Advances in Bioscience and Biotechnology
Vol.3 No.4(2012), Article ID:21867,3 pages DOI:10.4236/abb.2012.34046
SSDNA Cutter v0.0: A new in silico RFLP tool in C
![]()
1Department of Life Science and Biotechnology, Jadavpur University, Kolkata, India
2Astrophysics and Cosmology Research Unit, University of Kwazulu-Natal, Westville/Durban, South Africa
Email: #ratangachhui@yahoo.com
Received 6 June 2012; revised 25 June 2012; accepted 5 July 2012
Keywords: in silico RFLP; Restriction pattern segregation
ABSTRACT
Summary: SSDNA Cutter v0.0 is a new in silico RFLP tool written in C. It has a significant utility in studying population diversity as it has the capability of restriction digestion of a group of linear DNA sequences and then segregate the restriction patterns into separate groups as per similarity in the restriction maps. The software has an inbuilt database for 20 restriction enzymes and it is flexible so that the user can add up to 100 more restriction enzymes to the list. The interface is easy, simple and interactive which enables the user to obtain restriction pattern groups which are quite similar to Operational Taxonomic Units obtained from phylogenetic trees. Availability: The software and the source code are currently available from the authors on request without any cost. We intend to release it in a public domain soon. SSDNA Cutter v0.0 is licensed under the GNU General Public License.
1. INTRODUCTION
Restriction Fragment Length Polymorphism (RFLP) has been a very important tool in understanding populations from a diverse source of ecological niche [1]. But with availability of large numbers of sequenced DNA the requirement of performing the actual process is decreasing gradually [2,3]. Web servers like the NEBcutter V2.0. [4] is a very potent tool for such conceptual/in silico RFLP studies. However the lacuna remaining here is that a NEB cutter only provides the restriction map from a given sequence. Thus, if a large number of sequences are subjected to the conceptual RFLP the result would be in the form of equal number of restriction maps which needs to be grouped manually as very little bioinformatics tools are available for such purpose. The manual segregation into separate restriction patterns is both tedious and error prone.
In this paper we describe an in silico restriction digestion tool that digests linear DNA molecules and segregates them into separate groups according to the similarity of their restriction maps.
2. SOFTWARE DESCRIPTION
A new software SSDNA Cutter v0.0 has been developed in C. The program would digest the input DNA sequence with restriction enzymes as per the choice of the user. It could also distribute the DNA sequences as per the different restriction patterns. The software only requires the DNA sequence and it has an inbuilt database of 20 restriction enzymes: AciI, AluI, BfaI, BstUI, CviAII, CviQI, DpnI, DpnII, FatI, HaeIII, HhaI, HinfI, HpaII, MboI, MseI, MspI, PhoI, RsaI, Sau3AI and TaqI. The maximum number of DNA sequences that can be used in one go depends on the RAM of the computer. SSDNA Cutter is a logical program where it identifies the recognition sequence of the chosen enzymes on a particular DNA sequence by matching the base of the enzyme to every position in the DNA sequence from the beginning. Once a complete match is found it cleaves the DNA molecule at that position and continue to find the next match until it reaches the end of the DNA sequence. It then identifies the fragments as per length and also the cut sites and segregates the whole restriction map as similar or dissimilar to the restriction map of a second DNA sequence and group them together accordingly. In order to maintain accuracy we have also kept the option for the user to pre-determine the number of nucleotides (viz. 1, 2, 3, 4, 5, and 6 nucleotides) by which the DNA fragments may differ but still be considered as same. However this is not a default setting so user may take the raw data as well if they are confident that the DNA sequencing has been of high accuracy. We are slowly increasing the database and there is also provision for adding 100 more restriction enzymes and their recognition sequences as per requirement of the user. Any linear DNA sequence from any source can be used for in silico RFLP and the software is also adept to degenerate recognition sequences of various enzymes. The software can also be used to digest any linear DNA sequence, without opting for RFLP pattern segregation.
