Journal of Environmental Protection
Vol. 3 No. 3 (2012) , Article ID: 17941 , 8 pages DOI:10.4236/jep.2012.33034
H2 Gas Charging of Zero-Valent Iron and TCE Degradation
![]()
Department of Earth and Environmental Sciences, University of Waterloo, Waterloo, Canada.
Email: *czhao@uwaterloo.ca
Received January 10th, 2012; revised February 11th, 2012; accepted February 29th, 2012
Keywords: Granular zero-valent iron; Hydrogen; TCE remediation; groundwater
ABSTRACT
Granular zero-valent iron (ZVI) has been widely used to construct permeable reactive barriers (PRB) for the in situ remediation of groundwater contaminated with halogenated hydrocarbons. In the anaerobic condition of most groundwater flow systems, iron undergoes corrosion by water and results in hydrogen gas generation. Several studies have shown that some of the hydrogen gas generated at the iron/water interface can diffuse into the iron lattice. Hydrogen gas also can be an electron donor for dechlorination of chlorinated compounds. In this study, the possibility of hydrogen gas bound in the lattice of ZVI playing a role in dehalogenation and improving the degradation efficiency of ZVI was evaluated. Two different granular irons were tested: one obtained from Quebec Metal Powders Ltd (QMP) and the other from Connelly-GPM. Ltd. For each type of iron, two samples were mixed with water and sealed in testing cells. Since the rate of hydrogen entry varies directly with the square root of the hydrogen pressure, one sample was maintained for several weeks under near-vacuum conditions to minimize the amount of hydrogen entering the iron lattice. The other sample was maintained for the same period at a hydrogen pressure of over 400 kPa to maximize the amount of hydrogen entering the iron lattice. The degradation abilities of the reacted ironsand the original iron materials were tested by running several sets of batch tests. The results of this study show little to no improvement of inorganic TCE degradation reactions due to the presence of lattice-stored hydrogen in iron material. This is probably due to the high energiesrequired to release hydrogen trapped in the iron lattice. However, there are certain chemical compounds that can promote hydrogen release from the iron lattice, and there may be bacteria that can utilize lattice-bound hydrogen to carry out dechlorination reactions.
1. Introduction
Iron metal (Fe0) or zero-valent iron (ZVI), as often referred to in the literature on remediation of chlorinated hydrocarbons in groundwater, acts as an electron donor to reductively degrade halogenated compounds. The electron transfer occurs at the metal surface. The degradation pathways of trichloroethene (TCE) by granular iron include reductive β-elimination(primary), hydrogenolysis(minor) and catalytic hydrogenation(minor) [1]. For most compounds, more than one reaction may occur, and the partially halogenated compounds may be produced from more than one pathway. Ethene and ethane as the main products, with minoramounts of chlorinated intermediates (formed via hydrogenolysis) and acetylene (formed via β-elimination) [1,2]. TCE also can be degraded directly by Fe2+ ion, which is produced from anaerobic corrosion of Fe0 [2-4]:
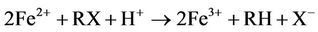
In the anaerobic condition of most groundwater flow systems, iron metal undergoes corrosion by water and results in hydrogen gas generation [5].

The hydrogen generated from iron corrosion at the iron/water interface can either diffuse into the water or enter the iron lattice. Surprisingly, the hydrogen that enters the iron diffuses faster through the iron lattice than it does through water [6]. A widely accepted mechanism for the entry and diffusion of hydrogen in iron is an electronchemical mechanism described by Carter and Cornish [7]. The mechanism involves the splitting of the H2 (g) molecule into separate hydrogen atoms or “adatoms” (Hads) at the gas/metal interface. The dissociation energy for hydrogen molecule is 432 kJ·mol–1, is smaller than the chemisorption energy, which varies between 500 and 600 kJ·mol–1 [7]. Thus the energy is available upon sorption for the hydrogen molecule to cleave into two separate atoms. These mobile hydrogen atoms can be adsorbed on the iron surface (Hads) as well as diffuse rapidly through the iron lattice. The diffused hydrogen atoms will become trapped in the iron at lattice defects sites (Habs). When the mono-atomic form hydrogen is trapped, it becomes immobile, and it is thought to remain as single atoms but can recombine to form hydrogen gas under certain conditions, such as when two H atoms encounter each other along a line defect [7]. The proportion of hydrogen produced from corrosion that enters the iron lattice depends on a number of factors such as the crystal structure of the iron and the presence and characteristics of surface oxide coatings, but most important is the ambient pressure of H2 (g). The entry rate is proportional to the square root of the pressure, i.e. “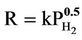 ” [5]. There is an analogous equilibrium constant expression
” [5]. There is an analogous equilibrium constant expression 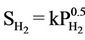 (Sieverts Law) that represents the solubility of in a variety of metals and metalalloys [8]. The
(Sieverts Law) that represents the solubility of in a variety of metals and metalalloys [8]. The 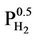 dependency in both expressions reflects the requirement that H2 molecules must split into individual H atoms in order to enter the iron lattice. Thus at 0 or low hydrogen pressure, there is little or no hydrogen taken up in the lattice and at high hydrogen pressure, a large proportion can be taken up.
