Journal of Modern Physics
Vol.4 No.3A(2013), Article ID:29345,10 pages DOI:10.4236/jmp.2013.43A059
DFT Calculations on the Effect of Solvation on the Tautomeric Reactions for Wobble Gua-Thy and Canonical Gua-Cyt Base-Pairs
1Department of Computer Science and Engineering, Toyohashi University of Technology, Toyohashi, Japan
2Department of Molecular and Quantum Biophysics, Institute of Molecular Biology and Genetics, National Academy of Sciences of Ukraine, Kyiv, Ukraine
Email: *kurita@cs.tut.ac.jp
Received January 5, 2013; revised February 6, 2013; accepted February 15, 2013
Keywords: DFT; Wobble Base Pair; Base Pair; Tautomerization; Transition State; Activation Energy; Solvation
ABSTRACT
To elucidate the reaction mechanism from wobble Guanine-Thymine (wG-T) to tautomeric G-T base-pairs, we investigate its transition state (TS) by density functional theory (DFT) calculations, in vacuum and in water approximated by continuum solvation model. From the comparison of these results, we attempt to elucidate the effect of solvation on the tautomeric reaction for wG-T. In addition, the same DFT calculations are performed for the canonical G-C base-pair, in order to reveal the difference in the activation energy for the reactions involving wG-T and G-C. The obtained TS structures between wG-T and G*-T/G-T* (asterisk is an enol-form of base) are almost the same in vacuum and in water. However, the activation energy is 16.6 and 19.1 kcal/mol in vacuum and in water, respectively, indicating that the effect of solvation enlarges the energy barrier for the reactions from wG-T to G-T*/G*-T. The activation energy for the tautomeric reaction from G-C to G*-C* is also evaluated to be 15.8 and 12.9 kcal/mol in vacuum and in water, respectively. Therefore, it is expected that the tautomeric reaction from wG-T to G*-T/G-T* can occur in vacuum with a similar probability as that from G-C to G*-C*. We furthermore investigate the TS structure for wG-BrU to reveal the effect of the BrU introduction into wG-T. The activation energy is 14.5 and 16.7 kcal/mol in vacuum and in water, respectively. Accordingly, the BrU introduction is found to increase the probability of the tautomeric reaction producing the enol-form G* and T* bases. Because G* prefers to bind to T rather than to C, and T* to G not A, our calculated results reveal that the spontaneous mutation from C to T or from A to G bases is accelerated by the introduction of wG-BrU base-pair.
1. Introduction
It was revealed from the studies of the genetic codes that the formation of the canonical Watson-Crick (W-C) basepairs (i.e. G-C and A-T) for DNA is strictly observed only for the first two base-pairs in the codon-anticodon interactions [1]. On the other hand, many types of basepairs including both the canonical W-C and non-canonical W-C base-pairs are involved at the third codon position. Accordingly, it was proposed that the base-pair at the 3’ end of the codon can deviate or wobble within arbitrary limits that are consistent with the additional pairing interactions suggested by the code, and the specific base-pairings in the codon-anticodon interactions have been investigated for understanding of the reason why particular base-pairs are selected at the wobble position [2].
In the high resolution nuclear magnetic resonance (NMR) and ethidium bromide binding studies [3], some polymers composed of G and T bases were found to form ordered double-helical structures. In these structures, G and T bases are hydrogen bonded together in a wobble base-pair form, while alternative hydrogen bonding schemes involving the tautomeric form of either G or T are eliminated. In addition, the X-ray diffraction analysis [4] at 2.5 Å resolution for the deoxyoligomer d(CGCGAATTTGCG) duplex showed that the G-T base-pairs in the duplex adopt a wobble structure. The wobble G-T (wG-T) basepair can also be accommodated in the octameric A-form DNA fragment [5] as well as the Z-form DNA with the sequence of d(CGCGTG) [6].
On the other hand, by the ab initio molecular orbital (MO) calculations [7], it was demonstrated that the double proton transfer (DPT) in the wG-T base-pair is a potential pathway for the generation of the rare tautomers of guanine. Physical-chemical mechanism of transformation of the wobble DNA base-pairs into the tautomeric base-pairs was suggested by Brovarets and Hovorun [8]. This transition mechanism, involving the tautomeric form of either G or T was investigated by means of ab initio MO and density functional theory (DFT) methods. The results indicated that the structure of the transition state (TS) between wG-T and the tautomeric G-T* (asterisk is an enol-form of base) base-pairs is planar. On the other hand, the TS between wG-T and enol-form G (G*)-T base-pairs is non-planar, and this TS has larger activation energy than that between wG-T and G-T*, indicating that the transition between wG-T and G*-T is unfavorable in DNA duplex structure energetically and structurally. In addition, Brovarets and Hovorun [9] performed the analogical MO and DFT calculations for the wobble G-BrU (wG-BrU) and G-BrU base-pairs to elucidate the effect of BrU on the transition mechanism. The effect of the other halogenated derivatives of U on the DNA replication errors was also investigated by ab initio MO calculations [10].
In previous ab initio MO and DFT study [11], we investigated in detail the transition mechanism between wG-T and G*-T/G-T* base-pairs, in order to search for more stable and planar TS structures for these transitions. In these calculations, we took into account correlation of electrons, which is important for determining stable structures and evaluating activation energies of reactions more accurately. The obtained TS between wG-T and G*-T is remarkably different from the TS obtained by the previous DFT calculation [8], and the activation free energy (17.9 kcal/mol) evaluated by our calculation is significantly smaller than that (39.21 kcal/mol) obtained by the previous calculation [8]. It is thus likely that the TS obtained by our calculations is more preferable than that by the previous calculation [8].
In the present study, to reveal the effect of solvation on the tautomeric reactions between the wG-T and G*-T/ G-T* base-pairs, we perform DFT calculations in vacuum and in water, which is approximately described by the continuum solvation model. In addition, the tautomeric reaction for the canonical G-C base-pair is investigated by the same DFT method to reveal the difference in the activation energy for the reactions involving wG-T and G-C. The activation energies evaluated by the DFT method indicate that the tautomeric reaction from wG-T to G*-T/G-T* can occur in vacuum with a similar probability as that from G-C to G*-C*.
