World Journal of Nano Science and Engineering
Vol.3 No.3(2013), Article ID:36676,10 pages DOI:10.4236/wjnse.2013.33010
Controlled Release of Diclofenac Sodium from Silica-Chitosan Composites
Chuiko Institute of Surface Chemistry, National Academy of Sciences of Ukraine, Kyiv, Ukraine
Email: kozakevych@gmail.com
Copyright © 2013 R. B. Kozakevych et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received July 8, 2013; revised August 12, 2013; accepted August 19, 2013
Keywords: Drug Incorporation; Diclofenac Sodium; Silica-Chitosan Composites
ABSTRACT
The release profiles of acidic form of diclofenac sodium adsorbed on mesoporous silicas (Silochrom and two samples of spherical silicas) were compared with the dissolution characteristics of the pure drug. Desorption of diclofenac sodium from impregnated silicas with various surface liophilicity and composites of silica with chitosan have been studied using rotating basket method in phosphate buffer, pH 6.8. Sedimentations of sodium diclofenac via adsorption and impregnation from alcohol solution on fumed silica and modified silicas with grafted aminopropyl and trimethylsilyl groups were carried out. Polymer-containing composites have been prepared by capsulation of silica particles with impregnated diclofenac sodium by protonated and deprotonated forms of chitosan. Effect of the silica surface nature on the active substance release rate was ascertained. Significant prolongation of diclofenac sodium release was detected in the case of application of hydrophobic silica as a carrier and protonated chitosan as a polymeric shell.
1. Introduction
Diclofenac sodium (as well as diclofenac) is a well-known non-steroidal anti-inflammatory drug (NSAID) with pronounced analgesic and antipyretic efficacy. It is widely used in the long-term treatment of degenerative joint diseases of rheumatoid and non-rheumatoid origin such as rheumatoid arthritis, osteoarthritis and ankylosing spondylitis [1].
Due to its clinical effect and short biological half-life [2], diclofenac-based pharmaceuticals are ideal candidates for prolonged release formulations. However, to provide an optimal biocompatibility (stable concentration during whole absorption period), a rapid release should be achieved at the first stage of drug uptake with following desorption prolongation. Thus, the development of preparations based on diclofenac (or diclofenac sodium) is focused on both an increasing and decreasing of the medicine release rate.
Despite the pharmaceutical effectiveness, a novel slow release drugs based on diclofenac or diclofenac sodium are insufficiently developed. Diclofenac sodium is an acidic compound (pKa about 4) with very low aqueous solubility [3]. Its solubility depends on the medium pH. Compound is practically insoluble in hydrochloric acid at pH 1.1, whereas in phosphate buffer at pH 6.8 the drug has higher dissolution rate [4-6]. Maybe due to this reason, there is insignificant number of scientific publications devoted to such system. However, a study of composites based on diclofenac and its derivatives has considerable practical significance. Controlled-release systems provide an increase in overall efficacy of the drug (i.e., therapeutic efficiency) due to maintaining constant drug concentration in a body within the optimum therapeutic range and under the toxicity threshold.
While it is well accepted that diclofenac sodium intake in doses of about 50 - 150 mg per day when administrated orally is sufficient to relieve acute pain [7], considerable controversy exists about the effects of so-called megadoses, i.e. “gram” doses.
An increase of diclofenac concentration [8] leads to the drug toxicity increase: non-controlled diclofenac administration can lead to severe damage or, sometimes cause lethal toxicity [9], which limits the therapeutic usage of the drug. Such diversion in biological action of the diclofenac (i.e. diclofenac sodium) can be overcome via development of sustainable release system to achieve constant release of active agent with time [10]. One of strategies to achieve such system exploits a change of the physicochemical properties of carrier in the drug delivery systems.
A target properties of drug carriers as it was shown elsewhere [11] could be achieved by change of matrix porosity in order to predict diffusion drug release rate [12]. Song et al. [13] found that ibuprofen and bovine serum albumin loading and release rate could be welladjusted by functionalization of mesoporous materials with organic matters. For instance, the aminopropylmodified SBA-15 shows larger drug loading capacity than that of pristine SBA-15 materials and drug (famotidine) release rate could be delayed significantly by grafting of the trimethylsilyl groups on the carrier (MCM-41) surface [14]. Silica particles for drug delivery systems with different pores size were obtained by applying a complex approach including both a sol-gel technology and a controlled polymerization [15-17]. Different initiators of the sol-gel process and a spray-drying technology were applied for producing of such system [18,19]. The control of the drug release rate in silica matrices can be achieved via adjustable limitation of the drug dissolution and diffusion within matrices.
Poor activity of convenient drug delivery systems in vivo is most often attributed to their low “bioavailability” —an extent and a rate of drugs penetration to target tissue which provides the desired effect [19]. This estimation is largely depends on the method of introducing, physiology of organ and metabolism [20].
Controlled delivery systems provide an alternative approach to regulate the bioavailability of therapeutic agents. In polymer-based controlled drug delivery systems, an active therapeutic is incorporated into polymeric network structure in a way that the drug releases from the material in a predefined manner [21,22]. Utilization of the natural polymers i.e. chitosan as drug delivery carriers [23,24] has attracted a great attention due to their biocompatibility, biodegradability and low toxicity that are well documented [25,26]. Chitin-silica represents a potentially useful super disintegrant in many pharmaceutical applications. These composites offer good compressibility and compactability characteristics that may allow it to function as a pharmaceutical filler [27].
This study aimed at the development of a more efficient slow-release system for diclofenac sodium attempting to explore the silica matrices in this field. In this study, a simple and novel experimental method formation of chitosan spheres was developed to simplify preparation of composite matrices for drug delivery. The present study provides a simple method to incorporate NSAID into various silicas followed by a systematic laboratory evaluation of the resultant compositions.
2. Materials and Methods
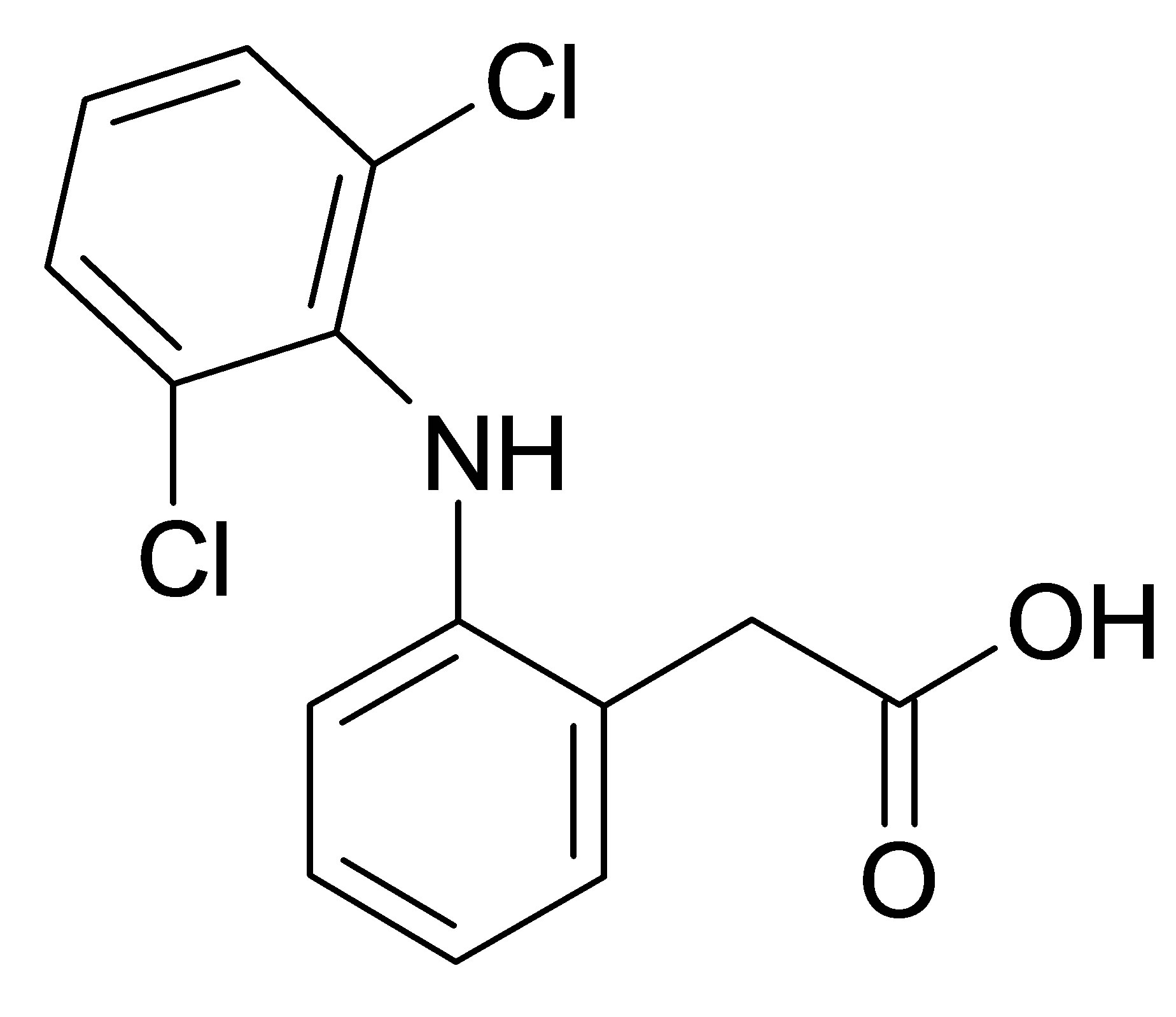
Active ingredient of diclofenac sodium (99.5%wt, Vaishali Pharmaceuticals, India) was used as received. Diclofenac sodium is 2-[(2,6-dichlorophenyl)amino] benzeneacetic acid monosodium salt, which has an empirical formula of C14H10Cl2NO2Na (Figure 1(a)). It is a white to yellow crystalline slightly hygroscopic powder, sparingly soluble in water and freely soluble in alcohols. It melts at about 280˚C with decomposition. Diclofenac is 2-[(2,6-dichlorophenyl)amino] benzeneacetic acid (Figure 1(b)) with pKa 3.80 at 25˚C and very low aqueous solubility in the unionized form.