dependency in both expressions reflects the requirement that H2 molecules must split into individual H atoms in order to enter the iron lattice. Thus at 0 or low hydrogen pressure, there is little or no hydrogen taken up in the lattice and at high hydrogen pressure, a large proportion can be taken up.
For hydrogen to leave the iron lattice (effusion), several steps are involved: 1) H atoms diffuses from trapping sites in the metal lattice to surface sites; 2) H atoms recombine to H2 molecules and 3) H2 moleculesthen desorbs from the surface into the contacting air or water phases. Therefore, all the parameters that influence the kinetics of hydrogen absorption and uptake in the lattice also affect the kinetics of hydrogen effusion and desorption from the iron surface [9].
The hydrogen accumulation at the metal surface can inhibit the continuation of corrosion [3]. Hydrogen can also act as electron donor in dehalogenation reactions (hydrogenolysis). Hydrogenolysis is one of the pathways of TCE degradation andcan occur in the presence of a catalyst such as Fe3O4 [3,10]. The surface of iron, its defects, or other solid phases present may also act as hydrogenolysis catalysts. The objective of this study is to determine if lattice-bound hydrogen can partake in dechlorination reactions independent of reaction with the iron surface; and if precharging granular iron with hydrogen by exposing the iron to high H2 (g). Pressures before emplacement in a permeable reactive barrier (PRB) can improve the dechlorination efficacy of the PRB.
2. Experimental
2.1. Materials
The 300,000 mg/L TCE stock solution used in the experiments was prepared using 99% TCE supplied by Fisher Scientific dissolved in HPLC grade methanol also supplied by Fisher Scientific. Two samples of granular iron: Quebec Metal Powders Ltd (QMP) and ConnellyGPM. Ltd. were used in this study. These irons were selected based on their ability to take up relatively high proportions of the hydrogen produced from anaerobic corrosion into their lattices at moderate H2 pressures [5].
The cells used to react the iron samples with H2 (g) were 250 cm3 stainless steel canisters with bolt-down caps outfitted with Vitor O-rings and Swagelok fittings to allow void space evacuation, water entry, and pressure measurements. The pressure measurements were made with Omega PX-302-015 0 - 500 kPa absolute vacuum pressure transducers. Pressure readings were logged at 30 s intervals using a 16-bit high-resolution data acquisition board and computer.
Sufficient water was added to the reaction cells to wet the iron granules but not to fully saturate them. The amounts of water and iron sample used in these tests are recorded in Table 1. The loaded cells were placed in a 25˚C water bath and evacuated by using a VacTorr Model 25 pump, with a 0.01 Pa vacuum rating. One sample of each iron was maintained for several weeks under nearvacuum conditions (QPL and CPL) to minimize the amount of hydrogen entering the iron lattice. The minimum pressure attainable with vacuum is 3.2 kPa, which is the water vapour pressure at 25˚C. Over time, the
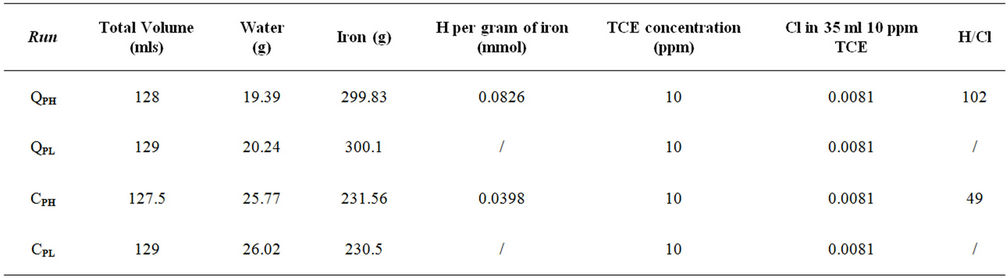
Table 1. The amounts of water and iron sample used in the experimental runs.
pressure in these cells would increase due to hydrogen production from iron corrosion and so the cells were periodically re-evacuated to maintain low pressure conditions. Over the same period, two other samples (QPH and CPH) were maintained at an over 400 kPa to promote hydrogen entry into the iron lattice. The cell pressure vs time plots for both irons and both pressure treatments are shown in Figures 1-2. The discontinuities in the pressure versus time curves for the low pressure runs (Figure 1) correspond to times when the cells were reevacuated. The pressure decreases in both high pressure cells over time (Figure 2) indicating that not only are the QMP and Connolly irons taking up hydrogen from the gas phase but that all the hydrogen produced from corrosion during this time has also entered the iron lattice. The sharp increase in pressure for the QMP iron at 100 h in Figure 2 was due to a repress urization of the cell with hydrogen gas.