2. Details of DFT Calculations
We first optimized the structures of the wG-T, G*-T and G-T* base-pairs by using the DFT method of the ab initio MO program Gaussian09 (G09) [12]. As exchange and correlation functionals, we employed the meta-hybrid M06 functional [13] based on generalized gradient approximation, because the M06 functional has good performance for calculating activation energies and thermochemical kinetics. Kohn-Sham orbitals were expanded in the 6-311++G(d,p) basis-set. The structures of the TSs between these base-pairs were searched for by the STQN method [14] of G09, and the obtained TS structures were confirmed to have a single imaginary frequency by the vibrational analysis of G09. Afterwards, the reaction path of proton transfer between bases was followed by performing an intrinsic reaction coordinate (IRC) calculation [15], in order to make sure that the obtained TS structure really is TS structure connecting the reactant and the product structures. The potential energy surface for the transitions between the G-T basepairs was evaluated by including the zero-point vibrational energy (ZPE), and the free energies at 298.15 K and 1 atmosphere of pressure were also evaluated. The same procedure was adapted for the analysis of the transition mechanism between the wG-BrU, G-BrU* and G*-BrU and between G-C and G*-C* base-pairs.
In addition, to investigate the effect of solvation on the reaction mechanism, we performed the same DFT calculations in water, which is approximately described by the continuum solvation model. As a self-consistent reaction field (SCRF) model, we employed the integral equation formalism for polarizable continuum model (IEFPCM) [16]. IEFPCM is the default SCRF method in G09 and creates the solute cavity via a set of overlapping spheres. We used IEFPCM, because it can describe accurately the charge distribution of solute outside of the PCM cavity.
3. Results and Discussion
3.1. Reaction Mechanism between wG-T, G-T*, G*-T in Vacuum
In our previous calculations [11], the structures of wG-T, G-T* and G*-T base-pairs were optimized by the MP2/ 6-31G(d,p) and M06/6-311++G(d,p) methods in vacuum. The structures optimized by the MP2 and M06 methods are planar and almost the same to each other, indicating that the M06 method is accurate enough for investigating stable structures of these base-pairs. Hereafter, we will describe only the results obtained by the M06 calculations. In addition, the optimized structures of wG-T, G-T* and G*-T were confirmed to fit into the B-form DNA duplex.
Their total energies including ZPE and free energies are listed in Table 1(a). Among the three structures, G*-T is the most stable. To obtain the TS structure between wG-T and G-T*, we performed the QST2 calculation using the optimized structures of wG-T and G-T*.
Table 1. Total energy (TE) (kcal/mol), total energy with zero-point vibrational energy (ZPE), and total free energy (FE) at 298.15 K and 1 atmosphere of pressure for the optimized structures of wG-T, G-T*, G*-T base-pairs, and the transition states between them evaluated by M06/6-311++G(d,p) method; (a) In vacuum [11] and (b) In water. Dipole moments (Debye) and stabilization energies (SE) (kcal/mol) by solvent are also listed for the structures optimized in water.
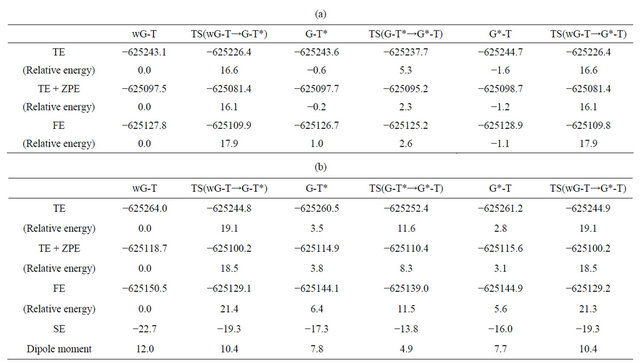
The obtained TS structure was confirmed to have only one imaginary frequency (−170 cm−1) and connect the wG-T and G-T* structures by the IRC analysis. The activation energy for this reaction was evaluated to be 16.1 kcal/mol including ZPE, while the activation free energy is 17.9 kcal/mol, as listed in Table 1(a).
By using the same procedure, we searched for the TS structure between wG-T and G*-T. The previous B3LYP/ 6-311++G(d,p) calculation [8] predicted a nonplanar TS structure. On the other hand, our M06/6-311++G(d,p) calculation obtained a planar TS structure, which is almost identical to the TS structure between wG-T and G-T*. We considered that this result may come from the close similarity of G-T* and G*-T structures in comparison with the wG-T structure. In fact, the positions of two protons contributing to the hydrogen bonds between the G and T bases are slightly different (about 0.1 Å), while the positions of the other atoms are the same to each other. Our obtained TS structure is more preferable than that obtained by the previous DFT study [8] in the viewpoint of activation energy. We furthermore performed the IRC analysis to elucidate the reaction path between wG-T and G*-T. The results elucidated that there is no direct reaction path between wG-T and G*-T, because G in wG-T can not easily release the proton bonded to the nitrogen to tautomerize into G*. Consequentlyit was elucidated in our previous DFT and MP2 study [11] that wG-T can be transferred into G-T* directly but not into G*-T in vacuum.
In addition, we investigated the transition mechanism between G-T* and G*-T by the QST3 calculation based on M06/6-311++G(d,p) method. The obtained TS structure was confirmed to have one imaginary frequency (−900 cm−1) and connect the structures of G-T* and G*-T by the IRC analysis. It should be noted that a double proton transfer easily occurs between G-T* and G*-T in vacuum, because the activation free energy was evaluated to be only 2.6 kcal/mol, as listed in Table 1(a).
