Journal of Behavioral and Brain Science
Vol.3 No.3(2013), Article ID:34792,11 pages DOI:10.4236/jbbs.2013.33032
GABA Attenuates L-DOPA-Induced Striatal and Nigral ERK1/2 Signaling in a Rat Model of Parkinson’s Disease
Department of Pharmacology and Toxicology, School of Medicine-Northwest, Indiana University, Gary, USA
Email: *ssivam@iun.edu, lynchsa@iun.edu
Copyright © 2013 Sarah Lynch, Subbiah P. Sivam. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received March 28, 2013; revised April 30, 2013; accepted May 15, 2013
Keywords: Dopamine; ERK1/2; Hemiparkinsonism; GABA; Striatum; Substantia Nigra
ABSTRACT
L-DOPA is the primary drug used to treat Parkinson’s disease (PD) symptoms, but motor side effects limit its long term use. Previous experimental studies show that L-DOPA acts on supersensitive D1 receptors in the basal ganglia to induce extracellular signal-regulated kinases 1 and 2 (ERK1/2), a pair of MAP-kinase proteins that may be involved in induction of motor side effects. Since GABA is known to be intimately involved in basal ganglia function, we investigated whether elevating GABA levels via a GABA-transaminase (GABA-T) inhibitor affects the L-DOPA-induced ERK1/2 phosphorylation in the striatum and substantia nigra (SN) using a rat model of PD. Unilateral dopaminergic lesions of median forebrain bundle neurons were done using the neurotoxin 6-hydroxydopamine. Rats were prescreened for the extent of the lesion by apomorphine-induced rotation test. Lesioned rats were treated with aminooxyacetic acid (AOAA, a GABA-T inhibitor), L-DOPA, or in combination. Immunohistochemistry of tyrosine hydroxylase (TH, a direct indicator of dopaminergic lesion), substance P (SP, an indirect marker that decreases after lesion), and phospho-ERK1/2 was done using slices at the level of striatum and SN. Unilateral dopaminergic lesioned rats, as expected, exhibited >90% TH loss and a modest SP loss in the striatum and SN. L-DOPA alone induced a 343% and 330% increase in phospho-ERK1/2 in the striatum and SN, respectively. We report here a novel finding that pretreatment with AOAA attenuated the L-DOPA induced increase in phospho-ERK1/2 by 62% and 68% in the striatum and SN, respectively, suggesting a DA-GABA-ERK1/2 link in the therapeutic and/or side effects of L-DOPA.
1. Introduction
In the basal ganglia, dopamine (DA) D1 and D2 receptors are segregated to the direct (striatonigral) and indirect (striatopallidal) pathways, respectively [1-4]. D1 recaptors belong to a subfamily of G-protein coupled receptors (GPCRs) [5], which influence extracellular signal-regulated kinases 1 and 2 (ERK1/2) in the DA-depleted striatum [6-8]. ERK-linked signaling pathways are a class of mitogen-activated protein kinase (MAPK) pathways implicated in the efficiency of transcription and translation [9-10]. In Parkinson’s disease (PD), degeneration of the dopaminergic neurons in the direct pathway of the basal ganglia results in an imbalance in DA function, leading to many motor deficits associated with the disease [7,11-12]. DA agonists, such as L-DOPA, have been shown to restore the balance and function of DA in the basal ganglia [1,6,11,13-17] . However, chronic L-DOPA administration leads to side effects such as dyskinesias that are characterized by aberrations in the motor response to L-DOPA as well as the emergence of uncontrolled movements [6,18-21] .
Numerous studies have reported that the stimulation of supersensitive D1 receptors in the DA-depleted striatum via a DA agonist leads to the phosphorylation of ERK1/2 in the striatum [6,8,16,22-24] . This phenomenon has been linked to the development of motor side effects in reports involving both hemiparkinsonian rodents [24-26] and non-human primates [16-17,21,27] . Moreno and Sivam (2012) recently reported that the D1 agonist, SKF- 38393 increased phospho-ERK1/2 levels in the striatum and in the substantia nigra (SN), and both responses were blocked by D1 antagonist SCH-23390 [28], further supporting the link between D1 receptor supersensitivity and ERK1/2 signaling in the striatum and SN.
GABA is the major inhibitory neurotransmitter of the basal ganglia. GABAergic pathways dominate most of the information processing of the basal ganglia [29]. For instance, about 95% of neurons comprising the striatum are GABAergic medium-sized spiny neurons (MSNs) [16], and 99% of all neurons in the basal ganglia are GABAergic [30]. GABA works cooperatively and interdependently with DA in the basal ganglia to facilitate motor control. DA modulates GABA release presynaptically in the striatum [30] as well as in the SN [31]. This GABAergic modulation in the direct DA pathway of the basal ganglia leads to the facilitation of movement via inhibition of MSNs in the SN followed by disinhibition of thalamocortical neurons, whereas activation in the indirect DA pathway leads to the suppression of movement via the inhibition of thalamocortical neurons [16,21, 29-30] . Numerous reports have found that DA imbalance indeed influences GABA transmission [32-38] . However, the role of GABA in DA-mediated effects as far downstream as ERK1/2 has yet to be evaluated.
Recent reports have found that ERK1/2 signaling plays a role in GABA release related to plasticity and learning in the hippocampus [39] and the effect of psychostimulant drugs such as cocaine and d-methamphetamine in the nucleus accumbens [40]. To our knowledge, however, there is a paucity of literature regarding the role of GABA in L-DOPA-induced ERK1/2 signaling. The present study employed a pharmacological approach to test whether elevating GABA levels via a GABA-transaminase (GABA-T) inhibitor would modify the L-DOPAinduced rotational response and ERK1/2 phosphorylation in the striatum and SN in a unilaterally lesioned rat model of PD.
2. Materials and Methods
2.1. Animals
Female Sprague-Dawley rats (Harlan Laboratories, Inc.) weighing 250 - 350 g were maintained on a 12 h light/12 h dark cycle at 22˚C ± 2˚C and 50% ± 10% humidity with ad libitum access to Wayne Lab Box chow and water. Experimental treatments and animal care protocols were approved by the Institutional Animal Care and Use Committee of Indiana University School of MedicineNorthwest.
2.2. Unilateral Dopaminergic Lesion with 6-Hydroxydopamine (6-OHDA)
Lesions of the nigrostriatal DA pathway were made unilaterally in the right median forebrain bundle (MFB) as previously described [28] by infusion of 9 µg of freebase 6-OHDA (Research Biochemicals, Inc.) in ascorbic acid (4 μl, 0.1% in saline, Sigma-Aldrich, Inc.) using ketamine HCl/xylazine HCl solution (80 mg/kg, i.p., Sigma-Aldrich, Inc.) for anesthesia. The stereotaxic coordinates of the MFB relative to the bregma were, A: 4.4L: 1.2, V: 7.8, and the I-bar was set at −2.5. Coornates were located using the atlas of [41]. Noradrenergic neurons were protected from the neurotoxin 6-OHDA with desipramine HCl (15 mg/kg, i.p., Sigma-Aldrich, Inc.) administered 60 min prior to neurotoxin infusion [42]. After anesthesia recovery, animals were returned to their cages and given ad libitum access to food and water. Meloxicam (1 mg/kg, s.c., Sigma-Aldrich, Inc.) was administered for two days post-surgery for pain relief.
2.3. Apomorphine-Induced Rotation Test to Screen Extent of Lesion
The extent of the unilateral 6-OHDA lesion was assessed by apomorphine-induced rotation test as previously described [28]. Two weeks post-surgery, an apomorphineinduced rotation test (apomorphine HCl, 0.1 mg/kg, i.p., Sigma-Aldrich, Inc., in 0.1% sodium bisulphite solution, Sigma-Aldrich, Inc., in saline) was applied. Animals were observed for contralateral rotation by placing each in a hemispherical bowl (14 inches in diameter) enclosed by transparent Plexiglas cylinder. After apomorphine injection, the number of rotations in 5-minute intervals at 15, 30, and 45 minutes were recorded. Animals with 5 or more average rotations/min were considered to have >90% DA depletion and were used for additional drug treatment.