In the batch tests, 10 g of high and low pressure treated iron were added to separate 40 ml glass hypovials. Then approximately 35 ml of 10 ppm TCE solution was added into each hypovial such that no headspace was present. The hypovials were sealed with aluminum crimp caps immediately upon filling. The vials were rotated

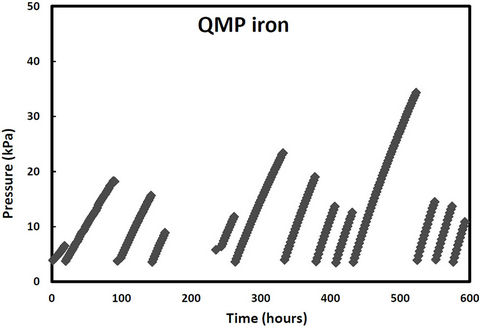
Figure 1. Cell pressure versus time for QMP and Connelly iron maintained at near vacuum conditions.
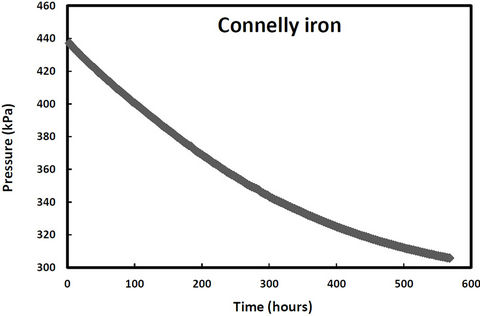
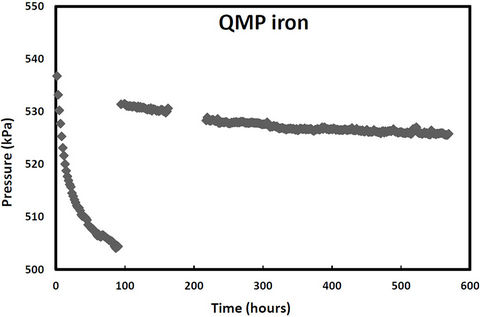
Figure 2. Cell pressure versus time for QMP and Connellyiron initially pressurized to over 500 kPa.
during the reaction period to promote mixing of the iron samples and TCE solutions. The solutions were sampled for analysis after 3 h, 6 h, 12 h, 24 h, 48 h, 4 d, and 6 d. At each sampling time, the reaction bottles were centrifuged to separate the solid particles from the solution before sampling the solution phase.
Based on the known corrosion rate for the QMP and Connolly irons and the H2 pressure decreases in the cells over time, the total amount of hydrogen uptake by the QPH and CPH samples was calculated and are recorded in Table 1. The amount of hydrogen available in each 10 g of hydrogen treated iron is many times more than required for complete degradation of the TCE used in the batch tests.
2.2. Characterization Techniques
The analyses for DCE isomers and VC were conducted using a Hewlett Packard 5890 Series II gas chromatograph (GC) equipped with an Hnu Photo ionization detector (PID) using an Hnu NSW-plot capillary column, while the TCE was analyzed using the same GC equipped with a Ni63 ECD detector.
Samples of the coatings of all the high and low pressure treated and original iron material for both types of iron were removed by gentle sieving through –120 mesh nylon screening. The mineralogical compositions of these coatings were analyzed by X-ray Diffraction (XRD) using an INEL powder diffractometer equipped with a position-sensitive detector covering 120˚ in 2θ and Cu Kα1 radiation was employed to characterize all products.
3. Results
3.1. After Connelly Iron TCE Degradation in Batch Test
The plot of TCE concentration against reaction time in the batch vials for high and low pressure treated and original Connelly iron are shown in Figure 3. Initially, all iron samples show a similar high efficiency for TCE degradation. However, after 12 h, the degradation efficiency of CPL and the original iron reduced slightly, while the CPH iron maintained a rapid reaction rate. Figure 7 shows that more than 99% of added TCE degraded in the CPH vials within 2 d, however it took 4 d and 6 d for the same amount of TCE to degrade for the CPL and original irons, respectively.