3.2. Reaction Mechanism between wG-T, G-T*, G*-T in Water
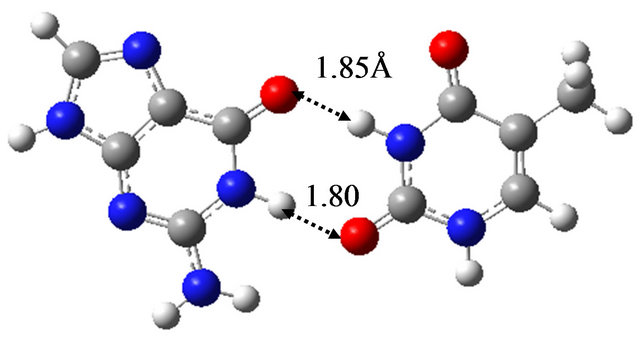
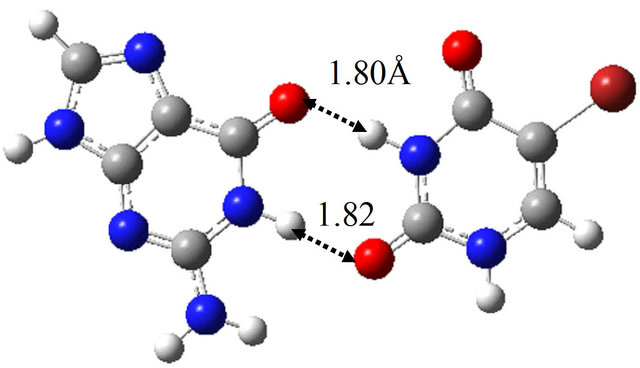
In order to elucidate the effect of solvation on the reaction mechanism between wG-T, G-T* and G*-T, we performed the same calculations in water approximated by the continuum solvation model IEFPCM [16]. The optimized structures are shown in Figure 1, being almost the same as those optimized in vacuum; the hydrogen bond distances between G and T are at most 0.06 Å changed by the optimization in water. The TS structure between wG-T and G-T* is the same as that between wG-T and G*-T. Therefore, the effect of solvation on the structures of the TS is confirmed to be rather small.
 (a)
(a)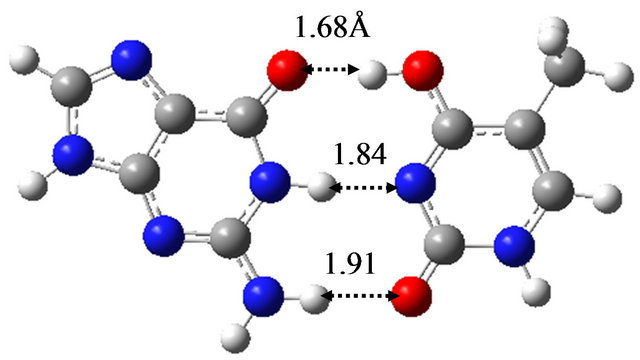 (b)
(b)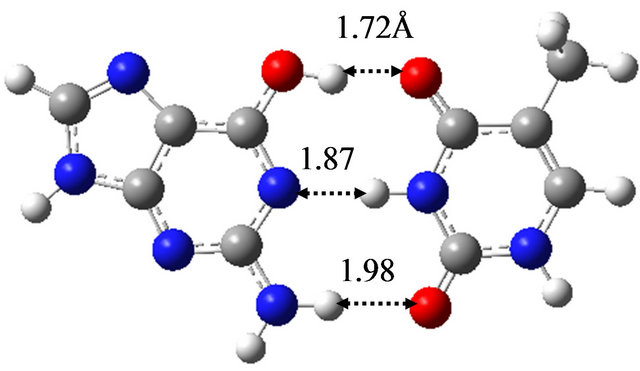 (c)
(c)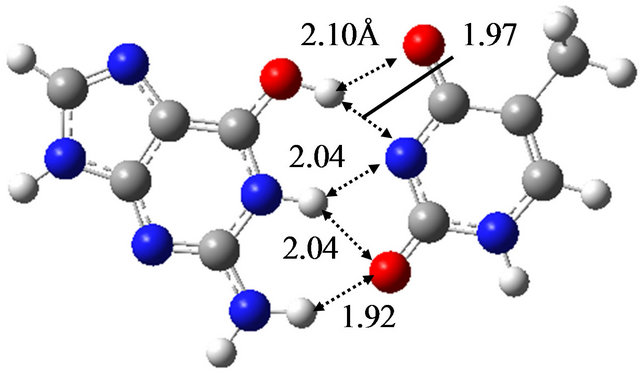 (d)
(d)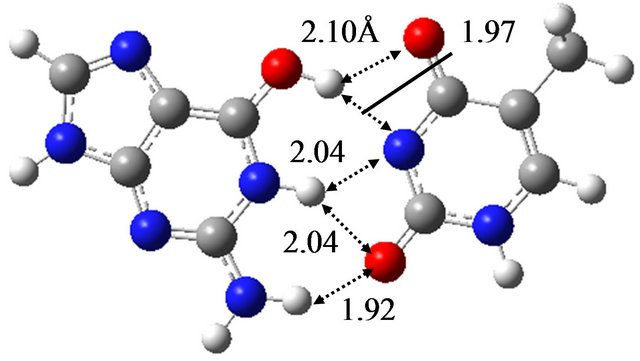 (e)
(e)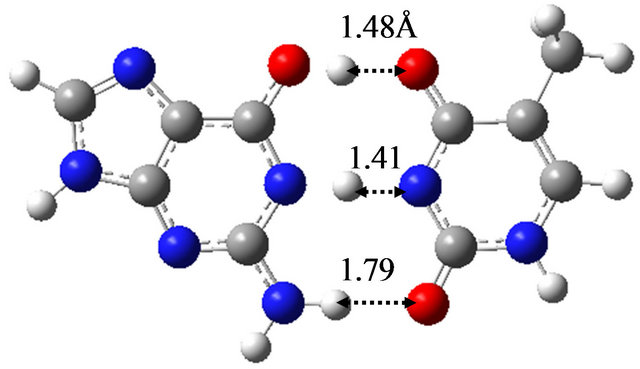 (f)
(f)
Figure 1. Structures of (a) wG-T; (b) G-T*; and (c) G*-T; and TS structures between them; (d) TS(wG-T→G-T*); (e) TS(wG-T→G*-T) and (f) TS(G-T*→G*-T) obtained in water by the M06/6-311++G(d,p) method.