3. HOW TO USE SSDNA CUTTER V0.0
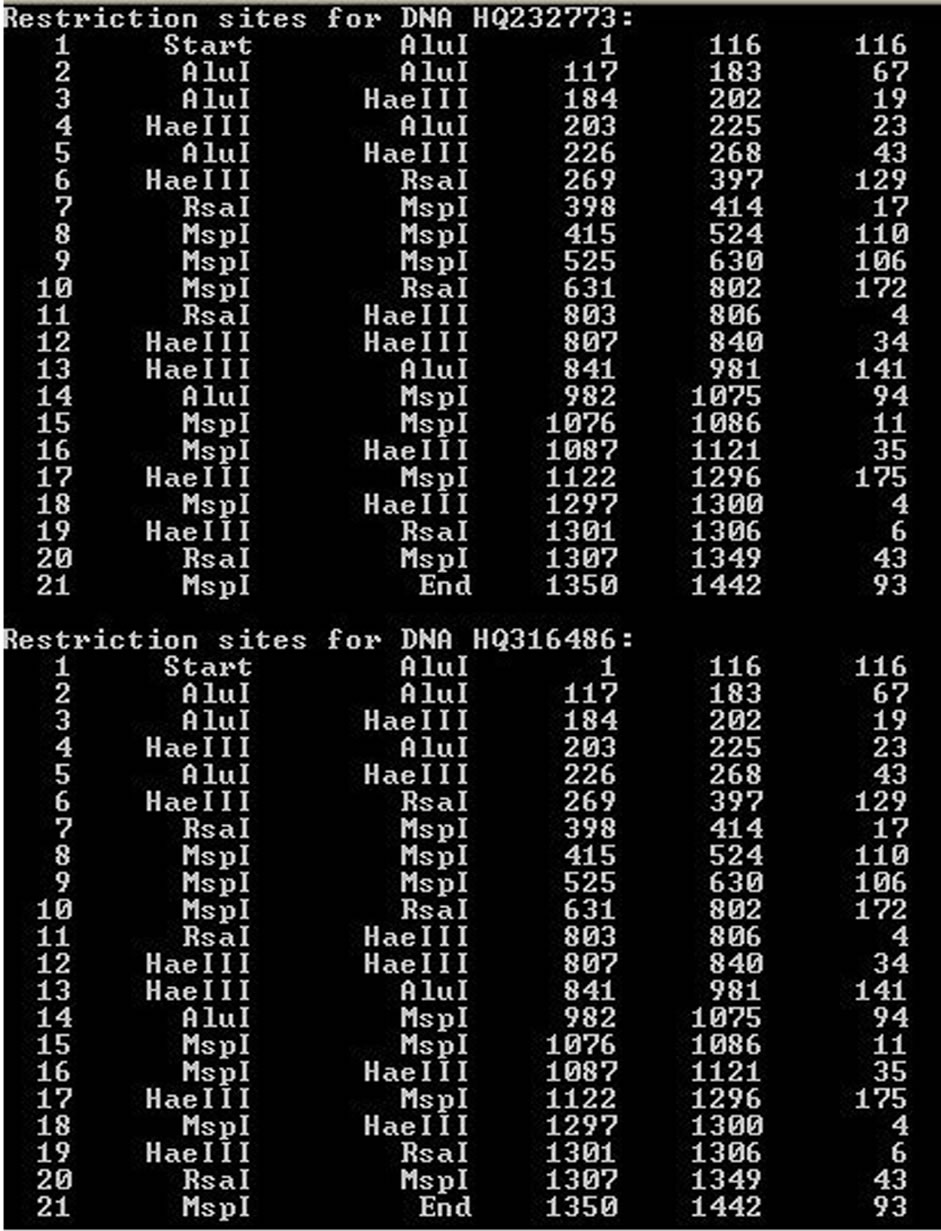
The program is currently available from the authors on request and we are planning to make it available on some public domain soon. In the package, we provide the main source code ssdnacutter.c along with a self executable (ssdnacutter.exe) that can be run in Windows platform. In any standard Linux platform (as well as in Windows having a C-compiler) one needs to compile the source code with any C compiler (i.e. cc, gcc etc.) to create the executable file. For example, “cc -o ssdnacutter.exe ssdnacutter.c” would create the executable ssdnacutter.exe. This executable can be run with the following command: ./ssdnacutter.exe. The interface of the software is an interactive dos command prompt. The user can choose whether they want to digest the DNA sequences and group them as well or they want to digest the DNA sequences only. In case of the former the program will then ask for the number of the DNA sequences to be digested. The software then shows the existing database of 20 restriction enzymes and will ask the user if they want any additional enzymes. If yes the software will then step by step ask for the number of enzymes the user would like to add, the name of each enzyme followed by the restriction recognition sequence and still followed by the restriction cut site. Once the details are complete the software would then ask the user to finally choose the restriction enzymes they want to use from the list. Finally it will ask for confirmation about the restriction enzymes chosen and then ask for the name of the DNA sequence. In case the user does not want to add any other enzyme to the inbuilt database the system will ask for the enzymes to be selected and perform as described in the preceding sentence. SSDNA Cutter does not require FASTA format as separate lines a have been delineated for the name of the sequence and the sequence itself. In this paper we have chosen 10 sequences from GenBank as an example to explain the utility of the software. The accession numbers of the sequences are: 1) HQ232773, 2) HQ316486, 3) HQ316487, 4) HQ232798, 5) HQ316490, 6) GQ867233, 7) HQ232762, 8) HQ232763, 9) HQ- 232765 and 10) GU198917. All 10 sequences have been cleaved with the following four enzymes: 1) AluI, 2) HaeIII, 3) MspI and 4) RsaI. The program will at first show the restriction map of the sequences and ask whether the user wants to delineate the number of bases by which each fragment may differ and still be considered as same. The final cut sites and grouped DNAs are shown on screen (Figures 1(a) and (b)), as well as they
 (a)
(a)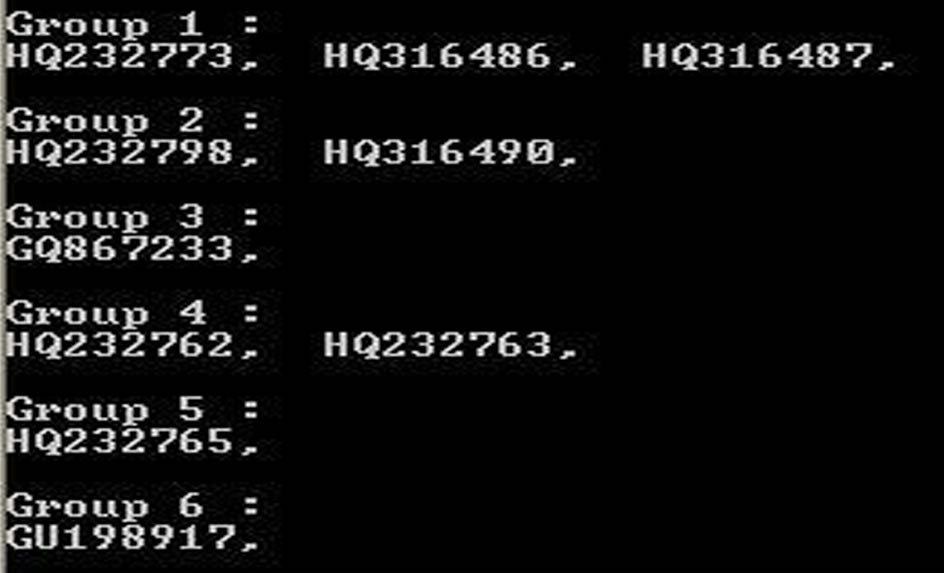 (b)
(b)
Figure 1. SSDNA Cutterv0.0 final interface. (a) Restriction map of two sequences HQ232773 and HQ316486; (b) Grouping of all 10 sequences after segregation according to restriction map similarity.