3.2. Connelly Iron TCE Degradation Products
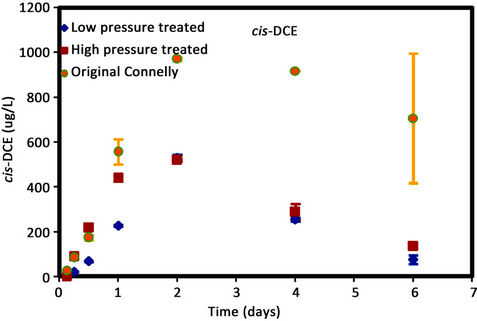
As shown in Figure 4, higher concentrations of cis-DCE
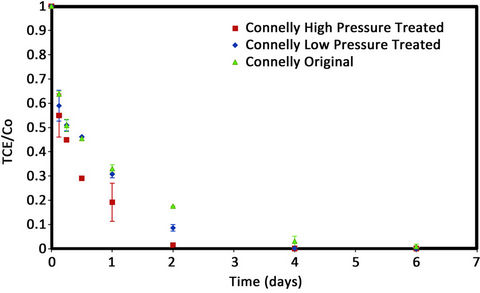
Figure 3. TCE degradation plot for Connelly iron.
 (a)
(a)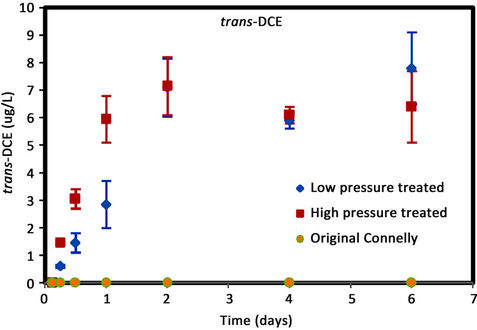 (b)
(b)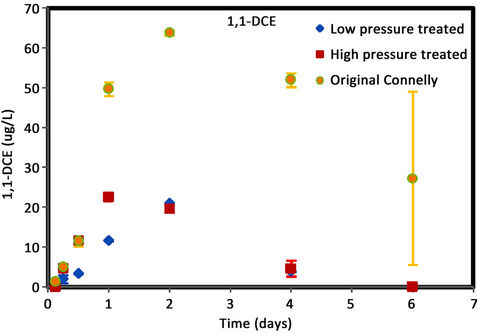 (c)
(c)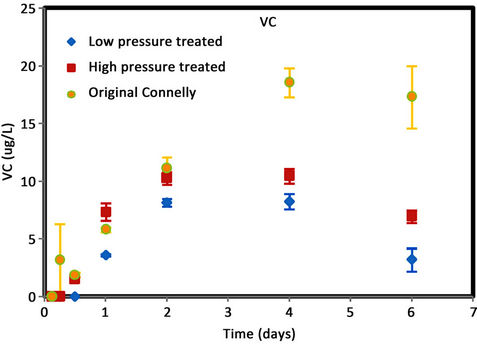 (d)
(d)
Figure 4. (a) cis-DCE concentration plot for Connelly iron; (b) trans-DCE concentration plot for Connelly iron; (c) 1,1-DCE concentration plot for Connelly iron; (d) VC concentration plot for Connelly iron.
were generated for the original iron. Its concentration reduced slowly and more than 700 ug/L was left after 6 d. No trans-DCE was generated for the original iron. However, similar concentrations were produced for the CPL and CPH irons. Figure 4 also shows the original iron produced three times the concentration of 1,1-DCE (60 ug/L) than either the CPL or CPH irons (20 ug/L). The results indicate a greater accumulation of VC after 4 d for the original iron compared to the CPL and CPH irons.
3.3. QMP Iron TCE Degradation in Batch Test
Figures 5 and 6 show no significant difference in TCE degradation rate for all QMP samples indicating no improvement in TCE degradation with hydrogen pre-treatment for QMP iron.
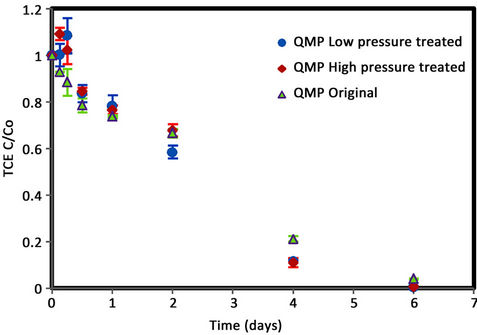
Figure 5. TCE degradation plot for QMP iron.