2.4. Aminooxyacetic Acid (AOAA) and L-DOPA Treatments
The following groups of 6-OHDA-lesioned animals were used: 1) vehicle + vehicle (control); 2) vehicle + AOAA; 3) vehicle + L-DOPA; 4) AOAA + L-DOPA; the numbers of animals in each group were 3, 3, 4, and 7, respectively. Previous studies have shown that the administration of a GABA-transaminase (GABA-T) inhibitor, aminooxyacetic acid (AOAA, 80 mg/kg, i.p., Sigma-Aldrich, Inc.), elevated GABA levels in the striatum as early as 30 min and rapidly increased by 2 h, with GABA levels peaking at 6 h [43]. In the present study we administered AOAA 2 h prior to L-DOPA and perfused the animals transcardially 30 minutes after L-DOPA administration. This paradigm ensured the maximum elevation of GABA levels during L-DOPA-induced effects. RO-4- 4602 (50 mg/kg, i.p., Hoffmann-La Roche, Inc.) was given 30 minutes prior to L-DOPA administration (94.5 mg/kg, i.p., Sigma-Aldrich, Inc.). Vehicle + vehicle served as the independent control group. The experimental design allowed the comparison of not only the relative changes from the lesioned versus intact side in the same animal, but also each drug treatment group versus the independent control group. Ten minutes after L-DOPA injection, rats were observed for 5 minutes to observe the rotational response to the agonist. An overdose of sodium pentobarbital (100 mg/kg, i.p., Sigma-Aldrich, Inc.) was used to kill animals 30 minutes after L-DOPA administration.
2.5. Immunohistochemistry
Immediately after sodium pentobarbital began to take effect, animals were transcardially perfused for 4 min with 10% sucrose (Sigma-Aldrich, Inc.) followed by phosphate-buffered paraformaldehyde (4% in 0.1 M sodium phosphate, Sigma-Aldrich, Inc.). Brains were removed and stored in the paraformaldehyde solution at 4˚C overnight. Brains were then stored in a 30% sucrose solution (in saline) at 4˚C for 24 h or until sunken. Sunken brains were removed and frozen, and sections through the striatumand SN-containing regions were cut (30 µm) coronally on a sliding microtome. Sections were collected into the sucrose solution for storage until processed according to the general protocol of [22], as modified previously [28]. Free-floating brain tissue sections were rinsed in phosphate-buffered saline (PBS) (0.15 M NaCl, 0.1 M sodium phosphate, pH 7.4), and subsequently incubated in 0.3% H2O2. Sections were then rinsed in PBS-0.3% Triton-X 100, and incubated in 10% normal goat serum with 0.2% Triton X-100 in PBS. Immunoreactivity was detected using affinity-purified monoclonal tyrosine hydroxylase (TH) (Affinity Bioreagents, Inc.), phospho-ERK1/2 (phospho-p44/42 MAP kinase (thr202/ tyr204); Cell Signaling Technology, Inc.), and substance P (SP, [44] ) were used to detect immunoreactivity.
Sections were incubated with agitation for 24 hours at 4˚C in 3% normal goat serum, 0.2% Triton X-100, and the 1˚ antibodies. Following incubation, sections were rinsed in PBS-0.3% Triton-X 100 and incubated for TH detection in affinity-purified biotinylated anti-mouse IgG and for SP and phospho-ERK1/2 detection in anti-rabbit IgG (Vector Laboratories, Inc.). Ready-to-use Vectastain Elite ABC kit (Vector Laboratories, Inc.) with nickelcobalt intensification of the diaminobenzidine reaction product was used for final antibody staining. Sections were rinsed, placed on slides, and soaked in a series of ethanol rinses and finally a xylene rinse. Permount (Fisher Scientific, Inc.) was used to cover-slip all slides.
2.6. Quantification and Statistical Analysis
Quantification of Immunostaining. The immunostaining intensities for TH, SP and phospho-ERK1/2 in the striatum and SN of the intact and lesioned sides were determined with ImageJ (NIH) and quantified using QuantiScan software (Biosoft, Inc.). The quantified values of TH, SP, and phospho-ERK1/2 immunoreactivity of the lesioned side were expressed as percent change from that of the intact side. For analysis of the effect of AOAA pretreatment, the percent change in AOAA + L-DOPA treatment group was compared to the percent change in the vehicle + L-DOPA group.
Quantification of Striatal Phospho-ERK1/2 Labeled Cell Counts. Counts of phospho-ERK1/2 immunostained cells were performed on tiled images of the striatum at 200X magnification using a Leica DM4000 microscope and digital camera and Image-Pro Plus 7.0 (Media Cybernetics, Inc.). Eight sample areas of 0.54 mm2 per striatum were counted manually, and values are expressed in cells/mm2 [45] .
Stastical Analysis. All values are depicted as mean ± SEM. Group means were compared using SigmaStat (Systat Software, Inc.) to apply one-way analysis of variance followed by Newman-Keuls multiple range test. P < 0.05 was considered significant.
3. Results
3.1. Assessment of Drug-Induced Rotations of Unilaterally 6-OHDA Lesioned Animals
In control and AOAA-treated animals, no rotations were observed. In animals treated with L-DOPA, an average of 13.3 ± 2.3 rotations per minute was observed. Finally, in animals pretreated with AOAA followed by L-DOPA, an average of only 1.0 ± 0.6 rotations was observed, indicating that AOAA pretreatment greatly attenuated L-DOPA-induced rotations (Figure 1).
3.2. Assessment of TH, SP, and Phospho-ERK1/2 Immunoreactivity in the Striatum and SN of Unilaterally 6-OHDA Lesioned Animals
Immunoreactivity of TH was assessed for all animals to
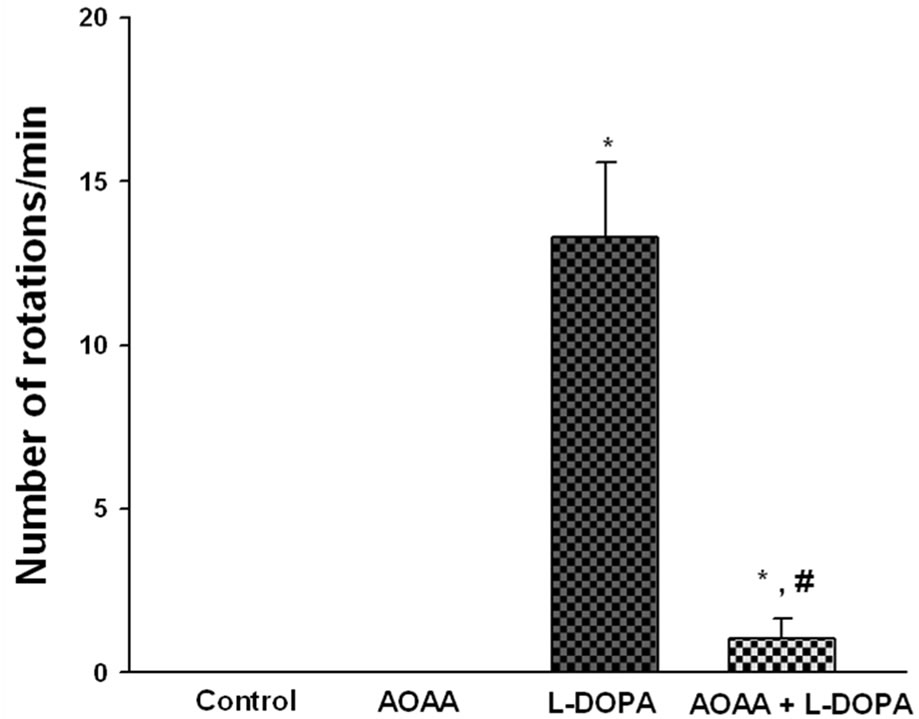
Figure 1. Pretreatment of AOAA greatly reduced the number of L-DOPA-induced rotations. Animals in control and AOAA groups did not rotate. Administration of L-DOPA induced a robust rotational response. Pretreatment of AOAA greatly reduced L-DOPA-induced rotations. *p < 0.05 as compared to the other groups. #p < 0.05 as compared to L-DOPA group.
verify an adequate loss of DA neurons in the lesioned striatum and SN due to unilateral 6-OHDA lesion. TH immunoreactivity decreased by greater than 90% in the lesioned striatum and SN in all groups (Figure 2, upper panel). Immunoreactivity of SP was determined as an indirect marker of DA neuron loss because near-total striatal DA denervation leads to a moderate decrease in SP in the lesioned striatum and SN [46-47] . A moderate decrease in SP immunoreactivity was observed in the striatum and SN in all groups (Figure 2, middle panel). Immunoreactivity of phospho-ERK1/2 was not apparent in lesioned control animals (Figure 2, bottom panel). The vehicle + vehicle control group served as the basal independent control.