are stored in dna_cut_info_XX.txt and dna_group_info_ XX.txt in the same directory, where XX is the number of time one runs SSDNA Cutter v0.0. In this example it was found that the 10 sequences were grouped into 6 groups such that HQ232773, HQ316486 and HQ316487 falls in Group 1, HQ232798 and HQ316490 falls in Group 2, Groups 3, 5 and 6 having solitary representation as GQ867233, HQ232765 and GU198917 respectively and Group 4 has HQ232762 and HQ232763. In case the user wants to use the software to cleave the DNA sequence only, he will have to do the same as described above. Only the final data presentation will be in the form of the restriction maps only, no group delineation will be done.
4. DISCUSSION
The program has its utility in cases where a large number of sequences are being dealt with. As the software provides the user to cluster identical restriction patterns into distinct groups a preliminary segregation of the sequences and hence the population is obtained very easily. Moreover the tool has the ability to determine polymorphism in any gene and segregate members of different polymorphisms, thus having the potential of being a powerful tool in forensic studies as well. There is a report where a similar program has been described [2] but in that report the software mainly provides a similarity coefficient matrix derived from the restriction patterns and thus a certain amount of human endeavor is required before the elucidation into members of same restriction patterns is made. This program allows the user to obtain precise groups on the basis of identical restriction patterns in a very short time. This feature is the major novelty of the algorithm as such a feature to the best of our knowledge is not present in any other in silico RFLP algorithms available. However the current version does not address the problem of methylation so users must take this fact into account while choosing the enzymes.
5. ACKNOWLEDGEMENTS
The publication of the paper has been funded by Indian Council of Medical Research, Senior Research Fellowship contingency grant (sanction no. 80/713/11-ECD-I). The authors would like to specially thank Mr. Arnab Pramanik (JU) and Mr. Writachit Chakraborty (JU) for their helpful comments.
REFERENCES
- Ficker, M., Krastel, K., Orlicky S. and Edwards, E. (1999) Molecular characterization of a toluene-degrading methanogenic consortium. Applied Environmental Microbiology, 65, 5576-5585.
- Wei, W., Davis, R. E., Lee I. M. and Zhao, Y. (2007) Computer-simulated RFLP analysis of 16S rRNA genes: Identification of ten new phytoplasma groups. International Journal of Systematic and Evolutionary Microbiology, 57, 1855-1867. doi:10.1099/ijs.0.65000-0
- Wei, W., Lee, I.M., Davis, R.E., Suo X. and Zhao, Y. (2008) Automated RFLP pattern comparison and similarity coefficient calculation for rapid delineation of new and distinct phytoplasma 16Sr subgroup lineages. International Journal of Systematic and Evolutionary Microbiology, 58, 2368-2377. doi:10.1099/ijs.0.65868-0
- Vincze, T., Posfai, J. and Roberts, R.J. (2003) NEBcutter: A program to cleave DNA with restriction enzymes. Nucleic Acids Research, 31, 3688-3691. doi:10.1093/nar/gkg526
NOTES
*Both contributed equally.
#Corresponding author.