 (a)
(a)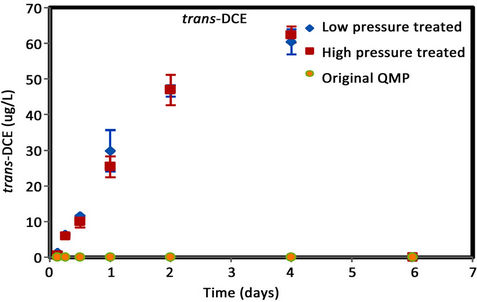 (b)
(b)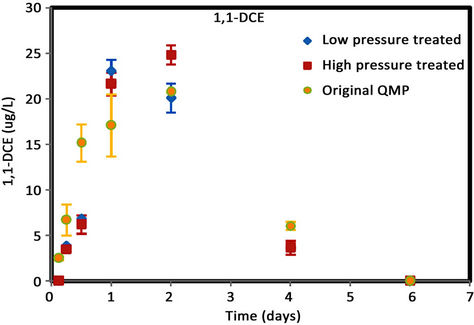 (c)
(c)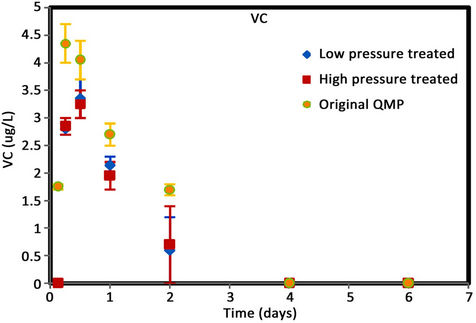 (d)
(d)
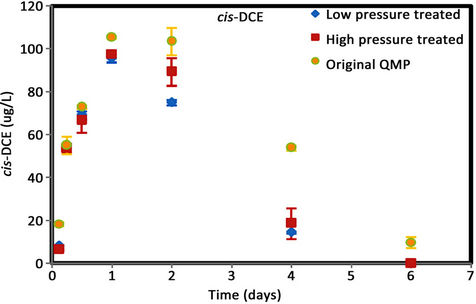
Figure 6. (a) cis-DCE concentration plot for QMP iron; (b) trans-DCE concentration plot for QMP iron; (c) 1,1-DCEconcentration plot for QMP iron; (d) VC concentration plot for QMP iron.
3.4. QMP Iron TCE Degradation Products
Similar to Connelly iron, cis-DCE is the main product of TCE degradation for QMP iron samples (Figure 6). Both H2-treated and original irons showed similar amounts of degradation products generated. The original iron had more cis-DCE generation than either of the reacted irons. cis-DCE decreased more gradually for the original iron compared to the reacted irons. Similar to the Connelly iron, there is no detectable trans-DCE generated for the original iron material, while both the high and low pressure treated samples showed similar concentrations and concentration variations with time. The original iron showed slightly more VC generation and a more gradual concentration decrease with time than either of the reacted irons. After 4 d, VC compound in all vials was below detection.
3.5. Surface Coating Analysis of Iron Samples
The X-ray diffraction spectra recorded for sieved samples of all QMP and Connelly irons are shown in Figure 7. Background intensity is high and there are very few peaks indicating the presence of mostly amorphous material. There is no substantial difference in the XRD results for the high or hydrogen gas free treated material compared to the original material for either QMP or Connelly iron. The prominent peaks indicate the principal components of the coatings to be magnetite or maghemite, along with some iron metal.
4. Discussion
The nature of the metal/metal oxide surface is one of the factors that will control the style of the TCE degradation reaction because of the electron transfer at the iron surface that is required. The first step in the reaction of Fe0 and a TCE solution is TCE sorption onto the iron surface [11]. From the XRD analysis of the surface coatings, the iron surfaces are covered by iron oxides, Fe3O4 (magnetite)/Fe2O3 (maghemite). Water-reacted irons presumably have more iron oxides on the surface than the original irons due to the long term anaerobic corrosion reaction with the water in the treatment cells. A previous study found that the passive film on the iron is an inner layer of Fe3O4 (magnetite) covered by an outer of γ-Fe2O3 (maghemite) [12]. Magnetite is electronically conductive and allows for charge transfer. Thus it can allow reductive dechlorination to proceed [13]. However, the degradation will proceed at a slower rate due to the slower charge transfer rate, compared to a bare metal surface [14]. Since hydrogen can be adsorbed on the magnetite, catalytic hydrogenation is possible at the magnetite/iron surface [15]. In contrast, other corrosion products like Fe2O3, FeOOH are semi-conductors or insulators. Thus they can passivate the iron surface and inhibit the electron transfer and catalytic hydrogenation as well [10]. In order to make commercial iron materials chemically active towards both water and organic contaminants, the breakdown of the protective oxide film, followed by the autoreduction of Fe2O3 is a required step [12]. Qin et al., [16]
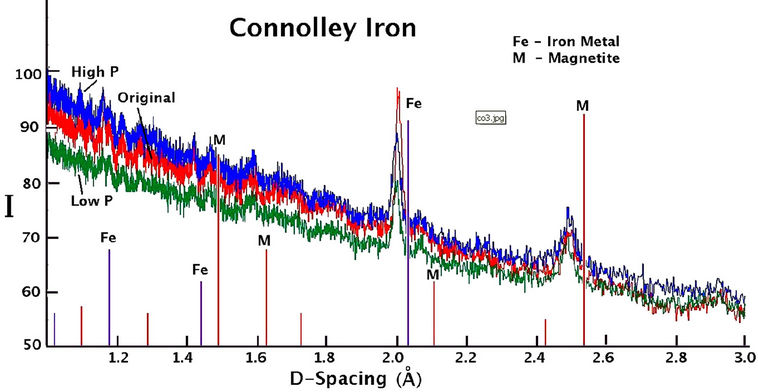
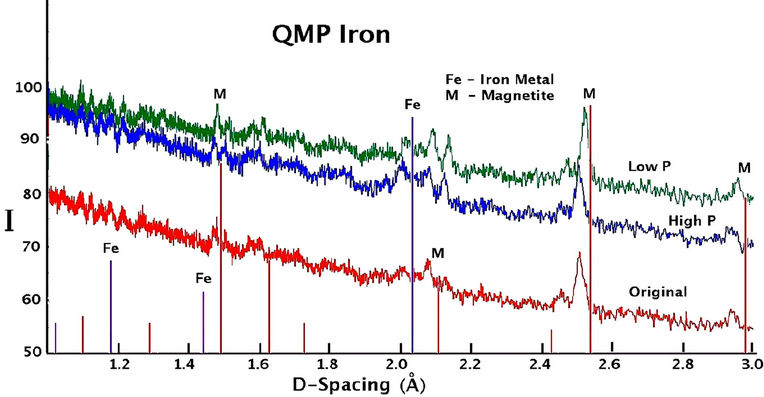