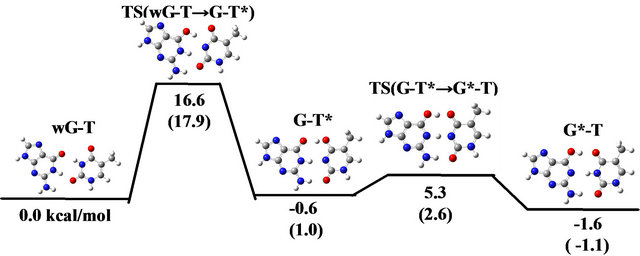
In contrast, as listed in Table 1, the change in energy and free energy during the reaction is remarkably affected by the solvation. The activation energy for the reaction from wG-T to G-T*/G*-T is evaluated 16.6 and 19.1 kcal/mol in vacuum and in water, respectively. Accordingly, the effect of solvation enlarges the energy barrier for the reactions from wG-T to G-T*/G*-T. To elucidate the reason for this effect, we investigated the dipole moment for the optimized and the TS structures of these base-pairs, because molecules with larger dipole moment are stabilized more significantly by solvation. As listed in the last line of Table 1(b), wG-T has larger dipole moment than the TS structure between wG-T and G-T*/G*-T, indicating that the energy of wG-T is stabilized more significantly by salvation in comparison with that of the TS structure. As a result, the activation energy is 2.5 kcal/mol enlarged by the solvation.
As for the reaction from G-T* to G*-T, the TS structure has a dipole moment smaller than those for G-T* and G*-T, resulting in that the energies of G-T* and G*-T are more largely stabilized by solvation than that of the TS. Consequently, the activation energy for the transition is enlarged significantly from 5.9 to 8.1 kcal/mol by the solvation. It is thus elucidated that the effect of solvation should be considered for evaluating the activation energy for the reactions. The similar results were obtained for the wG-BrU base pair as listed in Table 2.
The schematic diagrams of change in total energy and free energy during the reactions in vacuum and in water are compared in Figure 2. It is noted that wG-T is more stable than G-T* and G*-T in water, while G*-T is more stable in vacuum. The bottleneck of the reaction is from wG-T to G-T*, and its activation energy is significantly enhanced from 16.6 to 19.1 kcal/mol by the solvation, as shown in Figure 2. Therefore, by considering the effect of solvation, the reaction from wG-T to G-T*/G*-T is less preferable in the viewpoint of energy. Although the activation energy evaluated in vacuum is similar to that (15.8 kcal/mol) for the reaction from G-C to G*-C*, the energy evaluated in water is much larger than that (12.9 kcal/mol) for the G-C reaction, as will be described in Section 3.4.
Table 2. Total energy (TE) (kcal/mol), total energy with zero-point vibrational energy (ZPE), and total free energy (FE) at 298.15 K and 1 atmosphere of pressure for the optimized structures of wG-BrU, G-BrU*, G*-BrU, and the transition states between them evaluated by M06/6-311++G(d,p); (a) In vacuum [11] and (b) In water. Dipole moments (Debye) and stabilization energies (SE) (kcal/mol) by solvent are also listed for the structures optimized in water.
 (a)
(a)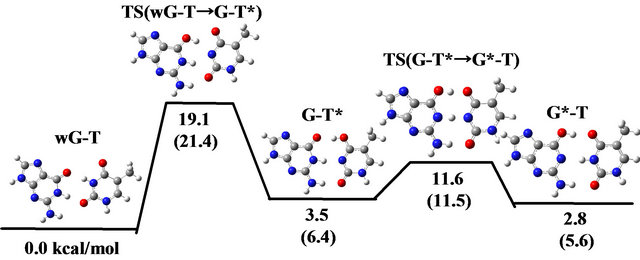 (b)
(b)
Figure 2. Schematic diagrams of change in total energy and free energy during the transitions between wG-T, G-T* and G*-T evaluated by M06/6-311++G(d,p); (a) in vacuum and (b) in water. The values shown in parenthesis are relative free energies.
3.3. Reaction Mechanism between wG-BrU, G-BrU*, G*-BrU
It is well known that the introduction of BrU base into base-pair enhances the provability of the spontaneous mutations in the sequences of bases of DNA and RNA. To elucidate the reason for this fact, we investigated the reaction mechanism between wG-BrU, G-BrU* and G*-BrU in water and compared the results with those for the wG-T base-pairs. The optimized structures in water for these base-pairs are shown in Figure 3. Compared with the wG-T structure, the hydrogen atoms contributing to the hydrogen bonds between G and BrU bases are shifted at most 0.05 Å in wG-BrU. The total energies and free energies for wG-BrU, G-BrU* and G*-BrU evaluated in vacuum [11] and in water are listed in Table 2. In vacuum, the free energies for the three base-pairs are identical to each other within the difference of 0.9 kcal/mol. By considering the solvation, wG-BrU structure is more stabilized than G*-BrU and G-BrU*, resulting in the 4.2 ~ 5.1 kcal/mol difference in free energy. This stabilization comes mainly from the large dipole moment of wG-BrU, as listed in Table 2(b).
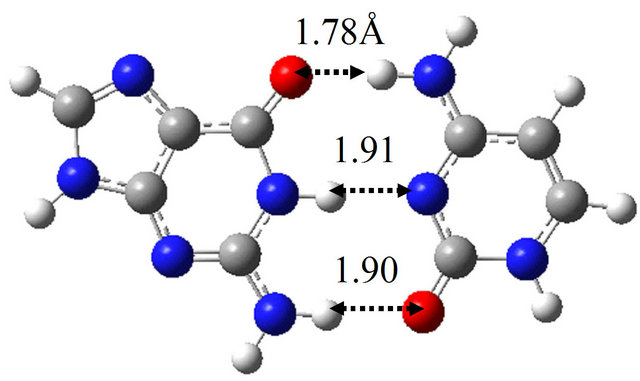
The TS structure between wG-BrU and G-BrU* obtained by the QST2 calculation based on the M06 method is shown in Figure 3(d). This structure has only one imaginary frequency (−125.0 cm−1), and the IRC calculation starting with the TS structure confirmed that this TS structure connects between the structures of wG-BrU and G-BrU*. Therefore, the structure of Figure 3(d) was confirmed to be a TS structure between wG-BrU and G-BrU*. This TS structure is identical to that obtained by the previous B3LYP/6-311++G(d,p) calculation [9] within a difference of 0.06 Å in hydrogen bonds.
In the same way, we obtained the TS structure between wG-BrU and G*-BrU. As shown in Figure 3(e) and Table 2(b), this TS structure and its electronic properties are almost the same as those calculated for the TS structure between wG-BrU and G-BrU*. The IRC analysis starting with this TS structure obtained the wG-BrU and G-BrU* structures. Therefore, it seems that there is no direct path between wG-BrU and G*-BrU in water. This result is the same as that for the transition between wG-T and G*-T.