Acidic form of diclofenac was prepared in laboratory by dissolution of sodium diclofenac in acetic acid with followed salting-out in distilled water. Precipitate of diclofenac was filtered and dried at 80˚C. Nanosized nonporous hydrophilic (Degussa) and hydrophobic fumed silicas with specific surface areas of 300 m2/g were used as carriers. Hydrophobic silica (methylated silica) was obtained by gas-phase modification with trimethylchlorosilane [28].
Porous spheres were obtained via spray-drying technique from highly dispersed non-porous silica particles (5 and 40 nm) and characterized by low temperature nitrogen adsorption/desorption.
Amine-functionalized silica samples (aminated silica) were prepared via treatment of the pristine fumed silica with 3-aminopropyltriethoxysilane (APTES, 99%, Fluka) according to procedure as follows: pristine silica was dehydrated at 200˚C for 2 h and then refluxed for 2 h at 60˚C with APTES in toluene (99.5 %wt, Aldrich) at the ratio 1 g SiO2/0.2 g APTES/10 mL toluene. Obtained product was filtered, washed subsequently with toluene, acetone and dried at 80˚C for 2 h in air. Content of the chemisorbed amino groups was determined using spectrophotometric method (UV spectrophotometer Specord М-40, Analytik-Jena AG, Germany, absorption wavelength 400 nm) [29,30] after reaction of the surface NH2- groups with the salicylic aldehyde. Quantity of the grafted amino groups was calculated as 0.67 mmol per 1 g of silica.
Chitosan of low viscosity and deacetylation degree 70 % (Fluka) was used without addition treatment.
Drug-loading procedure. Loading of the diclofenac into the porous silica spheres was performed by mixing of 1 g of calcined silica spheres with 1.5 mL of acetic
 (a)
(a)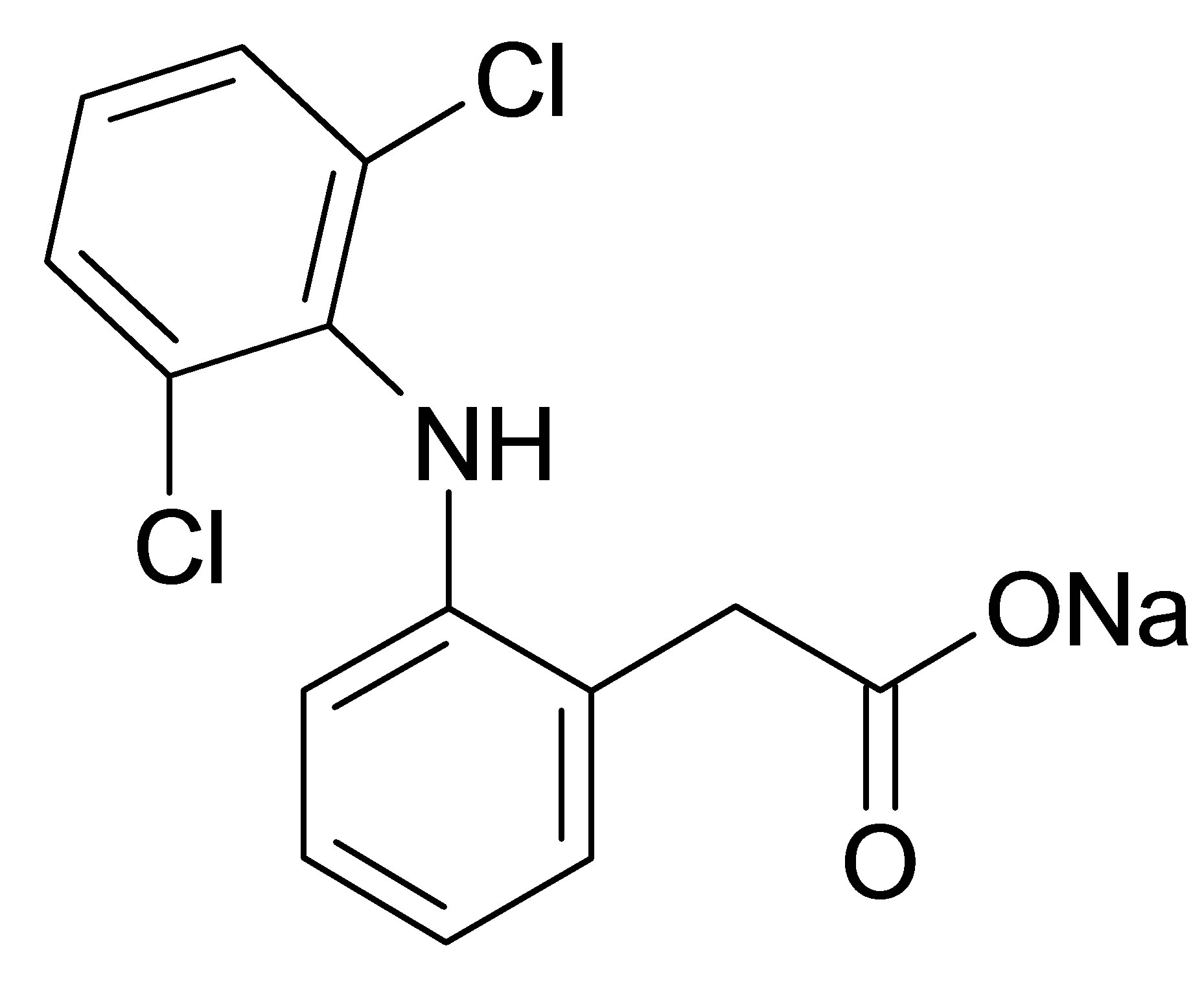 (b)
(b)
Figure 1. Structural formula of diclofenac sodium (а) and diclofenac (b).
acid solution (33.0 mg/mL). After impregnation for 24 h the drug-loaded mesoporous silica spheres were filtered, and dried at 80˚C for 2 h.
Preparation of chitosan composites. Two methods of composites preparation from chitosan were applied in this study: cross-linking of the polymeric matrices with glutaraldehyde [31] and using novel method presented in this paper.
Control samples were obtained according to first method where 3 wt% solution of chitosan was prepared by dissolving in 4 wt% aqueous acetic acid in glass beaker using magnetic stirrer, then sodium silicate (0.5 g) was added to this solution to prevent spheres aggregation. The spheres were formed using drop-by-drop technique; in brief: chitosan solution (3 wt%) was added dropwise through capillaries into the precipitation bath containing 0.1 M NaOH solution; a drop morphology was controlled by capillary diameter and regime of a peristaltic pump. Dispersion of chitosan spheres in alkaline solution was continuously stirred for 1 h at 25˚C to deprotonate chitosan [32]. Then polymeric spheres were thoroughly rinsed in DI water and immersed for 24 h in 200 mL of the aqueous solution of glutaraldehyde (2.5 wt%) for polymer cross-linking. Finally, polymeric spheres were washed with DI water and dried.
Novel method which proposed in this study simplifies the chitosan spheres preparation. The overall method consists in the following steps: chitosan solution (3 wt% in 4 wt% aqueous acetic acid) was introduced dropwise (through capillaries using peristaltic pump) into precipitation bath with layer of highly hydrophobic nanosized silica (methylated silica). The layer depth of the highly dispersed nanosized silica was designed in a way to prevent chitosan drops coalescence. Silica/chitosan drops were dried at 80˚C for 4 h. At these experimental conditions chitosan is protonated and sustained in the linear conformation of macromolecules. Moreover the proposed approach simplifies the synthetic procedure.
In vitro diclofenac drug release test (dissolution test) has been performed according to the requirements of the State Pharmacopoeia of Ukraine [33] using the rotating basket method [34]. Phosphate buffer with pH 6.8 has been used as a dissolution medium; for each formulation dissolution test has been conducted on 50 mg of active component (diclofenac) using 1000 mL of dissolution medium. Distance between inside bottom of dissolution vessel and basket is maintained at 25 ± 2 mm. The release experiment was performed at 37˚C under stirring rate of 100 r/min. Samples for measurement of drug release were taken in time intervals (shown in Experimental). Samples of 2 mL were taken and each sampling was followed by addition of 2 mL of fresh medium into dissolution vessels. Prior to determination of drug concentration, samples were filtered. Determination of drug concentration has been carried out using UV spectrophotometer Specord М-40 (absorption wavelength 276 nm).