Figure 7. X-ray diffraction spectra of iron coatings collected from the original and reacted QMP and Connelly iron samples.
described that cracks and stresses create ideal conditions for the auto-reduction processes, which can open up the porous structure of the oxide. These auto-reduction reactions can be written as
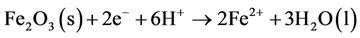
The cracked structure increases the possibility of local separation of anodic and cathodic sites, which would promote localized corrosion and degradation of organic contaminants [12].
Hydrogen can decrease the charge transfer resistance of the passive film and also decrease the passive film thickness, both of which result in an increase in the strength of the electric field across the film [17]. It is also observed that hydrogen increases the number of pitting sites on iron surfaces significantly and promotes the active pitting sites to stable pits [18]. Matheson and Tratnyek [3] claim that defects/pits present on the surface may act as catalysts for direct hydrogenation. The high pressure H2- treated iron should be more pitted than the original iron due to the long period of hydrogen sorption and entry into the iron lattice during its exposure to high pressures of hydrogen gas. As a result more active sites should be exposed to the TCE solution, thus promoting direct hydrogenation.
The hydrogen trap sites in the iron lattice can be classified by their physical and crystallographic properties. The possible trap sites include lattice vacancies and dislocations, grain boundaries or crystal interfaces, alloying element sites and microvoids [19]. Distinct energies are required to evolve hydrogen from each type of trapping site. The required activation energies increase in the order: crystal or grain boundary, lattice vacancy or dislocation, and microvoid. The evolution rate of trapped hydrogen increases with temperature and shows peaks at 385 K for hydrogen trapped in grain boundaries, and 488 K for dislocations and 578 K for microvoids [20]. The activation energies for evolution of trapped hydrogen for each peak temperature found to be 17.2 KJ/mol, 26.8 KJ/mol, and 35.2 KJ/mol, in grain boundaries, dislocations, and microvoids, respectively.
The temperatures of our tests (298 K) are too low for significant amounts of hydrogen to escape from various trapping sites in the iron lattice. As a result, the flux of hydrogen atoms or molecules to the surface is likely too low to contribute significantly to the TCE dechlorination reactions. This is presumably the reason for the similar performance of the high pressure H2-treated and hydrogen free irons in degrading TCE and its daughter products. There is a small improvement in TCE degradation performance observed for the hydrogen treated Connelly iron. This may suggest that a small amount of hydrogen can escape from lower energy trapping sites in this material and contribute to the TCE degradation process by either hydrogenation or hydrogenolysis reactions. In contrast, the exsolution of trapped hydrogen from QMP iron seems to be more difficult than from Connelly iron, and this may be due to a difference in iron surface morphology between these two irons. Hydrogen can be released during corrosion. However, the iron corrosion rate measured for both irons in this study are less than 1.0 mmol·kg–1·d–1 [5], which is too slow to account for any significant proportion of the TCE degradation observed in this study.