Furthermore, we searched for the TS structure between G-BrU* and G*-BrU by the QST3 calculation based on M06/6-311++G(d,p). The structure is shown in Figure
 (a)
(a)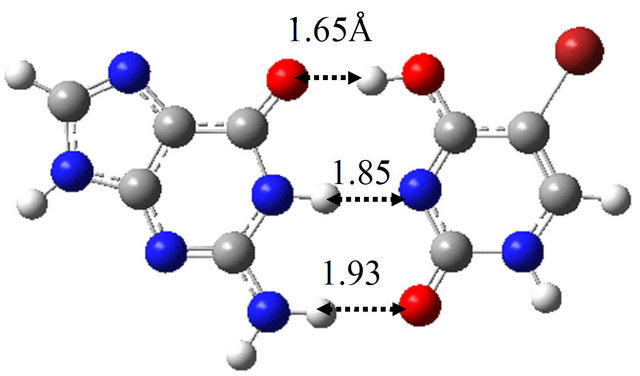 (b)
(b)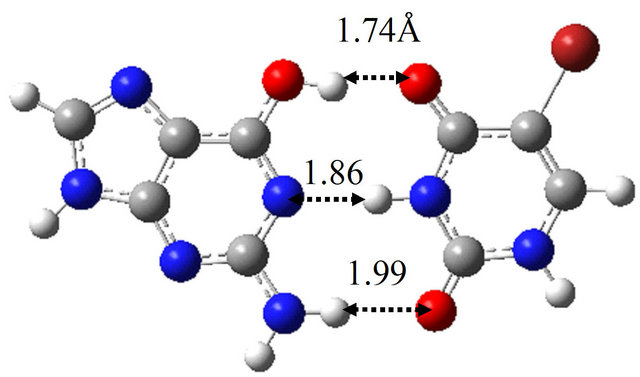 (c)
(c)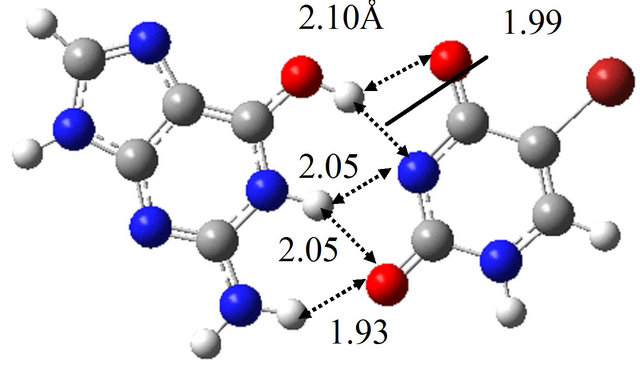 (d)
(d)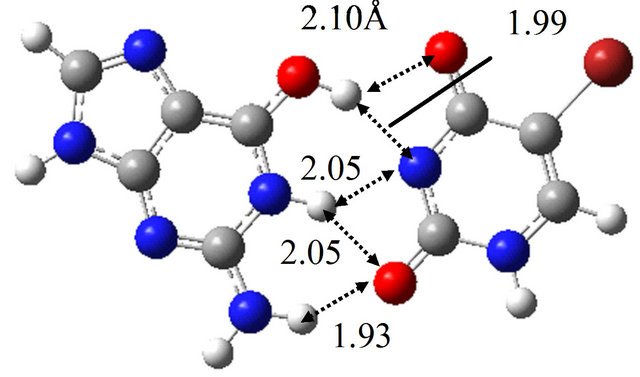 (e)
(e)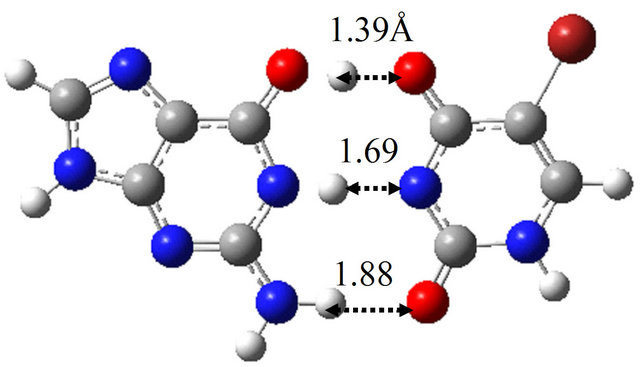 (f)
(f)
Figure 3. Structures of (a) wG-BrU; (b) G-BrU*; (c) G*-BrU and TS structures between them (d) TS(wG-BrU→G-BrU*); (e) TS(wG-BrU→G*-BrU) and (f) TS(G-BrU*→G*-BrU) obtained by M06/6-311++G(d,p) in water.
3(f), which has only one imaginary frequency (−901.4 cm−1) and was confirmed as a TS structure between G-BrU* and G*-BrU by the IRC analysis. From the comparison of Figures 1(f) and 3(f), it is elucidated that the introduction of Br atom into T base affects on the position of hydrogen atom contributing to the hydrogen bond between the nitrogen atoms of G and BrU. The other TS structures shown in Figures 1(d), 1(e), 3(d) and 3(e) are not affected significantly by the Br introduction.
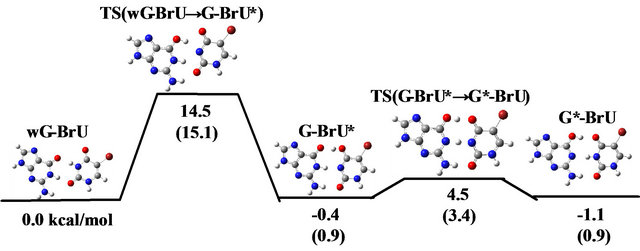
As shown in Figures 2 and 4, the change in energy during the reaction of wG-BrU is qualitatively similar to that for wG-T both in vacuum and in water. However, the activation energies are remarkably changed by the BrU introduction. The activation energy for the transition from wG-BrU to G-BrU* is evaluated to be 14.5 kcal/mol in vacuum and 16.7 kcal/mol in water, respectively. These values are significantly smaller than those (16.6 and 19.1 kcal/mol, respectively) for wG-T shown in Figure 2. In particular, the activation energy evaluated in water is 2.4 kcal/mol smaller than that for wG-T. Therefore, we can conclude that the introduction of BrU base into base-pair brings forward the transition reaction from wG-T to the tautomeric G-T*. Since G* is rather stable and pairs with T instead of C, T is introduced into the site complementary to G at the time of DNA replication. As a result, a mutation from G-C to A-T base-pair is generated at the subsequent duplex formation. The present DFT study reveals a possible origin for the spontaneous mutation induced by the introduction of BrU into wobble G-T base-pair.