Surface area and porosity were determined from nitrogen adsorption-desorption isotherms obtained at −196˚C using Micromeritics ASAP-2000 analyzer (USA). In all cases, 50 - 70 mg of material was degassed at 60˚C for 24 h under a vacuum (residual pressure <10−5 Torr) before nitrogen adsorption.
Infrared (FT-IR) spectra were collected using a Thermo Nicolet Nexus 450 from 4000 to 500 cm−1 and resolution of 4 cm−1, using KBr pellets in ratio sample:KBr as 1:20.
The X-ray diffractograms were obtained using DRON- 4-07 diffractometer with CuKα radiation (λ = 15.4 nm) at 25˚C. The relative intensity was registered in a range (2θ) of 0.6˚ - 60˚ with a step size of 0.02˚.
3. Results and Discussion
The porosity characteristics of samples were calculated from ad/desorption curves represented in Figure 2. For curves analysis the BET and BJH methods were applied. The specific surface area, total pore volume and values of average pore diameter of pristine silica matrices are collected in Table 1.
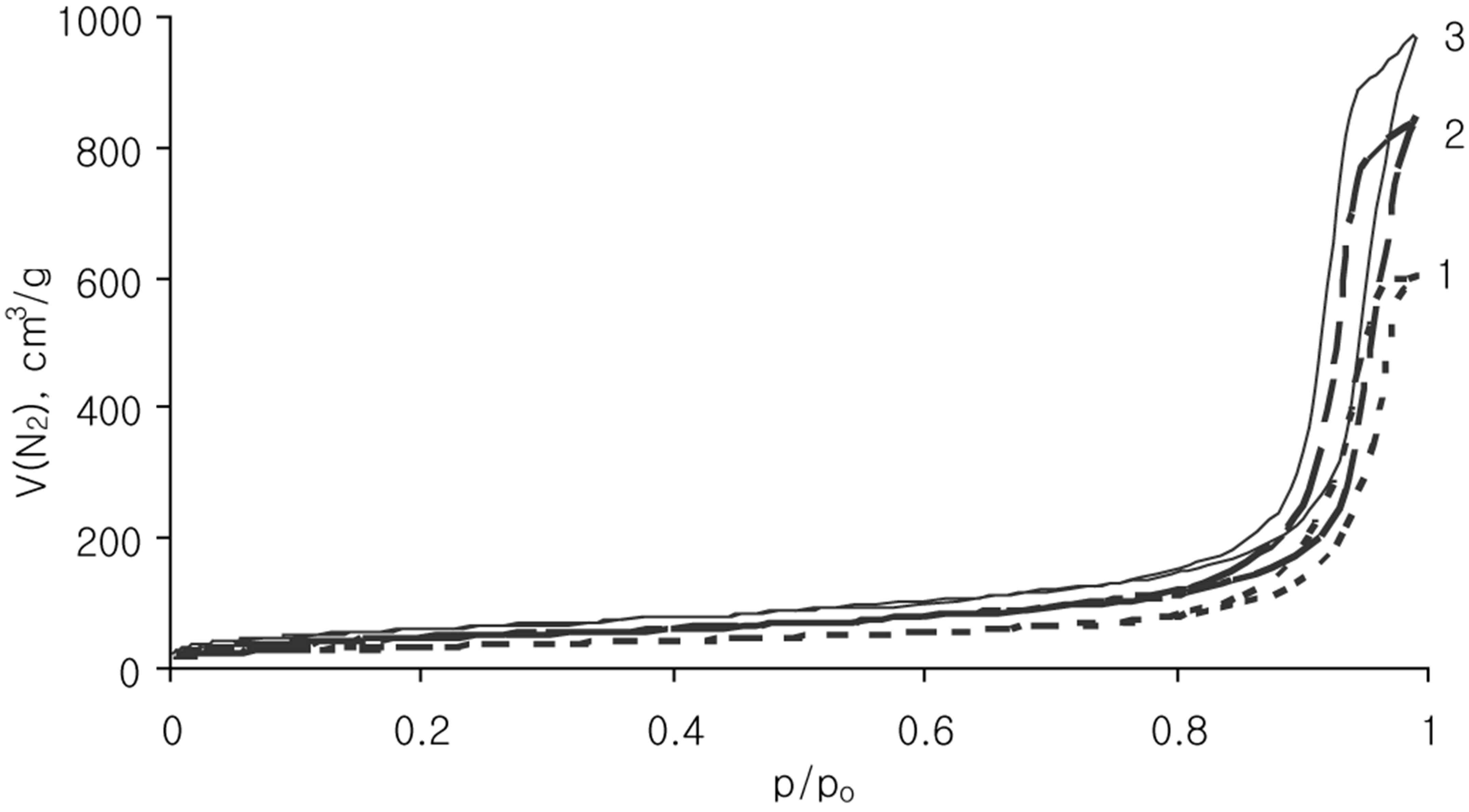
Figure 2. N2 adsorption/desorption isotherms for Silochrom (1), Spherical silica-1 (2), Spherical silica-2 (3) samples.

Table 1. Structural characteristics of the silica carriers.
The nitrogen ads/desorption isotherms (Figure 2) of all samples can be classified as type IV. The profile of the curves clear evidenced a porosity in mesoporous scale. It has been proved by hysteresis loop location at the high relative pressures (p/po > 0.8). The Silochrom and Spherical silica-1 samples have the same size of primary particles (Table 1). The Spherical silica-1 has higher specific surface area in comparison with Silochrom sample, while the pore volume follows the reverse trend. The Spherical silica-2 has the largest specific surface area, but much less a pore diameter compared with other samples under study. Such samples selection will allow to determine the effect of different morphological parameters of the carriers (particle size, pore size, specific surface area) on releasing of the loaded active substance.
In the FT-IR spectrum of the impregnated silica (Figure 3) the main characteristic bands of the diclofenac functional groups [35] are marked. Namely, broad absorption of the C-H bonds in the range 3020 - 2700 cm−1, bands of the stretching vibrations of C=O groups at 1691 cm−1, bands 1512 and 1450 cm−1 attributed to δ(NH) and δ(CH2, CH3), respectively. But the diclofenac characteristic band at 1570 cm−1 attributed to ν(C=C) vibration has a low intensity. Moreover, bands at 3422 and 3277 cm−1, related to ν(NH), combination band ‘five aromatic fingers’ at 2000 - 1600 cm−1, as well as carboxyl group vibration around 1690 cm−1 ν(COO-) and the ν(CCl) band at 745 cm−1 are not exhibited. Additionally, in range of conformation sensitive bands (below 1000 cm−1) the crystallinity bands of diclofenac in spectrum of impregnated silica are absent. These results suggest that the macrostructure of the drug had changed. A reduction of an intensity the vibration bands assigned to these groups can be attributed to an effect of intermolecular interacttion (mutual orientation the benzene rings of the neighbor molecules in the adsorption layer) or/and to influence of the active groups on the silica surface on interaction with diclofenac molecules (formation of the hydrogen bonds between the surface hydroxyl groups and the functional groups of diclofenac). The several different types of interactions could be realized. It can be polymorph transformation or chemical interaction involving carboxylic, chlorineor nitrogen-containing groups. The presence of the carbonyl stretching band in the 1800 - 1600 cm–1 spectral range without observed shift confirms the
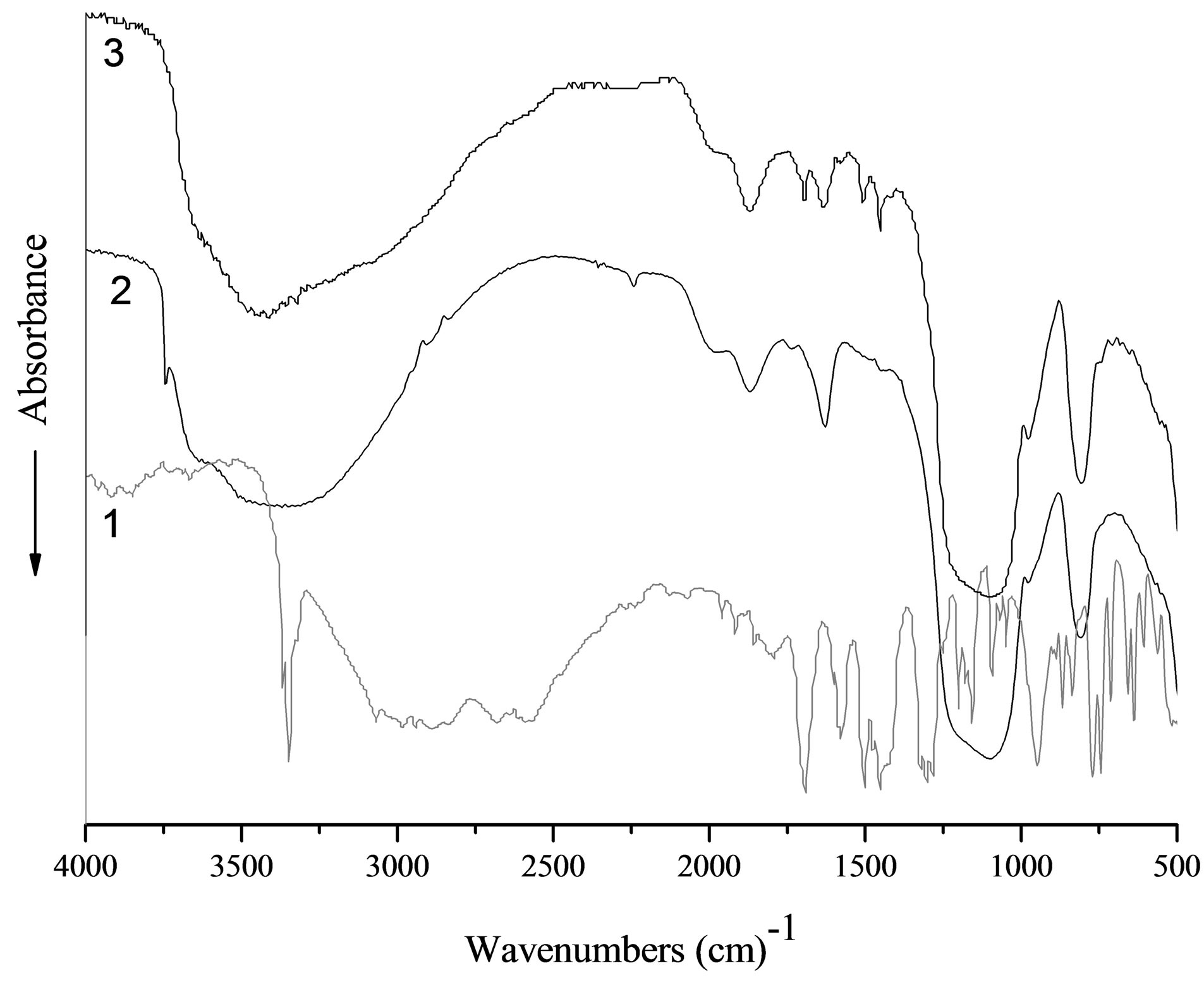
Figure 3. FT-IR-spectra of diclofenac (1), pristine silica (2), silica impregnated with diclofenac (3).