Another possible way to promote the release of hydrogen trapped in the iron lattice is through the addition of certain chemical species that have a higher binding energy to iron metal than does hydrogen. Such species can block sites for hydrogen adsorption and dissociation thus inhibiting hydrogen entry into the iron lattice [21]. A previous study reported that ethylenic type molecules can absorb on adjacent sites to sorbed hydrogen atoms and as a result block hydrogen movement and permeation through the lattice [22] (Roberson, 1980). Frandsen and Marcus [23] demonstrated that O2, CO2, and NO can prevent hydrogen entry into metal interaction. The extent, kinetics and bonding energies dissociative hydrogen adsorption on iron can be reduced by the presence of some electronegative elements such as carbon (C), oxygen (O), and the sulfur (S) on the iron surface. This is due to their ability to reduce the binding energy of hydrogen to the iron surface [24]. Another possibility is to combine the high H2 pressure reacted Fe0 with autotrophic hydrogenbacteria to see if the bacteria can utilize the hydrogen trapped in the iron lattice in TCE degradation reactions. Previous works [24-26] have found that the role of hydrogen as an electron donor is recognized as key factor governing the biologically-mediated dechlorination of chlorinated compounds in anaerobic systems. Several studies have examined the possibility of combining Fe0 and autotrophic hydrogen-bacteria under a hydrogen supplied environment to degrade chlorinated hydrocarbons [27-29]. The reaction of Fe0 with water is a hydrogen source and the cathodic hydrogen is utilized as an electron donor for respiratory dechlorination.
REFERENCES
- W. A. Arnold and A. L. Roberts, “Pathways and Kinetics of Chlorinated Ethylene and Chlorinated Acetylene Reaction with Fe(0) Particles,” Environmental Science & Technology, Vol. 34, No. 9, 2000, pp. 1794-1805. doi:10.1021/es990884q
- W. S. Orth and R. W. Gillham, “Dechlorination of Trichloroethene in Aqueous Solution Using Fe0,” Environmental Science & Technology, Vol. 30, No. 1, 1996, pp. 66-71. doi:10.1021/es950053u
- L. J. Matheson and P. G. Tratnyek, “Reductive Dehalogenation of Chlorinatedmethanes Byiron Metal,” Environmental Science & Technology, Vol. 28, No. 12, 1994, pp. 2045-2053. doi:10.1021/es00061a012
- D. R. Burris, T. J. Campell and V. S. Manoranjan, “Sorption of Trichloroethylene and Tetrachloroethylene in a Batch Reactive Metallic Iron-Water System,” Environmental Science & Technology, Vol. 29, No. 11, 1995, pp. 2850-2855. doi:10.1021/es00011a022
- E. J. Reardon, “Zerovalent Irons: Styles of Corrosion and Inorganic Control on HydrogenPressure Buidup,” Environmental Science & Technology, Vol. 39, No. 18, 2005, pp. 7311-7317. doi:10.1021/es050507f
- P. Kedzierzawski, R. A. Oriani, J. P. Hirth and M. Smialowski, “Hydrogen Trapping in Iron and Iron Alloys,” Acta Metallurgicaet Materialia, Vol. 39, 1985, pp. 271- 287.
- T. J. Carter and L. A. Cornish, “Hydrogen in Metals,” Engineering Failure Analysis, Vol. 8, No. 2, 2001, pp. 113-121. doi:10.1016/S1350-6307(99)00040-0
- A. Sieverts, “Absorption of Gases by Metals,” Zeitschrift für Metallkunde, Vol. 21, 1929, pp. 37-46.
- G. Schmitt, B. Sadlowsky and J. Noga, “Inhibition of Hydrogen Effusion from Steel—An Overlooked and Underestimated Phenomenon,” National Association for Corrosion Engineers, Orlando, 2000, p. 466.
- K. Ritter, M. S. Odziemkowski and R. W. Gillham, “An in Situ Study of the Role of Surface Films on Granular Iron in the Permeable Iron Wall Technology,” Contaminant Hydrology, Vol. 55, No. 1-2, 2002, pp. 87-111. doi:10.1016/S0169-7722(01)00187-5
- J. Gotpagar, S. Lyuksyutov, R. Cohn. E. Grulke and D. Bhattacharyya, “Reductive Dehalogenation of Trichloroethylene with Zero-Valent Iron: Surface Profiling Microscopy and Rate Enhancement Studies,” Langmuir, Vol. 15, No. 24, 1999, pp. 8412-8420. doi:10.1021/la990325x
- M. S. Odziemkowski and R. P. Simpraga, “Distribution of Oxides on Iron Materials Used for Remediation of Organic Groundwater Contaminants—Implications for Hydrogen Evolution Reactions,” Canadian Journal of Chemistry, Vol. 82, No. 10, 2004, pp. 1495-1506. doi:10.1139/v04-120
- M. L. Bonin, M. S. Odziemkowski and R. W. Gillham, “Influence of Chlorinated Solvents on Polarization and Corrosion Behaviour of Iron in Borate Buffer,” Corrosion Science, Vol. 40, No. 8, 1998, pp. 1391-1409. doi:10.1016/S0010-938X(98)00042-0
- J. W. Schultze, R. P. Frankenthal and J. Kruger, “Passivity of Metals,” The Electrochemical Society, 1978, p. 82.