The reason for the reduction of the activation energy for the transition from wG-BrU to G-BrU* can be explained by the size of dipole moments for wG-BrU and the TS between wG-BrU and G-BrU*. As listed in the last line of Table 2(b), wG-BrU and the TS have almost the same dipole moment; 13.2 and 13.1 (Debye), indicating that the stabilization by the solvation is similar to each other. As a consequence, the activation energy from wG-BrU to G-BrU*, which is estimated from the difference in total energies between wG-BrU and the TS, is not changed significantly by the solvation. On the other hand, for the wG-T base-pair, Table 1(b) indicates the larger stabilization by solvent for wG-T, resulting in the larger activation energy for the reaction from wG-T and G-T* in water.
3.4. Tautomeric Reaction Mechanism between G-C and G*-C*
In order to predict the possibility of the reaction from wG-T to G-T*/G*-T, we furthermore investigated the tautomeric reaction mechanism between G-C and G*-C* in vacuum and in water, by the M06/6-311++G(d,p) method. From the comparison of the activation energies for the reactions, we attempted to elucidate the possibility for the reaction from wG-T to G-T*/G*-T.
Figure 5 shows the optimized structures of G-C and G*-C*, as well as the TS between them in vacuum. The TS structure has only one imaginary frequency (−920.7 cm−1) and connects the G-C and G*-C* base-pairs. The activation energy and free energy evaluated by the M06 method is 15.8 and 13.7 kcal/mol, respectively, as listed
 (a)
(a)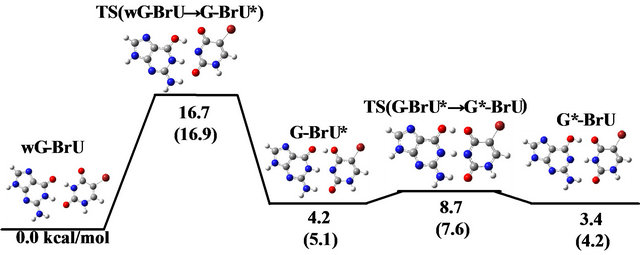 (b)
(b)
Figure 4. Schematic diagrams of change in total energy and free energy during the transitions between wG-BrU, G-BrU* and G*-BrU evaluated by M06/6-311++G(d,p); (a) in vacuum and (b) in water. The values shown in parenthesis are relative free energies.
 (a)
(a)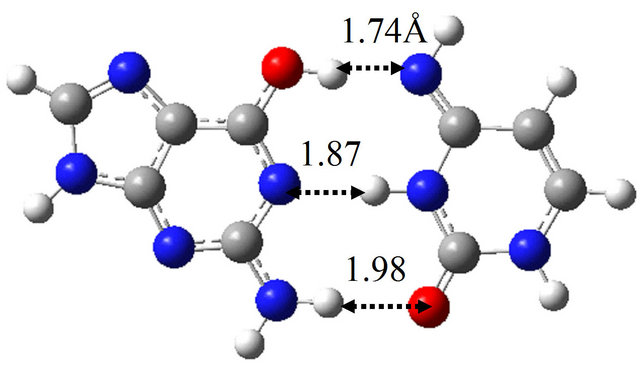 (b)
(b)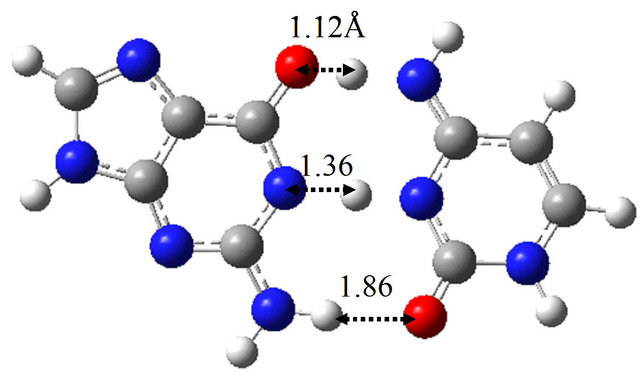 (c)
(c)
Figure 5. Structures of G-C, G*-C* and the TS between them obtained by M06/6-311++G(d,p) in vacuum. (a) G-C; (b) G*-C*; (c) TS(G-C→G*-C*).
in Table 3(a). The obtained structures are identical within the error of 0.04 Å to the previous results [17] obtained by the B3LYP/6-31G(d) method. In addition, the evaluated activation energy is comparable to that (17.3 kcal/mol) evaluated by the previous study [17]. The pathway of the proton transfer in G-C is confirmed to be a concerted double proton transfer by the IRC analysis.
To analyze the influence of the solvating water molecules around G-C on the reaction mechanism, we performed the same DFT calculation in water approximated by the continuum solvation model IEFPCM [16]. The reaction mechanism is more complicated and includes two steps through two TS structures and one intermediate state (IS) structure. The optimized structures in water for G-C, G*-C*, IS, and two TS are shown in Figure 6" target="_self"> Figure 6. Their relative energies and free energies are listed in Table 3(b). In the viewpoint of total energies, G-C is the most stable, while the TS between IS and G*-C* is the least stable. On the other hand, in the viewpoint of free energies, G*-C* is less stable than the TS between IS and G*-C*, indicating the disappearance of the local minimum structure corresponding to G*-C* on the surface of free energy. As a result, the G*-C* structure is spontaneously transferred into the IS structure shown in Figure 6(b). In the previous DFT study [17], such a complicated pathway was not obtained for the reaction between G-C and G*-C*. In our present M06/6-311++G(d,p) study, the activation energy is estimated from Table 3(b) to be 12.9 kcal/mol in water.