availability of carboxyl groups in sample without conversion into carboxylate moiety COO–, which has the absorption band below 1675 cm–1. The primary hydrogen bond between the two components, O···H–N (heterocycle), generates as usually two broad stretches at approximately 1900 and 2500 cm–1. At the composite spectrum only broad peak near 2500 cm–1 is recorded, the stretches near 1900 cm−1 due to its low intensities are overlapped with silica characteristic peak. Obtained results clearly suggest that there is interaction between diclofenac and silica surface without changes in the drug chemical structure.
XRD patterns of silica carrier without drug, pure diclofenac and diclofenac loaded-silica are shown in Figure 4. XRD pattern of diclofenac showed characteristic peaks indicating the presence of 98 wt% of HD2 phase (monocline, space group [SG] C2/c, a = 2.0226 nm, b = 0.6971 nm, c = 2.0061 nm, β = 109.64˚, Z = 8), while residual peaks are due to HD1 one (monocline SG P21/c, a = 0.8384 nm, b = 1.0898 nm, c = 1.4822 nm, β = 92.76˚, Z = 4 [36].
These peaks disappeared in the XRD pattern of drugloaded silica spheres (Figure 4), suggesting the presence of drug as molecular dispersion in the silica porous spheres. Consequently, the structure of the diclofenac after impregnation into silica carriers reversed to amorphous state.
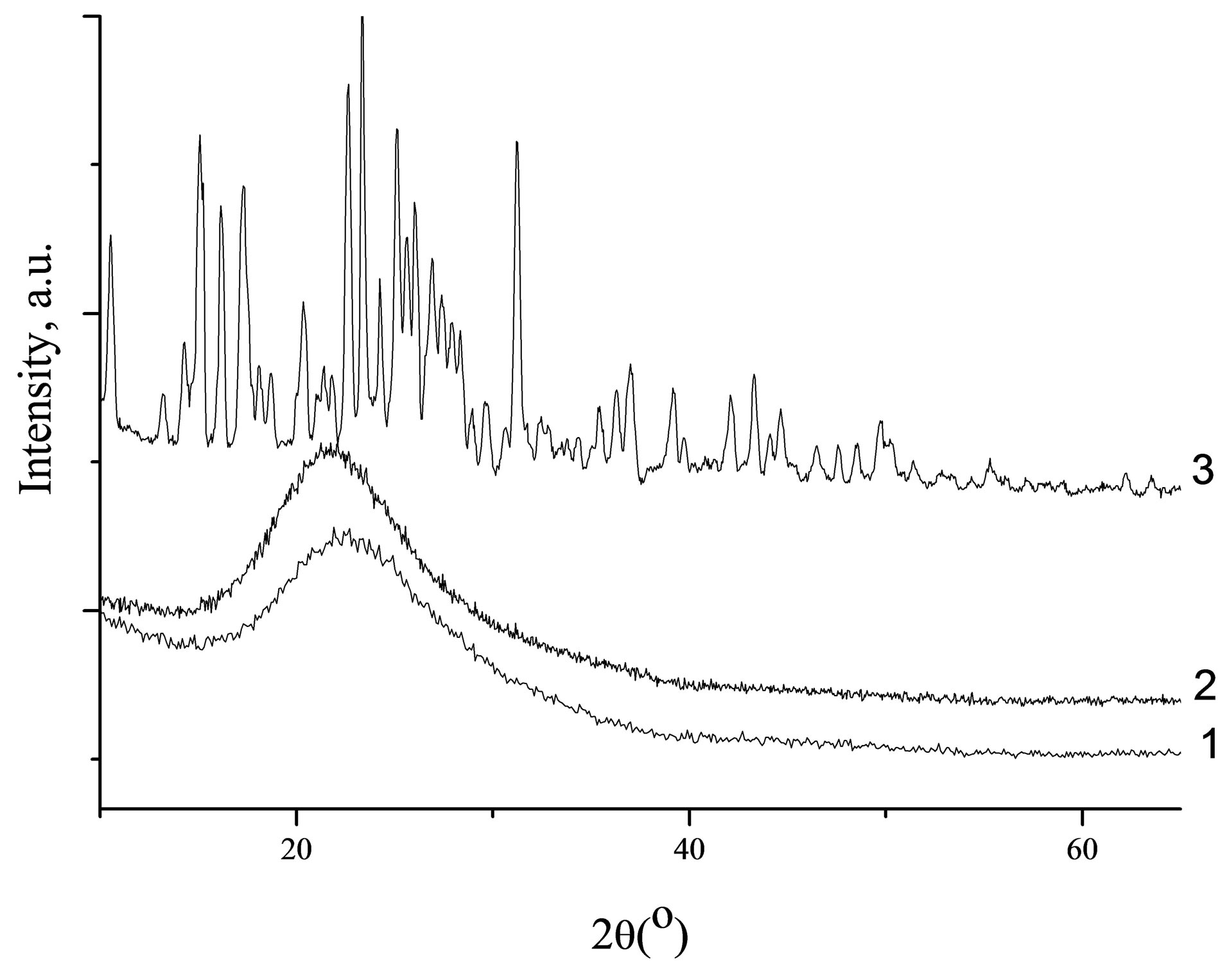
Figure 4. XRD patterns of Spherical silica-1 (1), Spherical silica-1 impregnated with diclofenac (2) and of crystalline diclofenac (3).
Drug release from silica carriers A dissolution profile of adsorbed diclofenac was found to be dependent on the porous structure of carriers, as well as crystalline or amorphous form of silica-supported drug. Therefore, the high drug capacity and fast release kinetics allow to rate porous silica matrices as potential drug carriers for the formulation of poorly soluble drug compounds. The profile of the crystalline diclofenac dissolution along with the diclofenac release profiles for mesoporous drug carriers are presented in Figure 5. The drug contents for all samples under study were similar. It was shown that the active matter release rate is decreased with the decreasing of the carrier’s pore size for the diclofenac/silica systems.
The observed dissolution rate for the pure diclofenac was quite low. The amount of dissolved diclofenac in the phosphate buffer solution, pH = 6.8 at typical sampling time of 15 min accumulated to 10 %, while the release of 99 % was reached during the total experiment time of 7 h. The drug release from Silochrom is characterized by high initial rate: 86 % of the drug impregnated was removed for 15 min. In the case of diclofenac loaded in the Spherical silica-1 after 15 min of the process, the diclofenac desorption was amounted to 84 %, and after 1 h the release amount reached about 98%. A release rate from the Spherical silica-2 during first 15 min was lower and quantity of dissolved drug was 60%. This behaviour is understood by assuming that more restrictive pores (less diameter) complicate the diclofenac diffusion from the carrier. In any case, the release rate of diclofenac from the mesoporous carriers was higher compared with the dissolution rate of pure diclofenac. Thus, the mesoporous silica carriers promotes the rapid diclofenac delivery than the pure form of the drug (Figure 6).