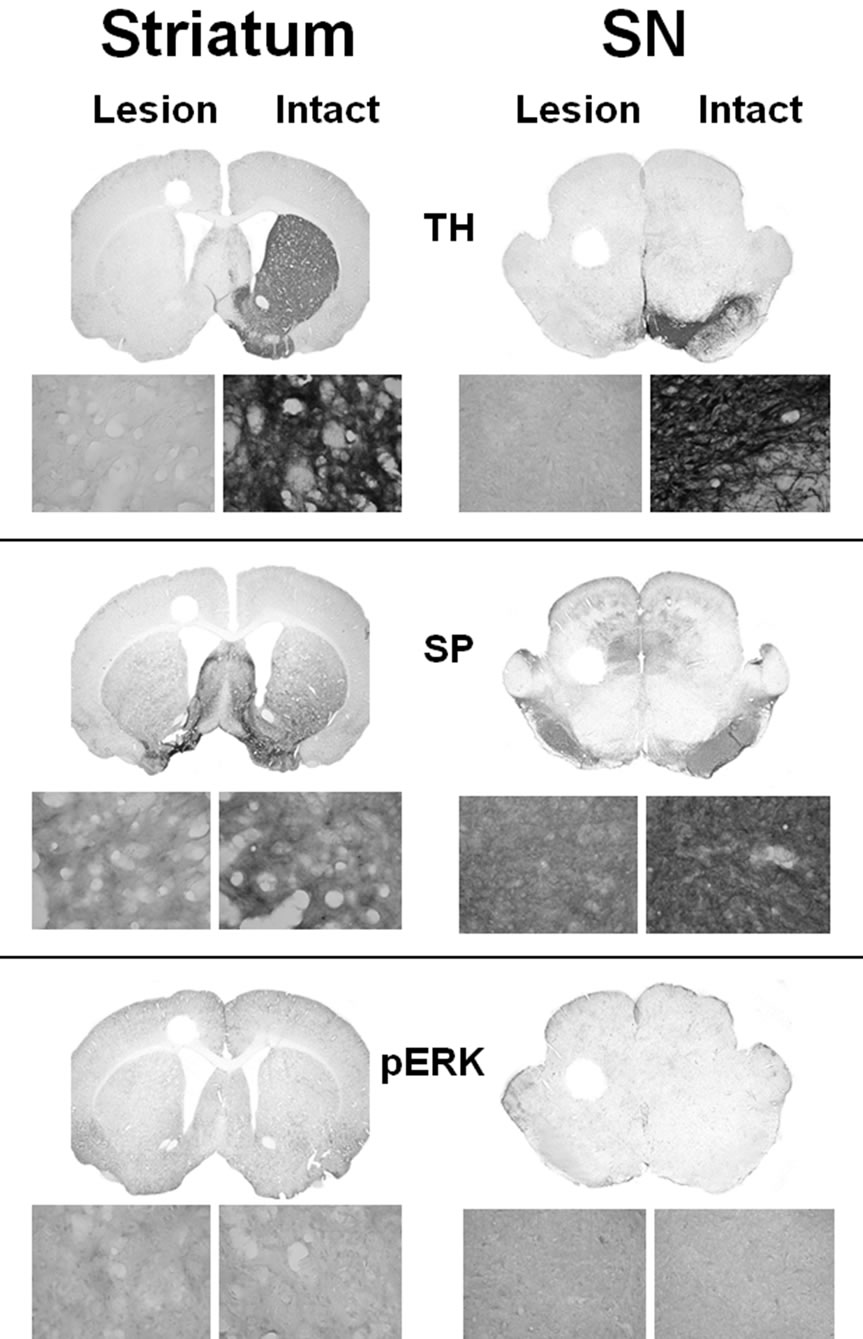
Figure 2. Assessment of TH, SP, and phospho-ERK1/2 immunoreactivity in the striatum and SN of unilaterally 6-OHDA lesioned animals. Immunohistochemistry for TH, SP, and phospho-ERK1/2 was done in coronal sections of the striatum and SN after unilateral DA depletion. Immunoreactivity of TH was severely decreased (upper panel) in the lesioned side as compared to the intact side. Immunoreactivity of SP was moderately decreased (middle panel) in the lesioned side as compared to the intact side. Immunoreactivity of phospho-ERK1/2 was not clearly apparent after DA loss (bottom panel). The vehicle + vehicle (control) group served as the basal independent control. The coronal slices represent 1.25X magnification and the higher power images represent 400X magnification.
3.3. Influence of AOAA and L-DOPA on TH, SP, and Phospho-ERK1/2 Levels in the Striatum of Unilaterally 6-OHDA Lesioned Animals (Figures 2 and 3)
Immunoreactive Intensity of TH, SP, and PhosphoERK1/2 in the Striatum. A greater than 90% decrease in TH immunoreactivity in the lesioned striatum was observed for control, AOAA, L-DOPA, and AOAA + LDOPA groups. The lesioned striatum of animals in all aforementioned groups demonstrated a moderate loss of SP immunoreactivity. AOAA, L-DOPA, or AOAA + L-DOPA treatments did not alter the basal loss of TH or SP immunoreactivity in the lesioned striatum. In the unlesioned side (intact striatum), none of the treatment groups showed phospho-ERK1/2 immunoreactivity. In control or AOAA treated rats, phospho-ERK1/2 immunoreactivity was not apparent in the lesioned or intact striatum (Figure 3). L-DOPA treatment resulted in a robust activation of phospho-ERK1/2 by 343% ± 17 in the striatum in the lesioned side as compared to the intact side (Figures 3 and 4). Pretreatment of AOAA to elevate GABA levels followed by L-DOPA administration resulted in 62% lower levels of phospho-ERK1/2 in the striatum as compared to phospho-ERK1/2 in the LDOPA group (Figures 3 and 4).
Cells Labeled with Phospho-ERK1/2 in the Striatum. The number of immunostained cells labeled with phospho-ERK1/2 was counted in the striatum. In control and AOAA treated animals, no phospho-ERK1/2 labeled cells were apparent. In L-DOPA treated animals, the number of phospho-ERK1/2 labeled cells was 1498 ± 59 cells/mm2. In AOAA + L-DOPA treated animals, the number of phospho-ERK1/2 labeled cells was 827 ± 79 cells/mm2. Thus, AOAA + L-DOPA treatment showed a 45% decrease in the amount of phospho-ERK1/2 labeled cells as compared to the rats treated with L-DOPA alone (Figure 5).
3.4. Influence of AOAA and L-DOPA on TH, SP, and Phospho-ERK1/2 Levels in the SN of Unilaterally 6-OHDA Lesioned Animals
Results similar to those seen in the striatum were observed within the SN with regard to changes in TH, SP, and phospho-ERK1/2 immunoreactivity. L-DOPA treatment resulted in the robust activation of phosphoERK1/2 by 330% ± 25 in the SN (Figures 3 and 4). Pretreatment of AOAA to elevate GABA levels followed by L-DOPA administration resulted in 68% lower levels of phospho-ERK1/2 in the SN as compared to phosphoERK1/2 in the L-DOPA group (Figures 3 and 4).