Finally, we would like to compare the activation energies for the reactions involving wG-T, wG-BrU or G-C base-pair and predict the probability for the reactions from wG-T (wG-BrU) to G-T* (G-BrU*). As shown in Figures 2(a) and 4(a), the activation energy in vacuum for the reaction from wG-T to G-T* and from wG-BrU to G-BrU* is 16.6 and 14.5 kcal/mol, respectively. These energies is similar to that (15.8 kcal/mol) for the tautomeric reaction from G-C to G*-C* in vacuum. It is thus expected that the reactions for wG-T and wG-BrU can occur with a similar probability as that for the G-C reaction in vacuum. On the other hand, in water, wG-T and wG-BrU are stabilized more significantly by the effect of salvation, resulting in the larger activation energies (19.1 kcal/mol for wG-T and 16.7 kcal/mol for wG-BrU) shown in Figures 2(b) and 4(b), in comparison with that (12.9 kcal/mol) for the tautomeric reaction from G-C to G*-C*. Consequently, it can be concluded that the reactions from wG-T to G-T* and from wG-BrU to G-BrU* are very rare events compared with the tautomeric reaction of G-C in water.
Since our present model is very simple compared to the real DNA duplex surrounded by water molecules and counter ions, the quantitative estimation for the rate constants of the above reactions and the equilibrium constants between the base-pairs will full of uncertainty and be meaningless. The DFT investigation for the wG-T and wG-BrU base-pairs including the backbone of DNA, hydrating water molecules and counter ions are underway now for quantitative investigation of activation free energy. In addition, since the present DFT study considered the influence of the solvating water molecules by the continuum salvation model, the dynamical effect of water molecules on the ZPE and free energy is not included explicitly. Accordingly, we did not discuss the free energies in detail here.
4. Conclusions
To elucidate the effect of solvation on the reaction between wG-T and G-T*/G*-T, we investigated the TS between these base-pairs by DFT calculations, in vacuum and in water approximated by continuum solvation model. The obtained TS between wG-T and G*-T/G-T* (asterisk is an enol-form of base) is almost the same in vacuum and in water. However, the activation energy is 16.6 and 19.1 kcal/mol in vacuum and in water, respectively, indicating that the effect of solvation enlarges the energy barrier for the reactions. The activation energy for the reaction between G-C and G*-C* was also evaluated to be 15.8 and 12.9 kcal/mol in vacuum and in water, respectively. Therefore, it is expected that the reaction from wG-T to G*-T/G-T* can occur in vacuum with a similar probability as that from G-C to G*-C*.
We furthermore investigated the reaction between wG-BrU and G-BrU*/G*-BrU to elucidate the effect of BrU. The activation energy is 14.5 and 16.7 kcal/mol in
Table 3. Total energy (TE) (kcal/mol), zero-point vibrational energy (ZPE) and total free energy (FE) at 298.15 K and 1 atmosphere of pressure for the optimized structures of G-C, G*-C*, intermediate structure (IS) and the transition state (TS) between them evaluated by M06/6-311++G(d,p); (a) in vacuum and (b) in water approximated by the continuum solvation model.
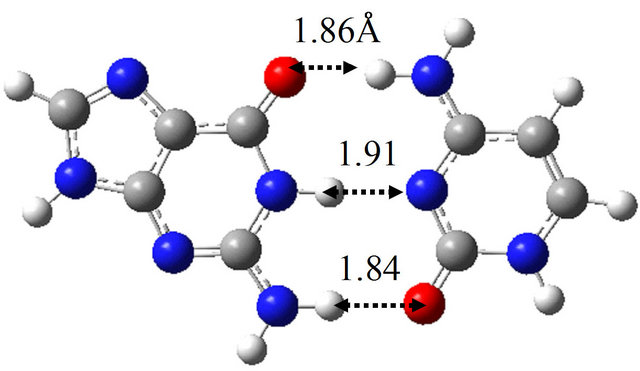 (a)
(a)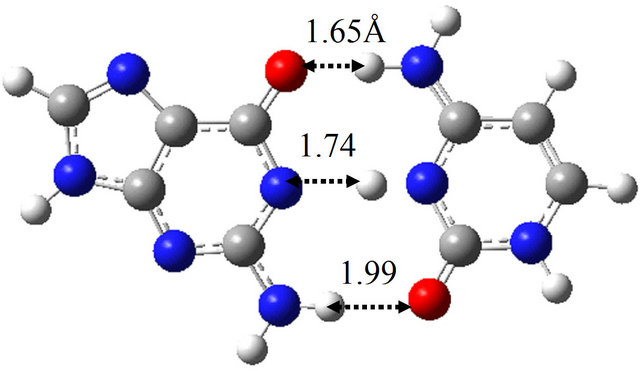 (b)
(b)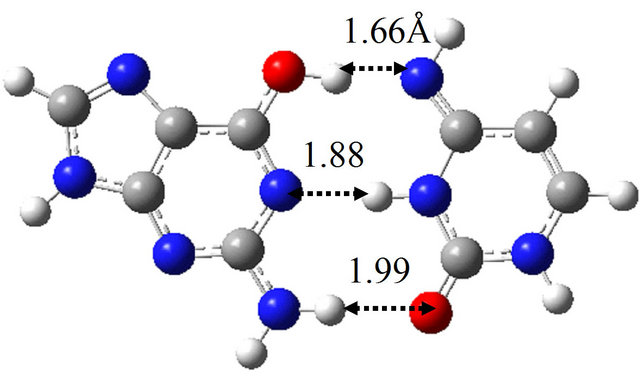 (c)
(c)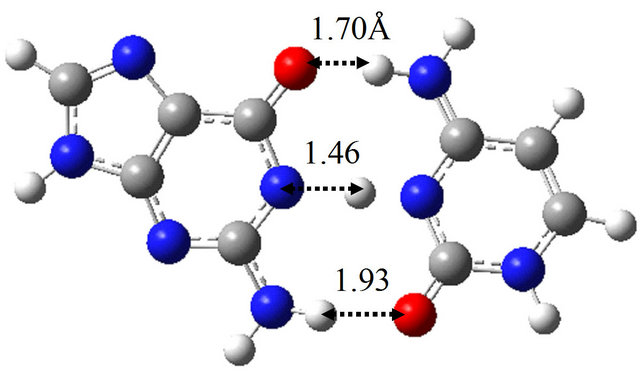 (d)
(d)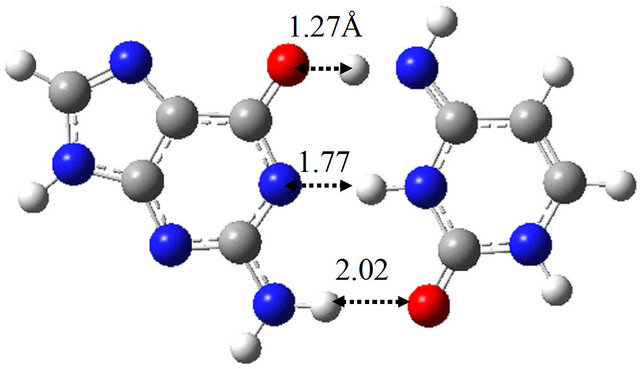 (e)
(e)
Figure 6. Structures of G-C, G*-C*, intermediate structure (IS), and the TS between them obtained by M06/6-311++G(d,p) in water approximated by the continuum solvation model. (a) G-C; (b) IS; (c) G*-C*; (d) TS(G-C→IS); (e) TS(IS→G*-C*).