The analysis of the profile release of the diclofenac
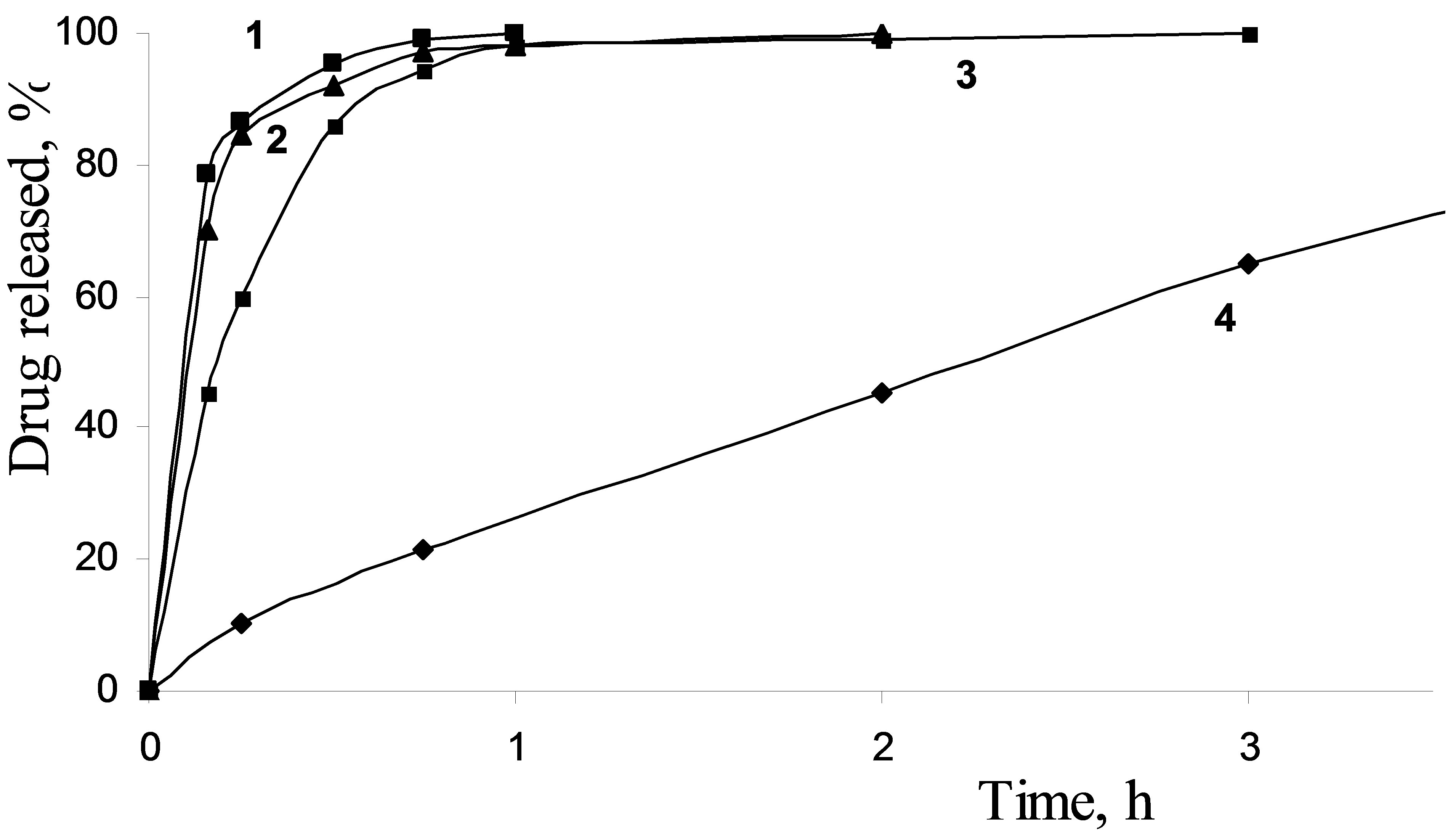
Figure 5. Impregnated diclofenac release from Silochrom (1), Spherical silica-1 (2), Spherical silica-2 (3) and dissolution profile for pure diclofenac (4) in the simulated intestinal fluid.
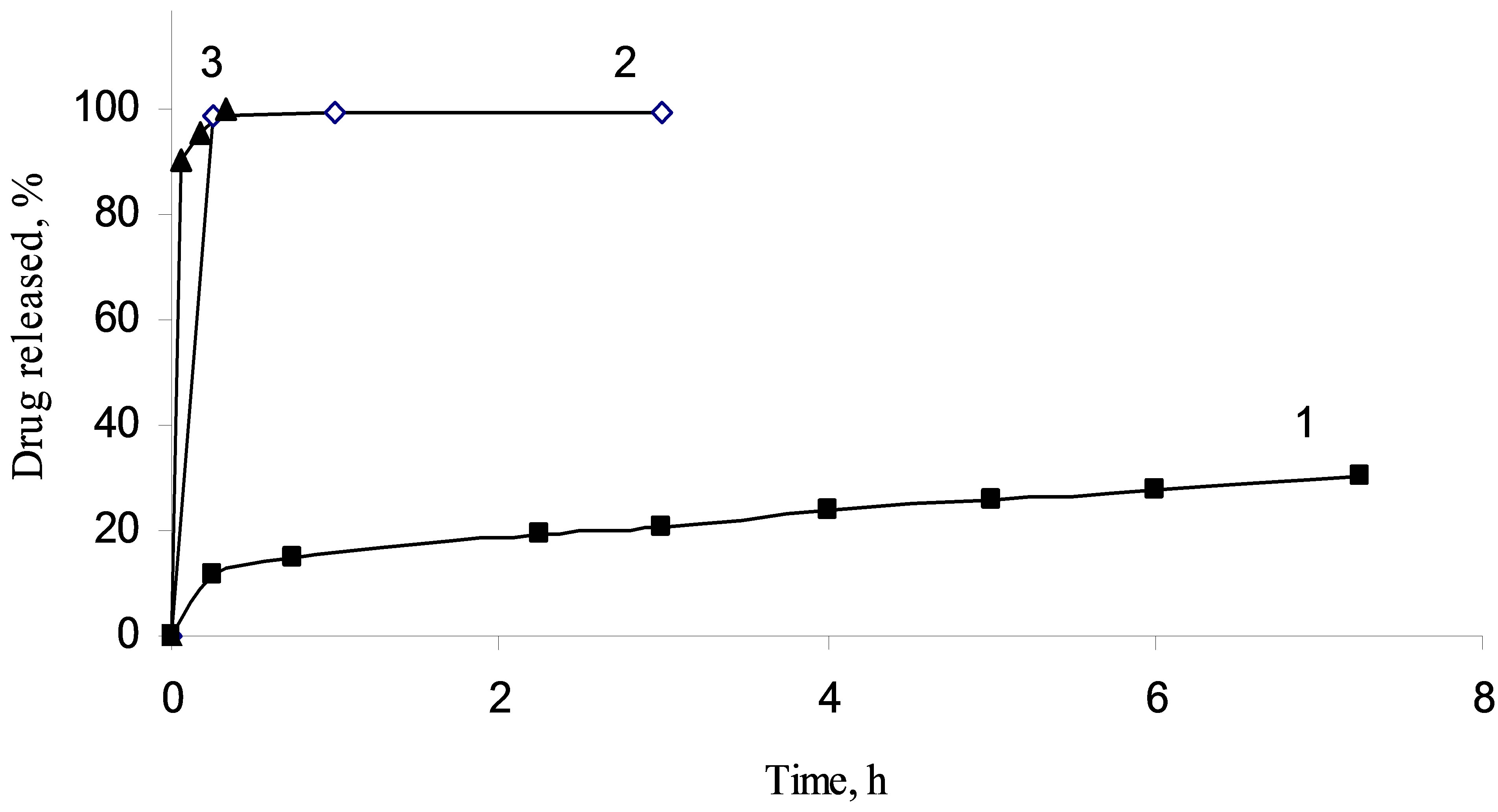
Figure 6. Impregnated diclofenac sodium release from methylated silica (1), aminated silica (2) and pristine fumed silica (3).
sodium from fumed silica carriers with different surface functionality has shown other regularities. For silica A-300 with immobilized diclofenac sodium the extremly fast release of the drug was observed. More than 95% of the loaded drug was desorbed from the composite within the first 15 min. The presence of the amino groups on the silica surface layer leads to slower elimination of the diclofenac sodium. Significant prolongation of diclofenac sodium release was achieved using the hydrophobic silica as a carrier. It may be specified by hydrophobic interactions of diclofenac sodium molecules with the organophilic silica surface, morphology of the silica aggregates and less wettability of the carrier by the release medium that are worsening washout of the loaded compound.
It has been found that two factors could affect drug/ matrix interaction, and, consequently, the release rate of the drug from the carrier: the structure of pore (shape and size of the pore) and a functionality of the pore wall surface that is activity of the carrier surface functional groups with respect to functional groups of organic compound.
Modelling of drug release kinetics. The dissolution profiles of all the batches were fitted to various models such as Higuchi and Korsmeyer-Peppas to ascertain the transport-kinetic model of drug release.