4. Discussion
The present study confirms and extends previous reports
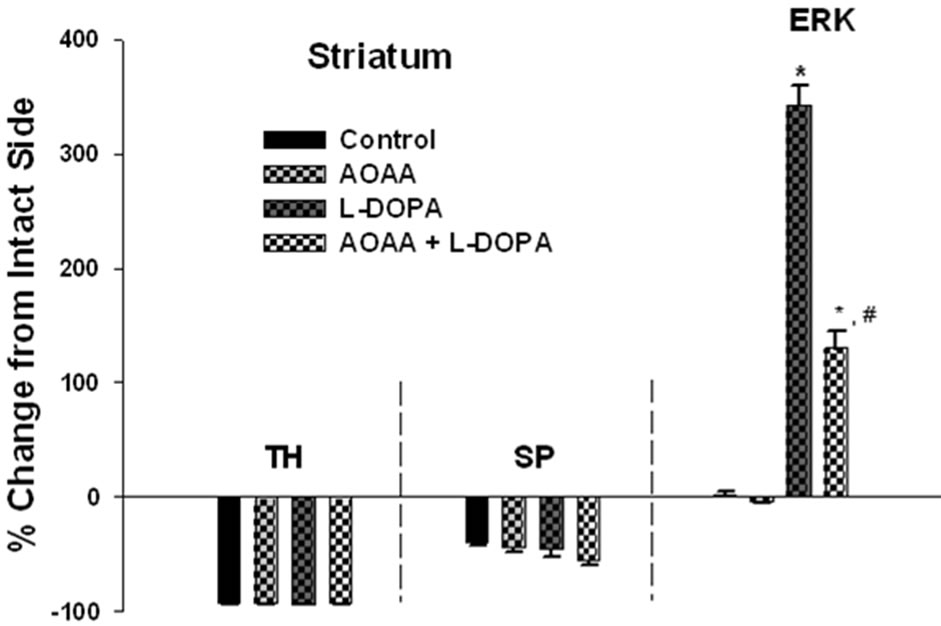
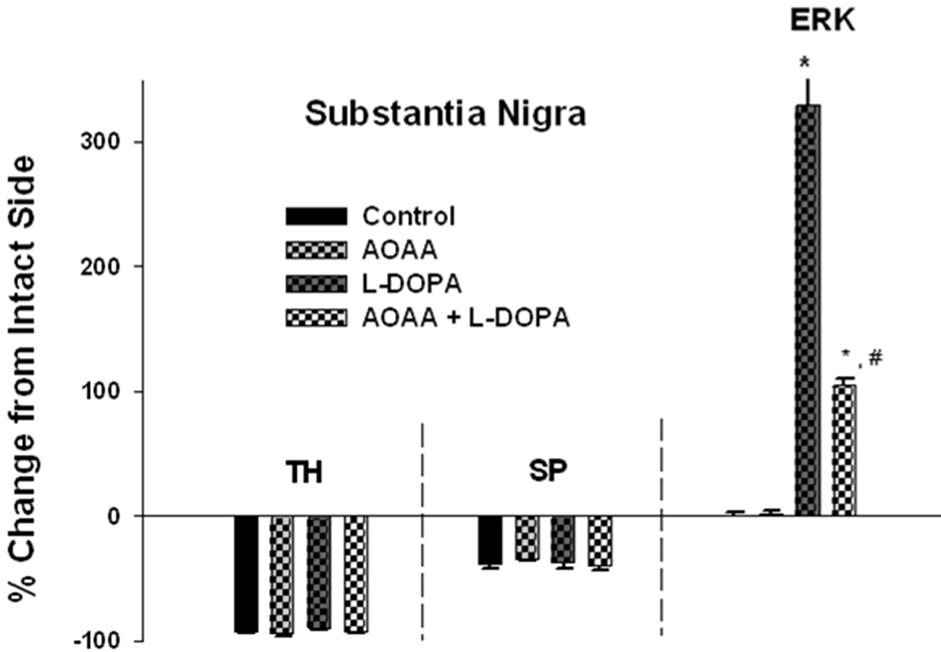
Figure 3. GABA attenuates L-DOPA-induced phosphoERK1/2 signaling: quantitative analysis of TH, SP, and phospho-ERK1/2 immunoreactivity following drug administration in the striatum (upper panel) and SN (lower panel) of unilaterally 6-OHDA lesioned animals. TH immunoreactivity in all groups was severely depleted in the lesioned side as compared to the intact side. SP immunoreactivity in all groups was moderately depleted in the lesioned side as compared to the intact side. In control and AOAA treated animals, phospho-ERK1/2 immunoreactivity was not apparent. In L-DOPA treated animals, phospho-ERK1/2 immunoreactivity increased robustly in the striatum and SN in the lesioned side as compared to the intact side. In AOAA + L-DOPA treated rats, phospho-ERK1/2 immunoreactivity was significantly attenuated in the striatum and SN when compared to the L-DOPA group. Data were expressed as percent change of the lesioned side compared to intact side. *p < 0.05 as compared to the other groups. #p < 0.05 as compared to L-DOPA group.
that DA agonists such as L-DOPA increase activation of phospho-ERK1/2 in the DA-depleted striatum and SN, suggesting L-DOPA administration affects the entire striatonigral pathway. We report a new finding that elevation of GABA by a GABA-transaminase inhibitor, AOAA, attenuated the L-DOPA-induced rotational response as well as increase in phospho-ERK1/2 levels both in the striatum and SN, indicating a GABA-DAERK1/2 interaction in the therapeutic and/or side effects of L-DOPA.
The direct (striatonigral) and indirect (striatopallidal) pathway activity, subserved by D1 and D2 receptors, respectively, is crucial to the normal functioning of the basal ganglia [11,48,49] . DA neuron degeneration in the direct pathway leads to an imbalance of direct and indirect projection pathway activity and is generally attributed to the motor deficits associated with PD [7,11,15] . It has been shown previously that, in the intact striatum, the indirect rather than direct striatal projection neurons are typically responsible for activating the ERK1/2 signaling pathway [7]. However, after nigrostriatal DA lesion, phospho-ERK1/2 is activated via the direct pathway D1 receptors [6-8,25,50,51] . Furthermore, D1 receptor supersensitivity associated with PD is characterized by changes in synaptic plasticity [9,52] and gene expression [11,53] , such as increased expression of D1 receptors at the plasma membrane [21,54,55] and a switch in the regulation of the ERK1/2 signaling pathway [6] . Other evidence indicates that D1 receptor agonists such as SKF-38393 can induce phospho-ERK1/2 levels in the striatum [6,8,22,28,52] . Blockade of D1 receptors with D1-antagonist, SCH-23390 can block L-DOPA-induced ERK1/2 phosphorylation in neonatal (bilateral dopaminergic lesion) [22] and adult (unilateral dopaminergic lesion) models of PD [24,56-57] , indicating that activation of D1 receptors induces effects in the striatum via the ERK1/2 signaling cascade. The degree of ERK1/2 phosphorylation has been correlated with the severity of the motor side effect known as dyskinesias [16,24,58] . These and other studies, along with the present results, support the conclusion that L-DOPA-induced ERK1/2 phosphorylation as well as alleviation of motor symptoms and occurrence of side effects occur through D1 receptor stimulation in the striatum. However, to our knowledge, the involvement of other major transmitter systems of the basal ganglia, such as GABA on L-DOPA, induced ERK1/2 phosphorylation levels and motor side effects in DA depleted basal ganglia have not been studied before.