vacuum and in water, respectively. Therefore, the introduction of wG-BrU base-pair increases the probability of the tautomeric reaction producing the enol-form G* base. Because G* prefers to bind to T rather than to C, our calculated results reveal that the spontaneous mutation from C to T bases is accelerated by the introduction of wGBrU base-pair.
5. Acknowledgements
This work was supported in part by grants from the JSPS Grant-in-Aid for Challenging Exploratory Research (No. 22650061), the Murata Science Foundation, the Iketani Science and Technology Foundation, the Tatematsu Foundation and the CASIO Science Promotion Foundation.
REFERENCES
- F. H. C. Crick, “The Croonian Lecture, 1966: The Genetic Code,” Proceedings of the Royal Society B, Vol. 167, No. 1009, 1967, pp. 331-347. doi:10.1098/rspb.1967.0031
- M. D. Topal and J. R. Fresco, “Complementary Base Pairing and the Origin of Substitution Mutations,” Nature, Vol. 263, 1976, pp. 285-289. doi:10.1038/263285a0
- T. A. Early, J. Olmsted III, D. R. Keams and A. G. Lezius, “Base Pairing Structure in the Poly d(G-T) Double Helix: Wobble Base Pairs,” Nucleic Acids Research, Vol. 5, No. 6, 1978, pp. 1955-1970. doi:10.1093/nar/5.6.1955
- W. N. Hunter, T. Brown, G. Kneale, N. N. Annad, D. Rabinovich and O. Kennard, “The Structure of GuanosineThymidine Mismatches in B-DNA at 2.5-A Resolution,” Journal of Biological Chemistry, Vol. 262, 1987, pp. 9962-9970.
- T. Brown, O. Kennard, G. Kneale and D. Rabinovich, “High-resolution Structure of a DNA Helix Containing Mismatched Base Pairs,” Nature, Vol. 315, 1985, pp. 604-606. doi:10.1038/315604a0
- P. S. Ho, C. A. Frederick, G. J. Quigley, G. A. van der Marel, J. H. van Boom, A. H.-J. Wang and A. Rich, “G-T Wobble Base-Pairing in Z-DNA at 1.0 A Atomic Resolution: The Crystal Structure of d(CGCGTG),” EMBO Journal, Vol. 4, 1985, pp. 3617-3623.
- A. Padermshoke, Y. Katsumoto, R. Masaki and M. Aida, “Thermally Induced Double Proton Transfer in GG and Wobble GT Base-pairs: A Possible Origin of the Mutagenic Guanine,” Chemical Physics Letters, Vol. 457, No. 1-3, 2008, pp. 232-236. doi:10.1016/j.cplett.2008.04.029
- O. O. Brovarets and D. M. Hovorun, “Physicochemical Mechanism of the Wobble DNA Base-pairs Gua-Thy and Ade-Cyt Transition into the Mismatched Base-Pairs Gua*- Thy and Ade-Cyt* Formed by the Mutagenic Tautomers,” Ukrainica Bioorganica Acta, Vol. 2, 2009, pp. 12-18.
- O. O. Brovarets and D. M. Hovorun, “The New Physicochemical Mechanism of the Mutagenic Action of 5- Bromouracil,” Ukrainica Bioorganica Acta, Vol. 2, 2009, pp. 19-23.
- O. O. Brovarets and D. M. Hovorun, “Whether 5-Bromouracile and Its Analogues Produce DNA Point Replication Errors? Results of Quantum-Chemical Analysis,” Bulletin of University of Kyiv Series: Physics & Mathematics, Vol. N2, 2010, pp. 239-242.
- K. Nomura, R. Hoshino, Y. Hoshiba, V. I. Danilov and N. Kurita, “Effect of BrU on the Transition Between Wobble Gua-Thy and Tautomeric Gua-Thy Base-pairs: Ab Initio Molecular Orbital Calculations,” IOP Journal of Physics: Conference Series (JPCS), the Proceedings of the AP-IRC 2012, 2013.
- M. J. Frisch, et al., “Gaussian09,” Gaussian Inc., Wallingford, 2009.
- Y. Zhao and D. G. Truhlar, “The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-class Functionals and 12 Other Functionals,” Theoretical Chemical Accounts, Vol. 120, No. 1-3, 2008, pp. 215-241. doi:10.1007/s00214-007-0310-x
- C. Peng and H. B. Schlegel, “Combining Synchronous Transit and Quasi-Newton Methods to Find Transition States,” Israel Journal of Chemistry, Vol. 33, 1993, pp. 449-454.
- C. Gonzalez and H. B. Schlegel, “An Improved Algorithm for Reaction Path Following,” Journal of Chemical Physics, Vol. 90, No. 4, 1989, pp. 2154-2161. doi:10.1063/1.456010
- J. Tomasi, B. Mennucci and E. Cancès, “The IEF Version of the PCM Solvation Method: An Overview of a New Method Addressed to Study Molecular Solutes at the QM Ab Initio Level,” Journal of Molecular Structure (Theochem), Vol. 464, 1999, pp. 211-226.
- L. Gorb, Y. Podolyan, P. Dziekonski, W. A. Sokalski and J. Leszczynski, “Double-proton Transfer in Adenine-Thymine and Guanine-Cytosine Base Pairs. A Post HartreeFock Ab Initio Study,” Journal of American Chemical Society, Vol. 126, No. 32, 2004, pp. 10119-10129. doi:10.1021/ja049155n
NOTES
*Corresponding author.