In order to compare the dissolution pure diclofenac and the drug release characteristics of mesoporous material data were fitted with the Korsmeyer–Peppas equation:
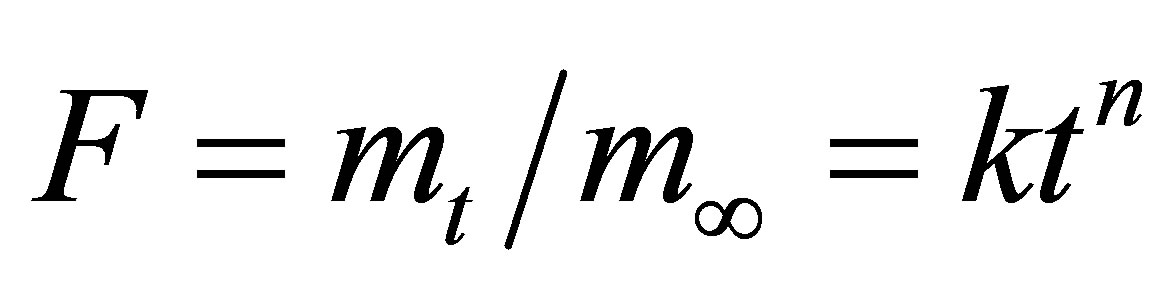 (1)
(1)
where F is the fractional release of drug, k is the kinetic release constant incorporating structural and geometrical characteristics of the dosage form (h−n), t is the elapsed time and n is the release exponent describing the drug release mechanism [37]. The release exponent n = 0.5 corresponds to a fully Fickian diffusion based on transport of drug to the dissolution medium. In such case, the Korsmeyer-Peppas model would be reduced to the Higuchi equation:
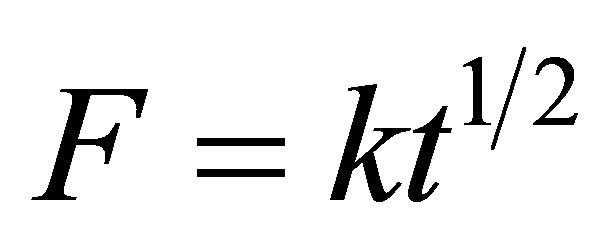 (2)
(2)
where, k is the Higuchi dissolution constant [38]. The Korsmeyer-Peppas models fitted to the diclofenac release from the silica and pure diclofenac are presented in Table 2.
The model was found to describe the initial 60% diclofenac release very well with high correlation coefficients (R2 > 0.99) in all cases (as a short time approximation the Korsmeyer–Peppas model cannot be applied beyond the 60% release). The clearly faster release of diclofenac from the silica carrier (kinetic constant k = 10.7) compared to silica (k = 4.7) demonstrated the unrestricted diffusion of the drug to the dissolution medium due to the high accessibility and stability of the silica pore network. The modelling of the dissolution curve of the pure crystalline form of diclofenac confirmed the much slower release of drug (k = 0.6) compared with the porous carriers.
The modelling of the Korsmeyer–Peppas exponent revealed that the diclofenac release mechanism of the spherical silica material was more diffusion based (n =
0.56) than for the Silochrom material (n = 0.22). It was evident that the highly accessible nanoreservoir of the silica material provided a relatively unrestricted release of the drug, whereas the long and narrow mesopore pathways of the Silochrom sterically hindered the free diffusion of diclofenac from the mesopores. On the other hand, the dissolution mechanism of pure diclofenac (n = 0.72) was close to a linear zero order type of release, typical for slowly dissolving drugs. The results emphasized the improving effect of the mesoporous carriers on diclofenac dissolution at the pH = 6.8 conditions, where the dissolution of pure diclofenac is otherwise very slow.
Composites based on chitosan and silica microgranules with immobilized diclofenac sodium.
Biodegradability and biocompatibility are the fundamental requirements that determine the possible therapeutic and surgical applications of a polymeric biomaterial. The one of the most utilized polysaccharides is chitosan. This attractive natural polysaccharide shares the benefits of other natural polymers, but does not induce an immune response. In order to achieve controlled release of active substances, the composites based on chitosan and silica impregnated with diclofenac sodium were created (Figure 7).
Desorption of the active compound from the granules based on protonated chitosan was found to be complicated due to low solubility of the diclofenac acid form (Figure 8). Another important factor is the formation of gel inside carriers due to polymer matrix swelling during contacting with release medium, which, undoubtedly, complicates diffusion both the solvent and the adsorbate. As was shown, the polymer matrix crosslinked with glutaraldehyde is also poorly permeable for the active compounds, whereas the granules based on deprotonated chitosan shown good results as carrier. The loaded diclofenac sodium is completely released during one hour in this case.
According to the obtained results (Figure 9), the release of diclofenac sodium from composite granules de-
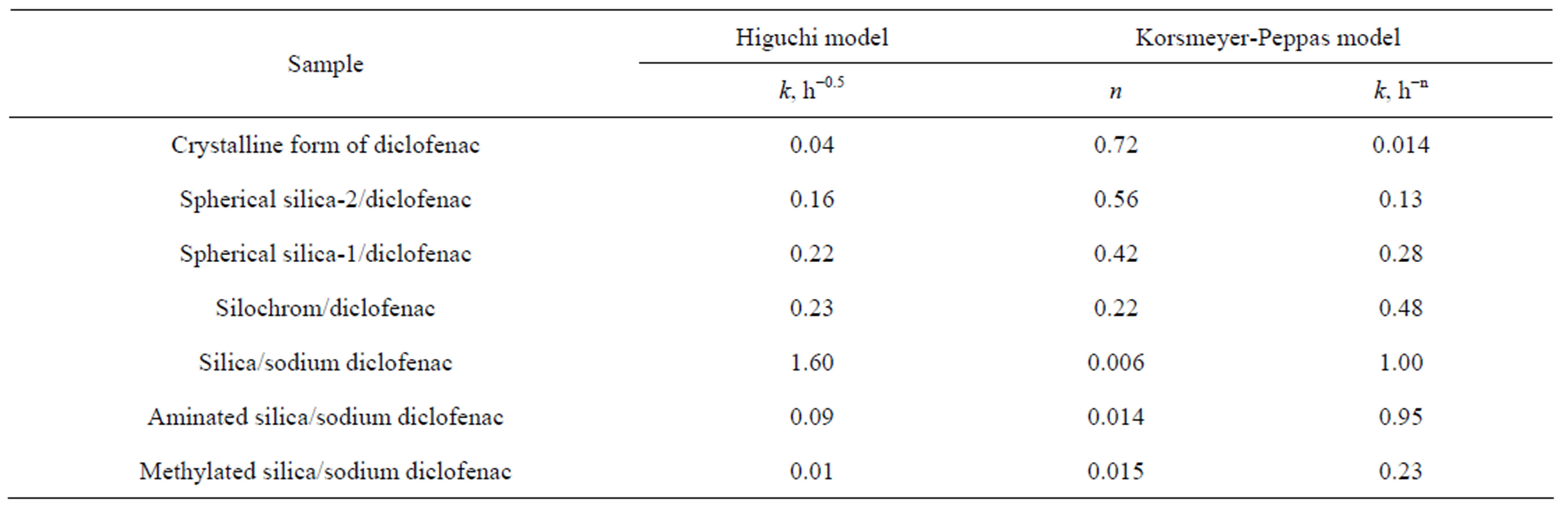
Table 2. Release kinetics constants for drug immobilized into silica carriers.
 1
1  2
2  3
3
1 - Composite based on chitosan and diclofenac sodium;
2 - Silica microgranules with immobilized diclofenac sodium;
3 - Composite based on chitosan and silica microgranules with immobilized diclofenac sodium
 2
2  3
3 Figure 7. Schematic structures of composites and their related optical images.
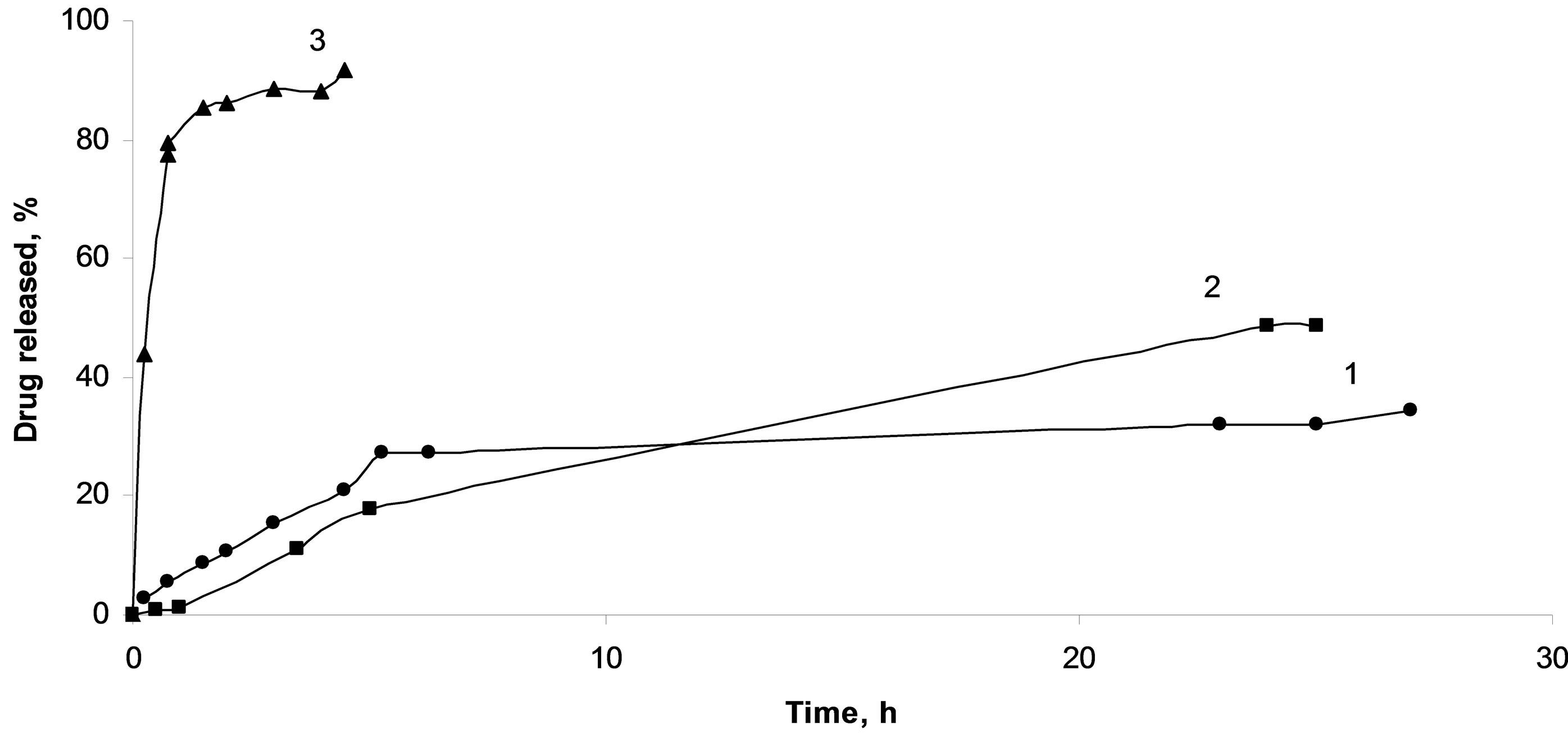
Figure 8. Diclofenac sodium release from composites based on crosslinked chitosan spheres (1), protonated chitosan spheres (2), deprotonated chitosan spheres (3).