Reports have shown that L-DOPA or D1 receptor agonist administration induce changes in striatal GABA transmission [32] . Morphologically, GABAergic medium-sized spiny neurons (MSNs) receive input from all areas of the basal ganglia and are the main type of neuron in the striatum [21,29,30,59] and the SN pars reticulata (SNr) [29,30,60] . DA regulates the activity of these GABAergic MSNs by acting on DA receptors, providing a DA-GABA link in the basal ganglia [21,61] . In unilaterally 6-OHDA lesioned rats, restored DA activity by L-DOPA administration leads to increased GABA transmission in the basal ganglia, which includes the striatum, via activation of the D1 receptor [62]. Increased levels of the GABA-synthesizing enzyme glutamic acid decarboxylase (GAD) in the striatum after acute and, to a greater extent, chronic administration of L-DOPA dem-
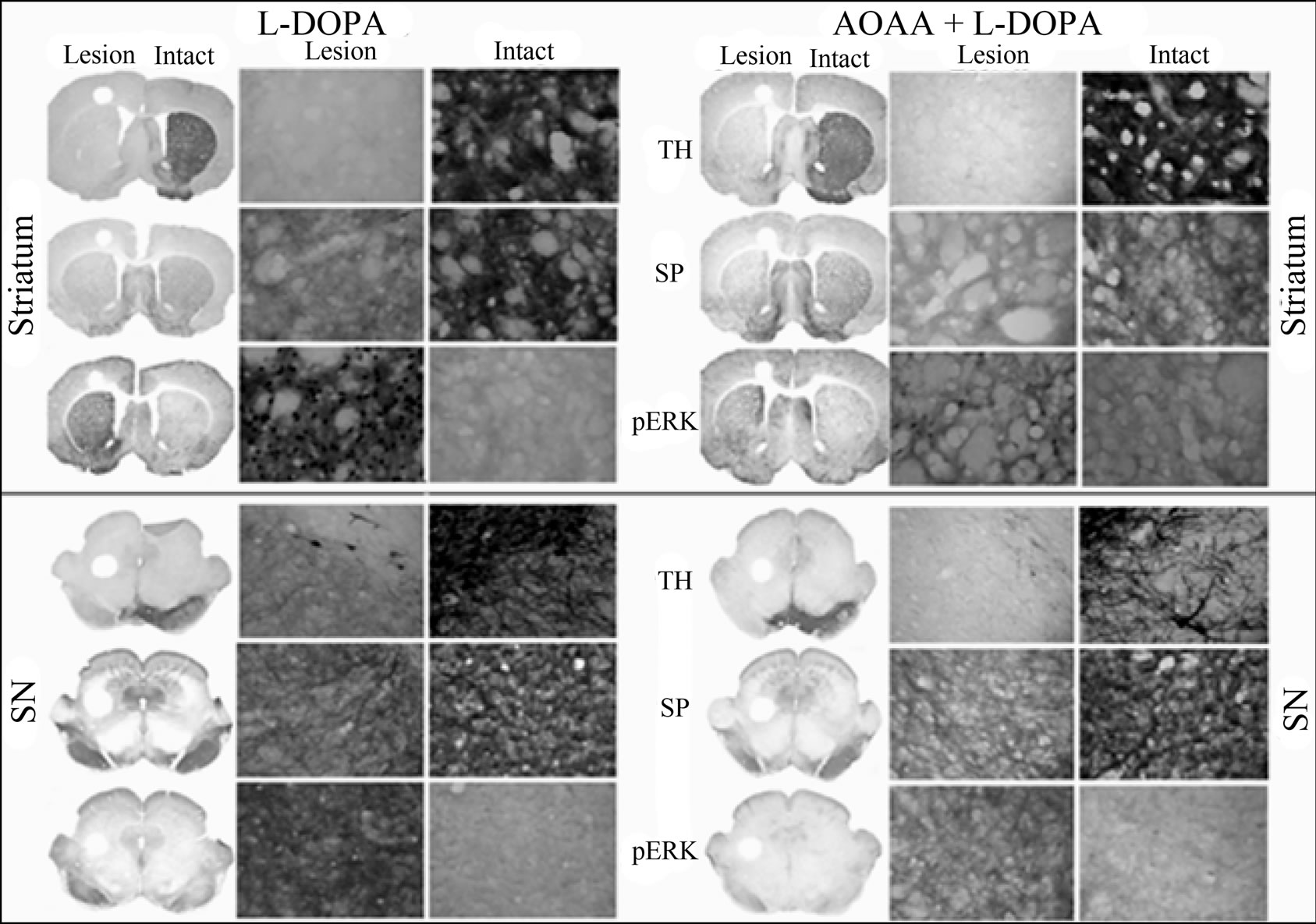
Figure 4. GABA attenuates L-DOPA-induced phospho-ERK1/2 signaling: representative micrographs showing immunohistochemistry analysis of TH, SP, and phospho-ERK1/2 following L-DOPA administration (left) and after AOAA + L-DOPA (right) in the striatum (upper panel) and SN (bottom panel) of unilaterally 6-OHDA lesioned animals. The loss of TH and SP immunoreactivity was not altered by the treatments. There was a robust increase of phospho-ERK1/2 immunoreactivity in the L-DOPA treatment group. AOAA treatment significantly attenuated L-DOPA-induced increase of phospho-ERK1/2 immunoreactivity. The coronal slices represent 1.25X magnification and the higher power images represent 400X magnification. A summary of changes in different groups is depicted in Figure 3.
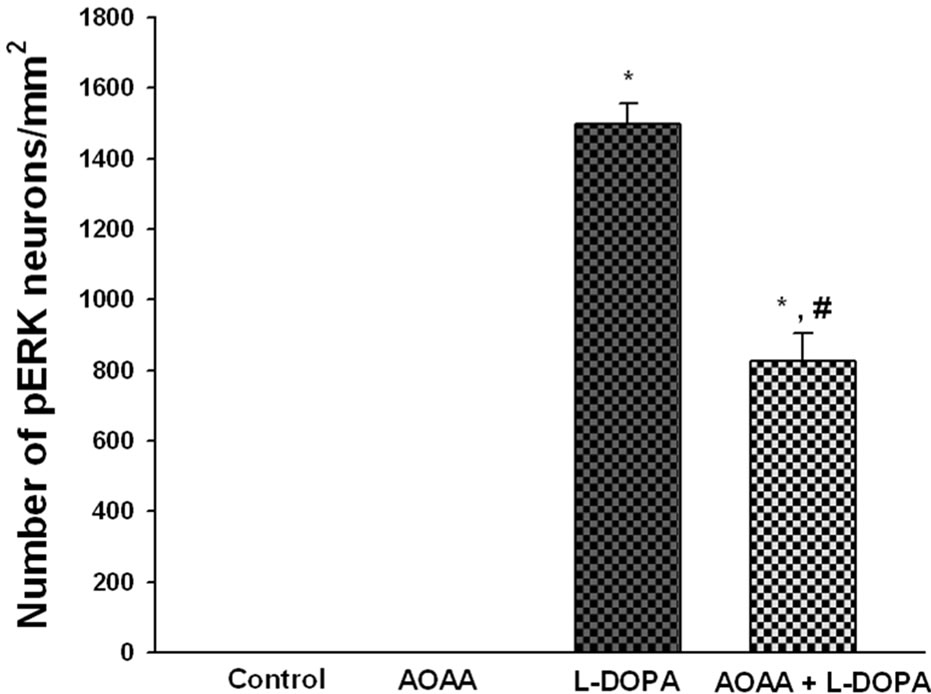
Figure 5. GABA attenuates the number of L-DOPA induced phospho-ERK1/2 labeled cells in the striatum of unilaterally 6-OHDA lesioned animals. In control animals and AOAA treated animals, no cells showing phospho-ERK1/2 immunoreactivity were observed. In L-DOPA treated animals, robust amounts of phospho-ERK1/2 labeled cells were observed. In AOAA + L-DOPA treated rats, significantly fewer phospho-ERK1/2 labeled cells were observed. *p < 0.05 as compared to the other groups. #p < 0.05 as compared to L-DOPA.
onstrate enhanced GABA transmission [34,63] . Similarly, vesicular GABA transporter (vGAT) mRNA codes for a protein responsible for regulating GABA release via synaptic vesicles; levels of vGAT mRNA were reported to be decreased in the DA denervated striatum, and increased following subchronic L-DOPA administration [35] .
The SN also displays changes in GABA transmission following D1 activation [33,36-38] . The substantia nigra pars compacta (SNc) contains the majority of DA neurons with dendrites extending to the SN pars reticulata (SNr), which primarily contains GABA projection neurons [60] lacking postsynaptic DA receptors [64]. D1 receptors were detected on presynaptic terminals onto GABAergic SNr neurons [65] . It was reported that L-DOPA administration stimulated GABA release in the SNr of 6-OHDA lesioned rats [62]. Trevitt, et al. (2002) showed similar results after administration of D1 agonist, SKF-82958 [32] . Also, the SKF-82958- or L-DOPA-induced effects were both completely blocked by coinfusion of D1 antagonist, SCH-23390 [32,62] . Both reports attributed the enhanced GABA release to the stimulation of D1 receptors. Western blot and immunofluorescence analyses showed that L-DOPA-induced ERK1/2 phosphorylation took place selectively on D1 receptorexpressing striatal MSNs [4,24,50,51] . These reports taken together suggest that the DA-GABA-ERK1/2 link is dependent on a D1 receptor mechanism.
According to neurochemical and behavioral studies, the D1 receptor-mediated facilitation of GABA transmission depends upon GABA-A receptor stimulation [32, 66] . GABA-A receptor stimulation resulted in increased DARPP-32 phosphorylation in the striatum and SN [67]. DARPP-32 is a phosphoprotein regulated by DA and cyclic AMP, and it is abundant in the striatum and SN [68]. Following L-DOPA administration, sensitized cAMP/cAMP-dependent protein kinase/DARPP-32 signaling in dyskinetic mice leads to the phosphorylation of ERK1/2 [26, 69] . Santini, et al. (2012) recently found that DARPP-32 is required for L-DOPA-induced ERK 1/2 phosphorylation in MSNs that express D1 receptors [70]. These reports suggest a DARPP-32 dependent mechanism for the involvement of DA, GABA, and ERK 1/2.