- P. Marcus and E. Protopopoff, “Thermodynamic Predictions of the Stability of Hydrogen and Oxygen Species Electroadsorbed on Transition Metals,” Electrochemical Surface Science of Hydrogen Adsorption, Montreal, 1997, pp. 211-224.
- Z. Qin, B. Demko, J. Noël, D. Shoesmith, F. King, R. Worthingham and K. Keith, “Localized Dissolution of Millscale-Covered Pipeline Steel Surfaces,” Corrosion, Vol. 60, No. 10, 2004, pp. 906-914. doi:10.5006/1.3287824
- J. G. Yu, J. L. Luo and P. R. Norton, “Electrchemical Investigation of the Effects of Hydrogen on the Stability of Passive Film on Iron,” Electrochimica Acta, Vol. 47, No. 10, 2002, pp. 1527-1536. doi:10.1016/S0013-4686(01)00882-9
- J. G. Yu, J. L. Luo and P. R. Norton, “Investigation of Hydrogen Induced Pitting Active Sites,” Electrochimica Acta, Vol. 47, No. 25, 2002, pp. 4019-4025. doi:10.1016/S0013-4686(02)00410-3
- G. M. Pressouyre, “A Classification of Hydrogen Traps in Steel,” Metallurgical Transactions A, Vol. 10, No. 10, 1979, pp. 1571-1573. doi:10.1007/BF02812023
- W. Y. Choo and J. Y. Lee, “Thermal Analysis of Trapped Hydrogen in Pure Iron,” Metallurgical and Materials Transactions A, Vol. 13A, No. 2, 1982, pp. 135-140.
- L. Raymond, “Hydrogen Embrittlement: Prevention and Control,” Chapter 6, ASTM Subcommittee, LA, 1988. doi:10.1520/STP962-EB
- W. M. Roberson, “Organic Inhibitors for Hydrogen Permeation in Iron,” Metallurgical Transactions A, Vol. 11, No. 7, 1980, pp. 1207-1212. doi:10.1007/BF02668144
- J. D. Frandsen and H. L. Marcus, “Environmentally Assisted Fatigue Crack Propagation in Steel,” Metallurgical Transactions A, Vol. 8, No. 2, 1977, pp. 265-272. doi:10.1007/BF02661639
- X. Maymo-Gatell, V. Tandoi, J. M. Gossett and S. H. Zinder, “Characterization of an H2-Utilizing Enrichment Culture That Reductively Dechlorinatestetrachloroethene to Vinyl Chloride and Ethene in the Absence of Methanogenesis and Acetogenesis,” Applied and Environmental Microbiology, Vol. 61, No. 11, 1995, pp. 3928-3933.
- J. M. Gossett and S. H. Zinder, “Microbiological Aspects Relevant to Natural Attenuation of Chlorinated Ethenes,” Symposium on Natural Attenuation of Chlorinated Organics in Ground Water, Washington DC, 1997, pp. 12- 14.
- C. R. Smatlak, J. M. Gossett and S. H. Zinder, “Comparative Kinetics of Hydrogen Utilization for Reductive Dechlorination of Tetrachloroethene and Methanogenesis in an Anaerobic Enrichment Culture,” Environmental Science & Technology, Vol. 30, No. 9, 1996, pp. 2850- 2858. doi:10.1021/es9602455
- L. J. Weathers, G. F. Parkin and P. J. Alvarez, “Utilization of Cathodic Hydrogen as Electron Donor for Chloroform Cometabolism by a Mixed, Methanogenic Culture,” Environmental Science & Technology, Vol. 31, No. 3, 1997, pp. 880-885. doi:10.1021/es960582d
- T. Lee, T. Tokunaga, A. Suyama and K. Furukawa, “Efficient Dechlorination of Tetrachloroethlene in Soil Slurry by Combined Use of an Anaerobic Desulfitobacterium sp. Strain Y-51 and Zero-Valent Iron,” Bioscience and Bioengineering, Vol. 92, No. 5, 2001, pp. 453-458.
- S. M. Wang and S. K. Tseng, “Dechlorination of Trichloroethylene by Immobilized Autotrophic HydrogenBacteria and Zero-Valent Iron,” Bioscience and Bioengineering, Vol. 107, No. 3, 2009, pp. 287-292. doi:10.1016/j.jbiosc.2008.11.010
NOTES
*Corresponding author.