Figure 9. Composites based on chitosan and methylated silica microgranules with immobilized diclofenac sodium (1), aminated silica microgranules with immobilized diclofenac sodium (2), silica microgranules with immobilized diclofenac sodium (3).
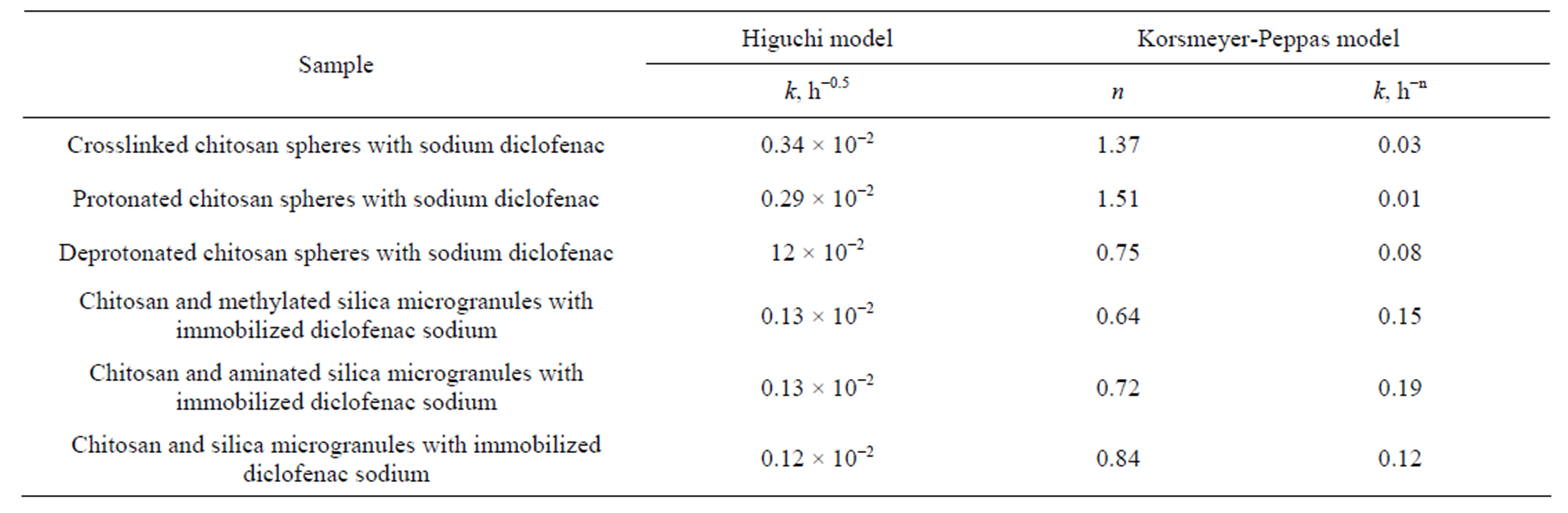
Table 3. Release kinetics constants for drug immobilized into silica-chitosan composites.
pends on the nature of the silica filler surface.
Introducing the hydroxylated (as obtained) silica accelerates the release rate of the active substance. In the case of composites containing hydrophobic silica desorption of diclofenac sodium was significantly slower (Table 3).
For chitosan/diclofenac sodium composite, the structure of the polymer was found to effect also the active substance release rate. The combination of the protonated chitosan affinity with methylated silica and compatibility of this composition and active compound allows significantly prolongate the releasing of the drug.
4. Conclusions
The structure of active compound was shown to become amorphous after impregnation on the silica surface. Amorphization of the structure provides an increase of dissolution rate of diclofenac. The diclofenac release rate depends on the porous structure of carriers and increases with the increase of pore diameter. Another regularity was obtained for carriers with different surface functionalities. The presence amino groups in the silica surface layer lead to slower eliminating of the diclofenac sodium from the carrier. Significant prolongation of diclofenac sodium release was achieved using the hydrophobic silica as a carrier.
The highest release rate (30 mg/h) was observed for composites containing deprotonated chitosan. Significant rate reduction (1.5 mg/h) was obtained for spheres in which chitosan remained in the protonated form. Crosslinking of chitosan with glutaraldehyde provides controlled rate of the diclofenac sodium removal (2.5 mg/h), nevertheless part of the impregnated substance remains in the bulk of the composite.
Introducing of the hydroxylated silica into polymer granules improves the rate of active compound release. In composites containing hydrophobic silica, desorption of diclofenac sodium is decelerated. Surface chemistry of the silica carrier has an influence on diclofenac sodium release.
In the case of composites with hydrophilic silica carrier, the release parameters are determined by characteristics of the polymer shell. For aminated or methylated silicas, the release is controlled by the combined properties of both the carrier and the polymeric shell.
REFERENCES
- N. Gaddam and J. Aukunuru, “Systemic Delivery of Diclofenac Sodium after Topical Application of Gels Incorporated with Drug-Loaded Solid Lipid Nanoparticles (SLN),” Journal of Pharmaceutical Health Services Research, Vol. 2, No. 2, 2010, pp. 177-187.
- K. M. Manjanna, B. Shivakumar and T. M. P. Kumar, “Diclofenac Sodium Microbeads for Oral Sustained Drug Delivery,” International Journal of PharmTech Research, Vol. 1, No. 2, 2009, pp. 317-327.
- A. Chiarini, A. Tartarini and A. Fini, “pH-Solubility Relationship and Partition Coefficients for Some Anti-Inflammatory Arylaliphatic Acids,” Archiv der Pharmazie, Vol. 317, No. 3, 1984, pp. 268-273. doi:10.1002/ardp.19843170314
- A. S. Bravo, M. C. Lamas and C. J. Salomón, “In-Vitro Studies of Diclofenac Sodium Controlled-Release from Biopolymeric Hydrophilic Matrices,” Journal of Pharmaceutical Sciences, Vol. 5, No. 3, 2002, pp. 213-219.
- M. Bartolomei, P. Bertocchi, E. Antoniella and A. Rodomonte, “Physico-Chemical Characterisation and Intrinsic Dissolution Studies of a New Hydrate form of Diclofenac Sodium: Comparison with Anhydrous Form,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 40, No. 5, 2006, pp. 1105-1113. doi:10.1016/j.jpba.2005.09.009
- B. M. Al-Taani and B. M. Tashtoush, “Effect of Microenvironment pH of Swellable and Erodable Buffered Matrices on the Release Characteristics of Diclofenac Sodium,” AAPS PharmSciTech, Vol. 4, 2003, pp. 110-115. doi:10.1208/pt040343
- S. L. Collins, R. A. Moore, H. J. McQuay and P. J. Wiffen, “Oral Ibuprofen and Diclofenac in Post-Operative Pain: A Quantitative Systematic Review,” European Journal of Pain, Vol. 2, No. 4, 1998, pp. 285-291.