In other reports, Adenosine A2A receptor antagonist, KW-6002, has demonstrated prominent therapeutic effects in both animal models and human patients of PD [71,72] without inducing dyskinesias [73,74] . The receptor antagonist has also been shown to increase GABA release in the SNr of unilaterally 6-OHDA lesioned rats [75] , which suggests that increased GABA release could be linked to the amelioration rather than induction of motor side effects.
While the possible mechanism(s) underlying the involvement of GABA in L-DOPA therapy and ERK1/2 phosphorylation have not been verified, speculations about the ultimate consequence of GABA release in the striatum and SN are varied. The most common speculation is that the relationship between L-DOPA administration and enhanced GABA transmission could contribute to the development of motor side effects [35,38] . This opinion has likely formed due to numerous reports that L-DOPA administration in unilaterally 6-OHDA lesioned rats resulted in enhanced GABA levels in the SNr [36,38] . It was also reported that changes in GABA release mediated by L-DOPA are correlated with the behavioral effects of L-DOPA [38] . Similarly, GABA release in the SNr was increased after acute and subchronic L-DOPA administration [76] . L-DOPA also increased the levels of vesicular GABA transporter (vGAT) in the SNr ADDIN EN.CITE ADDIN EN.CITE.DATA [35] . Aceves, et al. (1991) reported stimulation of GABA release through L-DOPA-induced activation of presynaptic D1 receptors in the basal ganglia [62]. However, our results suggest that enhanced GABA transmission after L-DOPA administration could play a different role.
The aforementioned studies have suggested an involvement of GABA in L-DOPA-induced motor side effects, but according to the present results, the effect of GABA may be the opposite. Huot, et al. (2013) suggested that GABAergic modulation may be a useful tool in alleviating behavioral deficits associated with LDOPA treatment due to its centrality to basal ganglia physiology [77]. Furthermore, it is possible to speculate that enhanced GABA transmission could potentially compensate for motor side effects by reducing the impact of L-DOPA-induced D1-receptor-mediated increases in phospho-ERK1/2. Therefore, the administration of exogenous GABA may further reduce this impact by dampening ERK1/2 phosphorylation, as found in this study. Ochi, et al. (2004) speculated that the increase in GABA release in the SNr following L-DOPA administration may be attributed to the amelioration of PD symptoms by L-DOPA rather than the induction of the dyskinetic side effect [33]. Additional reports show that, independent of L-DOPA, local activation of GABA-A receptors in the striatum or SNr provides beneficial effects. Injections of the GABA-A receptor agonist muscimol into the SNr of parkinsonian monkeys have been reported to alleviate motor symptoms [78] . Similar results were observed after the implantation of genetically-engineered GABA-releasing cells in the SNr of 6-OHDA-lesioned rats [79] . Behavioral recovery was observed in 6-OHDA-lesioned rats after injection of benzodiazepines, which are believed to act via GABA-A receptors [80] , and after transplantation of GABA-rich grafts [81] , both into the striatum. These studies taken together suggest that GABAergic stimulation could lead to the amelioration of PD symptoms.
As previously noted in this discussion, it is tempting to speculate that the reported L-DOPA induced GABA release in DA depleted striatum is likely a compensatatory mechanism to inhibit motor side effects. GABAergic drugs have minor success in relieving the motor symptoms of PD in clinical trials [29]. The present results showing that a pharmacological increase in GABA levels attenuated the phospho-ERK1/2 levels and rotational response, which is characteristic of L-DOPA-induced motor deficits [25] , lend further support to the concept. Our results suggest that GABA is indeed closely involved in striatonigral D1 receptor supersensitivity, ERK1/2 phosphorylation, and motor manifestations, but that it is not likely a causative factor in the side effect itself. Instead, our data suggests that at high enough levels, GABAergic agents may help alleviate motor side effects.
In summary, we report for the first time that GABAlevel enhancement via AOAA attenuates the L-DOPAinduced phospho-ERK1/2 levels in the striatum and SN and also L-DOPA induced rotations. Our study suggests that a combination of L-DOPA and a GABA level-enhancing agent may potentially be employed to reduce L-DOPA induced motor side effects and enhance the therapeutic benefits of L-DOPA.
5. Acknowledgements
This work was supported in part by a Research Enhancement Award from IUPUI, a summer research internship award from IUSM-NW, and an award from IUN Research Fund. The generous supply of SP antiserum used in this study from Dr. J.-S. Hong, NIEHS, Research Triangle Park is greatly appreciated.
REFERENCES
- C. R. Gerfen, T. M. Engber, L. C. Mahan, Z. Susel, T. N. Chase, F. J. Monsma and D. R. Sibley, “D1 and D2 Dopamine Receptor-Regulated Gene Expression of Striatonigral and Striatopallidal Neurons,” Science, Vol. 250, No. 8986, 1990, pp. 1429-1432.
- C. R. Gerfen, “The Neostriatal Mosaic: Multiple Levels of Compartmental Organization in the Basal Ganglia,” Annual Review of Neuroscience, Vol. 15, No. 4, 1992, pp. 285-320.
- C. Le Moine and B. Bloch, “D1 and D2 Dopamine Receptor Gene Expression in the Rat Striatum: Sensitive cRNA Probes Demonstrate Prominent Segregation of D1 and D2 mRNAs in Distinct Neuronal Populations of the Dorsal and Ventral Striatum,” Journal of Computational Neurology, Vol. 355, No. 3, 1995, pp. 418-426.
- S. Gong, C. Zheng, M. L. Doughty, K. Losos, N. Didkovsky, U. B. Schambra, N. J. Nowak, A. Joyner, G. Leblanc, M. E. Hatten and N. Heintz, “A Gene Expression Atlas of the Central Nervous System Based on Bacterial Artificial Chromosomes,” Nature, Vol. 425, No. 6961, 2003, pp. 917-925.
- C. B. Brink, B. H. Harvey, J. Bodenstein, D. P. Venter and D. W. Oliver, “Recent Advances in Drug Action and Therapeutics: Relevance of Novel Concepts in G-Protein-Coupled Receptor and Signal Transduction Pharmacology,” British Journal of Clinical Pharmacology, Vol. 57, No. 4, 2004, pp. 373-387. doi:10.1111/j.1365-2125.2003.02046.x
- C. R. Gerfen, S. Miyachi, R. Paletzki and P. Brown, “D1 Dopamine Receptor Supersensitivity in the Dopamine-Depleted Striatum Results from a Switch in the Regulation of ERK1/2/MAP Kinase,” Journal of Neuroscience, Vol. 22, No. 12, 2002, pp. 5042-5054.
- C. R. Gerfen, “D1 Dopamine Receptor Supersensitivity in the Dopamine-Depleted Striatum: Animal Model of Parkinson’s Disease,” Neuroscientist, Vol. 9, No. 6, 2003, pp. 455-462.
- D. S. Kim, R. D. Palmiter, A. Cummins and C. R. Gerfen, “Reversal of Supersensitive Striatal Dopamine D1 Receptor Signaling and Extracellular Signal-Regulated Kinase Activity in Dopamine-Deficient Mice,” Neuroscience, Vol. 137, No. 4, 2006, pp. 1381-1388. doi:10.1016/j.neuroscience.2005.10.054
- G. M. Thomas and R. L. Huganir, “MAPK Cascade Signalling and Synaptic Plasticity,” Nature Reviews Neuroscience, Vol. 5, No. 3, 2004, pp. 173-183. doi:10.1038/nrn1346
- M. Costa-Mattioli, W. S. Sossin, E. Klann and N. Sonenberg, “Translational Control of Long-Lasting Synaptic Plasticity and Memory,” Neuron, Vol. 61, No. 1, 2009, pp. 10-26. doi:10.1016/j.neuron.2008.10.055
- C. R. Gerfen, “Molecular Effects of Dopamine on Striatal Projection Pathways,” Trends in Neurosciences, Vol. 23, Suppl. 10, 2000, pp. S64-S70.
