1515. https://doi.org/10.1063/1.1696963
- 18. Dickinson, A.S. and Certain, P.R. (1968) The Journal of Chemical Physics, 49, 4209-4211. https://doi.org/10.1063/1.1670738
- 19. Lill, J.V., Parker, G.A. and Light, J.C. (1982) Chemical Physics Letters, 89, 483. https://doi.org/10.1016/0009-2614(82)83051-0
- 20. Heather, R.W. and Light, J.C. (1983) The Journal of Chemical Physics, 79, 147. https://doi.org/10.1063/1.445574
- 21. Baye, D. and Heenen, P.H. (1986) Journal of Physics A, 19, 2041. https://doi.org/10.1088/0305-4470/19/11/013
- 22. Machtoub, G. and Zhang, C. (2002) International Journal of Theoretical Physics, 41, 293. https://doi.org/10.1023/A:1014010923548
- 23. Dunseath, K.M., Launay, J.M., Terao-Dunseath, M. and Mouret, L. (2002) Journal of Physics B, 35, 3539. https://doi.org/10.1088/0953-4075/35/16/313
- 24. Deb, B.M. and Chattaraj, P.K. (1989) Physical Review A, 39, 1696. https://doi.org/10.1103/PhysRevA.39.1696
- 25. Roy, A.K. and Chu, S.-I. (2002) Journal of Physics B: Atomic, Molecular and Optical Physics, 35, 2075-2086.
- 26. Roy, A.K. (2011) Journal of Mathematical Chemistry, 49, 1687-1699. https://doi.org/10.1007/s10910-011-9851-2