- J. Schwaiger, H. Ferling, U. Mallow, H. Wintermayr and R. D. Negele, “Toxic Effects of the Non-Steroidal AntiInflammatory Drug Diclofenac. Part I: Histopathological Alterations and Bioaccumulation in Rainbow Trout,” Aquatic Toxicology, Vol. 68, No. 2, 2004, pp. 141-150. doi:10.1016/j.aquatox.2004.03.014
- E. J. Hickey, R. R. Raje, V. E. Reid, S. M. Gross and S. D. Ray, “Diclofenac Induced in Vivo Nephrotoxicity May Involve Oxidative Stress-Mediated Massive Genomic DNA Fragmentation and Apoptotic Cell Death,” Free Radical Biology and Medicine, Vol. 31, No. 2, 2001, pp. 139-152. doi:10.1016/S0891-5849(01)00560-3
- C. C. Kao, S. C. Chen and M. T. Sheu, “Lag Time Method to Delay Drug Release to Various Sites in the Gastrointestinal Tract,” Journal of Controlled Release, Vol. 44, No. 2-3, 1997, pp. 263-270. doi:10.1016/S0168-3659(96)01529-5
- C. Barbé, J. Bartlett, L. Kong, K. Finnie, H. Q. Lin, M. Larkin, S. Calleja, A. Bush and G. Calleja, “Silica Particles: A Novel Drug-Delivery System,” Advanced Materials, Vol. 16, No. 21, 2004, pp. 1959-1966. doi:10.1002/adma.200400771
- Y. Zhou and X. Y. Wu, “Finite Element Analysis of Diffusional Drug Release from Complex Matrix Systems. I. Complex Geometries and Composite Structures,” Journal of Controlled Release, Vol. 49, No. 2-3, 1997, pp. 277‑288. doi:10.1016/S0168-3659(97)00103-X
- S. W. Song, K. Hidajat and S. Kawi, “Functionalized SBA-15 Materials as Carriers for Controlled Drug Delivery: Influence of Surface Properties on Matrix-Drug Interactions,” Langmuir, Vol. 21, No. 21, 2005, pp. 9568- 9575. doi:10.1021/la051167e
- W. J. Xu, Q. Gao, Y. Xu, D. Wu, Y. H. Sun, W. L. Shen and F. Deng, “Controlled Drug Release from Bifunctionalized Mesoporous Silica,” Journal of Solid State Chemistry, Vol. 181, No. 10, 2008, pp. 2837‑2844. doi:10.1016/j.jssc.2008.07.011
- M. Manzano, V. Aina, C. O. Arean, F. Balas, V. Cauda, M. Colilla, M. R. Delgado and M. Vallet-Regi, “Studies on MCM-41 Mesoporous Silica for Drug Delivery: Effect of Particle Morphology and Amine Functionalization,” Chemical Engineering Journal, Vol. 137, No. 1, 2008, pp. 30-37. doi:10.1016/j.cej.2007.07.078
- M. Aw, S. Simovic, Y. Yu, J. Addai-Mensah and D. Losic, “Porous Silica Microshells from Diatoms as Biocarrier for Drug Delivery Applications,” Powder Technology, Vol. 223, 2012, pp. 52-58. doi:10.1016/j.powtec.2011.04.023
- M. D. Popova, Á. Szegedi, I. N. Kolev, J. Mihály, B. S. Tzankov, G. T. Momekov, N. G. Lambov and K. P. Yoncheva, “Carboxylic Modified Spherical Mesoporous Silicas as Drug Delivery Carriers,” International Journal of Pharmaceutics, Vol. 436, No. 1-2, 2012, pp. 778-785. doi:10.1016/j.ijpharm.2012.07.061
- P. Kortesuo, M. Ahola, M. Kangas, I. Kangasniemi, A. Yli-Urpo and J. Kiesvaara, “In Vitro Evaluation of SolGel Processed Spray Dried Silica Gel Microspheres as Carrier in Controlled Drug Delivery,” International Journal of Pharmaceutics, Vol. 200, No. 2, 2000, pp. 223-229. doi:10.1016/S0378-5173(00)00393-8
- K. Czarnobaj, “Preparation and Characterization of Silica Xerogels as Carriers for Drugs,” Drug Delivery, Vol. 15, No. 8, 2008, pp. 485-492. doi:10.1080/10717540802321495
- R. Jayakumar, D. Menon, K. Manzoor, S. V. Nair and H. Tamura, “Biomedical Applications of Chitin Nanomaterials: A Short Review,” Carbohydrate Polymers, Vol. 82, No. 2, 2010, pp. 227-232. doi:10.1016/j.carbpol.2010.04.074
- E. Hodgson and J. A. Goldstein, “Metabolism of Toxicants: Phase I Reactions and Pharmacogenetics,” In: E. Hodgson and R. C. Smart, Eds., Introduction to Biochemical Toxicology, 3rd Edition, Wiley-Interscience, New York, 2001, pp. 67-113.
- L. S. Fonseca, R. P. Silveira, A. M. Deboni, E. V. Benvenutti, T. M. Costa, S. S. Guterres and A. R. Pohlmann, “Nanocapsule@xerogel Microparticles Containing Sodium Diclofenac: A New Strategy to Control the Release of Drugs,” International Journal of Pharmaceutics, Vol. 358, No. 1-2, 2008, pp. 292-295. doi:10.1016/j.ijpharm.2008.02.005
- E. J. Lee, S. H. Jun, H. E. Kim, H. W. Kim, Y. H. Koh and J. H. Jang, “Silica Xerogel-Chitosan Nano-Hybrids for Use as Drug Eluting Bone Replacement,” Journal of Materials Science: Materials in Medicine, Vol. 21, No. 1, 2010, pp. 207-214. doi:10.1007/s10856-009-3835-9
- M. Prokopowicz, “Silica-Polyethylene Glycol Matrix Synthesis by Sol-Gel Method and Evaluation for Diclofenac Diethylammonium Release,” Drug Delivery, Vol. 14, 2007, pp. 129-138.
- J. H. Park, G. Saravanakumar, K. Kim and I. C. Kwon, “Targeted Delivery of Low Molecular Drugs Using Chitosan and Its Derivatives,” Advanced Drug Delivery Reviews, Vol. 62, No. 1, 2010, pp. 28-41. doi:10.1016/j.addr.2009.10.003
- M. Dash, F. Chiellini, R. M. Ottenbrite and E. Chiellinial, “Chitosan: A Versatile Semi-Synthetic Polymer in Biomedical Applications,” Progress in Polymer Science, Vol. 36, No. 8, 2011, pp. 981-1014. doi:10.1016/j.progpolymsci.2011.02.001
- I. Rashid, M. Al-Remawi, A. Eftaiha and A. Badwan, “Chitin-Silicon Dioxide Coprecipitate as a Novel Super Disintegrant,” Journal of Pharmaceutical Sciences, Vol. 97, No. 11, 2008, pp. 4955-4969. doi:10.1002/jps.21354
- Y. M. Bolbukh, P. O. Kuzema, V. A. Tertykh and B. Gawdzik, “Effect of Surface Hydride, Vinyl, and Methyl Groups on Thermal Stability of Modified Silica-Divinylbenzene-Di(methacryloyloxymethyl)naphthalene Composites,” International Journal of Polymeric Materials and Polymeric Biomaterials, Vol. 56, No. 3, 2007, pp. 803-823. doi:10.1080/00914030601163464
- R. B. Kozakevych, Y. M. Bolbukh and V. A. Tertykh, “Desorption of Diclofenac Sodium from Silica-Chitosan Composites,” Pharmaceutical Journal, Vol. 3, 2009, pp. 103-111.
- K. V. Katok, Y. Bolbukh and V. A. Tertykh, “Chemical Modification of Silica Surface by Immobilization of Amino Groups for Synthesis of Silver Nanoparticles, NATO Science Series II. Mathematics, Physics and Chemistry,” In: M. Vaclavikova, K. Vitale, G. Gallios and L. Ivanicova, Eds., Water Treatment Technologies for Removal of High-Toxicity Pollutants, Springer, New York, 2009, pp. 283-287. doi:10.1007/978-90-481-3497-7_29
- V. L. Gonçalves, M. C. M. Laranjeira, V. T. Fávere and R. C. Pedrosa, “Effect of Crosslinking Agent on Chitosan Microsheres in Controlled Release of Diclofenac Sodium,” Polímeros, Vol. 15, No. 1, 2005, pp. 6-12. doi:10.1590/S0104-14282005000100005
- F. Pati, B. Adhikari and S. Dhara, “Development of Chitosan-Tripolyphosphate Fibers through pH-Dependent Ionotropic Gelation,” Carbohydrate Research, Vol. 346, No. 16, 2011, pp. 2582-2588. doi:10.1016/j.carres.2011.08.028
- Ukrainian State Pharmacopoeia, “REREIG,” Kharkiv, Ukraine, 2001.
- J. C. McElnay and A. C. Nicol, “The Comparison of a Novel Continuous-Flow Dissolution Apparatus for Suppositories with the Rotating Basket Technique,” International Journal of Pharmaceutics, Vol. 19, No. 1, 1984, pp. 89-96. doi:10.1016/0378-5173(84)90135-2
- I. M. Kenawi, B. N. Barsoum and M. A. Youssef, “Cetirizine Dihydrochloride Interaction with Some Diclofenac Complexes,” European Journal of Pharmaceutical Sciences, Vol. 26, No. 3-4, 2005, pp. 341-348. doi:10.1016/j.ejps.2005.07.007
- M. L. Manca, M. Zaru, G. Ennas, D. Valenti, C. Sinico, G. Loy and A. M. Fadda, “Diclofenac-β-Cyclodextrin Binary Systems: Physicochemical Characterization and in Vitro Dissolution and Diffusion Studies,” AAPS PharmSciTech, Vol. 6, No. 3, 2005, pp. 464-472. doi:10.1208/pt060358
- J. Siepmann and F. Siepmann, “Mathematical Modeling of Drug Delivery,” International Journal of Pharmaceutics, Vol. 364, No. 2, 2008, pp. 328-343. doi:10.1016/j.ijpharm.2008.09.004
- S. Dash, P. N. Murthy, L. Nath and P. Chowdhur, “Kinetic Modeling on Drug Release from Controlled Drug Delivery Systems,” Acta Poloniae Pharmaceutic, Vol. 67, 2010, pp. 217-223.