- J. W. Mink, “The Basal Ganglia and Involuntary Movements: Impaired Inhibition of Competing Motor Patterns,” Archives of Neurology, Vol. 60, No. 10, 2003, pp. 1365-1368. doi:10.1001/archneur.60.10.1365
- C. D. Marsden and J. D. Parkes, “Success and Problems of Long-Term Levodopa Therapy in Parkinson’s Disease,” The Lancet, Vol. 1, No. 8007, 1977, pp. 345-349. doi:10.1016/S0140-6736(77)91146-1
- T. M. Engber, Z. Susel, S. Kuo, C. R. Gerfen and T. N. Chase, “Levodopa Replacement Therapy Alters Enzyme Activities in Striatum and Neuropeptide Content in Striatal Output Regions of 6-Hydroxydopamine Lesioned Rats,” Brain Research, Vol. 552, No. 1, 1991, pp. 113- 118.
- M. Lebel, P. Robinson and M. Cyr, “Canadian Association of Neurosciences Review: The Role of Dopamine Receptor Function in Neurodegenerative Diseases,” Canadian Journal of Neurological Sciences, Vol. 34, No. 1, 2007, pp. 18-29.
- A. Berthet and E. Bezard, “Dopamine Receptors and L- DOPA-Induced Dyskinesia,” Parkinsonism & Related Disorders, Vol. 15, Suppl. 4, 2009, pp. S8-12. doi:10.1016/S1353-8020(09)70827-2
- A. Nadjar, C. R. Gerfen and E. Bezard, “Priming for L- DOPA-Induced Dyskinesia in Parkinson’s Disease: A Feature Inherent to the Treatment or the Disease?” Progress in Neurobiology, Vol. 87, No. 1, 2009, pp. 1-9. doi:10.1016/j.pneurobio.2008.09.013
- T. N. Chase, M. M. Mouradian and T. M. Engber, “Motor Response Complications and the Function of Striatal Efferent Systems,” Neurology, Vol. 43, No. 12, Suppl. 6, 1993, pp. S23-S27.
- J. A. Obeso, C. W. Olanow and J. G. Nutt, “Levodopa Motor Complications in Parkinson’s Disease,” Trends in Neurosciences, Vol. 23, Suppl. 10, 2000, pp. S1-S7.
- J. E. Ahlskog and M. D. Muenter, “Frequency of Levodopa-Related Dyskinesias and Motor Fluctuations as Estimated from the Cumulative Literature,” Movement Disorders, Vol. 16, No. 3, 2001, pp. 448-458. doi:10.1002/mds.1090
- M. Feyder, A. Bonito-Oliva and G. Fisone, “L-DOPA- Induced Dyskinesia and Abnormal Signaling in Striatal Medium Spiny Neurons: Focus on Dopamine D1 Receptor-Mediated Transmission,” Frontiers in Behavioral Neuroscience, Vol. 5, 2011, p. 71.
- S. T. Papadeas, B. L. Blake, D. J. Knapp and G. R. Breese, “Sustained Extracellular Signal-Regulated Kinase 1/2 Phosphorylation in Neonate 6-Hydroxydopamine-Lesioned Rats after Repeated D1-Dopamine Receptor Agonist Administration: Implications for NMDA Receptor Involvement,” Journal of Neuroscience, Vol. 24, No. 26, 2004, pp. 5863-5876. doi:10.1523/JNEUROSCI.0528-04.2004
- E. Bezard, C. E. Gross, L. Qin, V. V. Gurevich, J. L. Benovic and E. V. Gurevich, “L-DOPA Reverses the MPTP-Induced Elevation of the Arrestin2 and GRK6 Expression and Enhanced ERK Activation in Monkey Brain,” Neurobiology of Disease, Vol. 18, No. 2, 2005, pp. 323-335. doi:10.1016/j.nbd.2004.10.005
- J. E. Westin, L. Vercammen, E. M. Strome, C. Konradi and M. A. Cenci, “Spatiotemporal Pattern of Striatal ERK1/2 Phosphorylation in a Rat Model of L-DOPA-Induced Dyskinesia and the Role of Dopamine D1 Receptors,” Biological Psychiatry, Vol. 62, No. 7, 2007, pp. 800-810. doi:10.1016/j.biopsych.2006.11.032
- N. Pavon, A. B. Martin, A. Mendialdua and R. Moratalla, “ERK Phosphorylation and FosB Expression Are Associated with L-DOPA-Induced Dyskinesia in Hemiparkinsonian Mice,” Biological Psychiatry, Vol. 59, No. 1, 2006, pp. 64-74. doi:10.1016/j.biopsych.2005.05.044
- E. Santini, E. Valjent, A. Usiello, M. Carta, A. Borgkvist, J. A. Girault, D. Herve, P. Greengard and G. Fisone, “Critical Involvement of cAMP/DARPP-32 and Extracellular Signal-Regulated Protein Kinase Signaling in LDOPA-Induced Dyskinesia,” Journal of Neuroscience, Vol. 27, No. 26, 2007, pp. 6995-7005. doi:10.1523/JNEUROSCI.0852-07.2007
- M. A. Cenci, K. E. Ohlin and D. Rylander, “Plastic Effects of L-DOPA Treatment in the Basal Ganglia and Their Relevance to the Development of Dyskinesia,” Parkinsonism & Related Disorders, Vol. 15, Suppl. 3, 2009, pp. S59-63. doi:10.1016/S1353-8020(09)70782-5
- C. Moreno and S. Sivam, “The Time Course of D1 Agonist Induced Striatonigral ERK1/2 Signaling in a Rat Model of Parkinson’s Disease,” Journal of Behavioral and Brain Science, Vol. 2, No. 1, 2012, pp. 1-9. doi:10.4236/jbbs.2012.21001
- A. Galvan and T. Wichmann, “GABAergic Circuits in the Basal Ganglia and Movement Disorders,” Progress in Brain Research, Vol. 160, No. 3, 2007, pp. 287-312.
- J. M. Tepper, E. D. Abercrombie and J. P. Bolam, “Basal Ganglia Macrocircuits,” Progress in Brain Research, Vol. 160, 2007, pp. 3-7.
- U. Misgeld, G. Drew and Y. Yanovsky, “Presynaptic Modulation of GABA Release in the Basal Ganglia,” Progress in Brain Research, Vol. 160, 2007, pp. 245-259.
- T. Trevitt, B. Carlson, M. Correa, A. Keene, M. Morales and J. D. Salamone, “Interactions between Dopamine D1 Receptors and Gamma-Aminobutyric Acid Mechanisms in Substantia Nigra Pars Reticulata of the Rat: Neurochemical and Behavioral Studies,” Psychopharmacology, Vol. 159, No. 3, 2002, pp. 229-237. doi:10.1007/s002130100908
- M. Ochi, S. Shiozaki and H. Kase, “L-DOPA-Induced Modulation of GABA and Glutamate Release in Substantia Nigra Pars Reticulata in a Rodent Model of Parkinson’s Disease,” Synapse, Vol. 52, No. 2, 2004, pp. 163- 165. doi:10.1002/syn.20006
- J. Katz, K. M. Nielsen and J. J. Soghomonian, “Comparative Effects of Acute or Chronic Administration of Levodopa to 6-Hydroxydopamine-Lesioned Rats on the Expression of Glutamic acid Decarboxylase in the Neostriatum and GABAA Receptors Subunits in the Substantia Nigra, Pars Reticulata,” Neuroscience, Vol. 132, No. 3, 2005, pp. 833-842. doi:10.1016/j.neuroscience.2004.12.032
- H. Wang, J. Katz, P. Dagostino and J. J. Soghomonian, “Unilateral 6-Hydroxydopamine Lesion of Dopamine Neurons and Subchronic L-DOPA Administration in the Adult Rat Alters the Expression of the Vesicular GABA Transporter in Different Subsets of Striatal Neurons and in the Substantia Nigra, Pars Reticulata,” Neuroscience, Vol. 145, No. 2, 2007, pp. 727-737. doi:10.1016/j.neuroscience.2006.12.001
- C. Rangel-Barajas, I. Silva, M. Garcia-Ramirez, E. Sanchez-Lemus, L. Floran, J. Aceves, D. Erlij and B. Floran, “6-OHDA-Induced Hemiparkinsonism and Chronic L-DOPA Treatment Increase Dopamine D1-Stimulated [3H]-GABA Release and [3H]-cAMP Production in Substantia Nigra Pars Reticulata of the Rat,” Neuropharmacology, Vol. 55, No. 5, 2008, pp. 704-711. doi:10.1016/j.neuropharm.2008.06.002
- N. Yamamoto and J. J. Soghomonian, “Time-Course of SKF-81297-Induced Increase in Glutamic Acid Decarboxylase 65 and 67 mRNA Levels in Striatonigral Neurons and Decrease in GABAA Receptor alpha1 Subunit mRNA Levels in the Substantia Nigra, Pars Reticulata, in Adult Rats with a Unilateral 6-Hydroxydopamine Lesion,” Neuroscience, Vol. 154, No. 3, 2008, pp. 1088- 1099. doi:10.1016/j.neuroscience.2008.04.015
- C. Rangel-Barajas, I. Silva, L. M. Lopez-Santiago, J. Aceves, D. Erlij and B. Floran, “L-DOPA-Induced Dyskinesia in Hemiparkinsonian Rats Is Associated with Up-Regulation of Adenylyl Cyclase Type V/VI and Increased GABA Release in the Substantia Nigra Reticulata,” Neurobiology of Disease, Vol. 41, No. 1, 2011, pp. 51-61. doi:10.1016/j.nbd.2010.08.018
- Y. Cui, R. M. Costa, G. G. Murphy, Y. Elgersma, Y. Zhu, D. H. Gutmann, L. F. Parada, I. Mody and A. J. Silva, “Neurofibromin Regulation of ERK Signaling Modulates GABA Release and Learning,” Cell, Vol. 135, No. 3, 2008, pp. 549-560. doi:10.1016/j.cell.2008.09.060
- G. Gangarossa, J. Espallergues, A. de Kerchove d’Exaerde, S. El Mestikawy, C. R. Gerfen, D. Herve, J. A. Girault and E. Valjent, “Distribution and Compartmental Organization of GABAergic Medium-Sized Spiny Neurons in the Mouse Nucleus Accumbens,” Front Neural Circuits, Vol. 7, 2013, p. 22. doi:10.3389/fncir.2013.00022
- G. Paxinos and C. Watson, “The Rat Brain in Stereotaxic Coordinates,” 5th Edition, Elsevier Academic Press, New York, 2005.
- G. R. Breese, A. A. Baumeister, T. J. McCown, S. G. Emerick, G. D. Frye, K. Crotty and R. A. Mueller, “Behavior Differences between Neonatal and Adult 6-Hydrocydopamine-Treated Rats to Dopamine Agonists: Relevance to Neurological Symptoms in Clinical Syndromes with Reduced Brain Dopamine,” Journal of Pharmacology and Experimental Therapeutics, Vol. 231, No. 2, 1984, pp. 343-354.
- S. P. Sivam and H. J. Hong, “GABAergic Regulation of Enkephalin in Rat Striatum: Alterations in Met5-Enkephalin Level, Precursor Content and Preproenkephalin Messenger RNA Abundance,” Journal of Pharmacology and Experimental Therapeutics, Vol. 237, No. 1, 1985, pp. 326-331.
- S. J. Li, S. P. Sivam, J. F. McGinty, Y. S. Huang and J. S. Hong, “Dopaminergic Regulation of Tachykinin Metabolism in the Striatonigral Pathway,” Journal of Pharmacology and Experimental Therapeutics, Vol. 243, No. 2, 1987, pp. 792-798.
- M. Andersson, C. Konradi and M. A. Cenci, “cAMP Response Element-Binding Protein Is Required for Dopamine-Dependent Gene Expression in the Intact but Not the Dopamine-Denervated Striatum,” Journal of Neuroscience, Vol. 21, No. 24, 2001, pp. 9930-9943.
- L. K. Nisenbaum, W. R. Crowley and S. T. Kitai, “Partial Striatal Dopamine Depletion Differentially Affects Striatal Substance P and Enkephalin Messenger RNA Expression,” Brain Research. Molecular Brain Research, Vol. 37, No. 1-2, 1996, pp. 209-216. doi:10.1016/0169-328X(95)00317-L
- S. P. Sivam, G. R. Breese, J. E. Krause, T. C. Napier, R. A. Mueller and J. S. Hong, “Neonatal and adult 6-Hydroxydopamine-Induced Lesions Differentially Alter Tachykinin and Enkephalin Gene Expression,” Journal of Neurochemistry, Vol. 49, No. 5, 1987, pp. 1623-1633. doi:10.1111/j.1471-4159.1987.tb01036.x
- A. I. Levey, S. M. Hersch, D. B. Rye, R. K. Sunahara, H. B. Niznik, C. A. Kitt, D. L. Price, R. Maggio, M. R. Brann and B. J. Ciliax, “Localization of D1 and D2 Dopamine Receptors in Brain with Subtype-Specific Antibodies,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 90, No. 19, 1993, pp. 8861-8865. doi:10.1073/pnas.90.19.8861
- C. R. Gerfen, “Dopamine-Mediated Gene Regulation in Models of Parkinson’s Disease,” Annals of Neurology, Vol. 47, No. 4, 2000, pp. S42-S50.
- J. Bertran-Gonzalez, C. Bosch, M. Maroteaux, M. Matamales, D. Herve, E. Valjent and J. A. Girault, “Opposing Patterns of Signaling Activation in Dopamine D1 and D2 Receptor-Expressing Striatal Neurons in Response to Cocaine and Haloperidol,” The Journal of Neuroscience, Vol. 28, No. 22, 2008, pp. 5671-5685. doi:10.1523/JNEUROSCI.1039-08.2008
- E. Santini, C. Alcacer, S. Cacciatore, M. Heiman, D. Herve, P. Greengard, J. A. Girault, E. Valjent and G. Fisone, “L-DOPA Activates ERK Signaling and Phosphorylates Histone H3 in the Striatonigral Medium Spiny Neurons of Hemiparkinsonian Mice,” Journal of Neurochemistry, Vol. 108, No. 3, 2009, pp. 621-633. doi:10.1111/j.1471-4159.2008.05831.x
- C. R. Gerfen, R. Paletzki and P. Worley, “Differences between Dorsal and Ventral Striatum in Drd1a Dopamine Receptor Coupling of Dopamineand cAMP-Regulated Phosphoprotein-32 to Activation of Extracellular Signal-Regulated Kinase,” The Journal of Neuroscience, Vol. 28, No. 28, 2008, pp. 7113-7120. doi:10.1523/JNEUROSCI.3952-07.2008
- J. D. Berke, R. F. Paletzki, G. J. Aronson, S. E. Hyman and C. R. Gerfen, “A Complex Program of Striatal Gene Expression Induced by Dopaminergic Stimulation,” Journal of Neuroscience, Vol. 18, No. 14, 1998, pp. 5301- 5310.
- C. Guigoni, E. Doudnikoff, Q. Li, B. Bloch and E. Bezard, “Altered D1 Dopamine Receptor Trafficking in Parkinsonian and Dyskinetic Non-Human Primates,” Neurobiology of Disease, Vol. 26, No. 2, 2007, pp. 452-463. doi:10.1016/j.nbd.2007.02.001
- A. Berthet, G. Porras, E. Doudnikoff, H. Stark, M. Cador, E. Bezard and B. Bloch, “Pharmacological Analysis Demonstrates Dramatic Alteration of D1 Dopamine Receptor Neuronal Distribution in the Rat Analog of L-DOPA-Induced Dyskinesia,” Journal of Neuroscience, Vol. 29, No. 15, 2009, pp. 4829-4835. doi:10.1523/JNEUROSCI.5884-08.2009
- S. Sivam and C. Moreno, “D1 Antagonist Blocks L-DOPA-Induced Striatal and Nigral ERK1/2 Phosphorylation in a Rat Model of Parkinson’s Disease,” Parkinsonism & Related Disorders, Vol. 15, Suppl. 2, 2009, p. S113. doi:10.1016/S1353-8020(09)70442-0
- H. S. Lindgren, K. E. Ohlin and M. A. Cenci, “Differential Involvement of D1 and D2 Dopamine Receptors in L-DOPA-Induced Angiogenic Activity in a Rat Model of Parkinson’s Disease,” Neuropsychopharmacology, Vol. 34, No. 12, 2009, pp. 2477-2488. doi:10.1038/npp.2009.74
- S. Schuster, A. Nadjar, J. T. Guo, Q. Li, C. Ittrich, B. Hengerer and E. Bezard, “The 3-Hydroxy-3-Methylglutaryl-CoA Reductase Inhibitor Lovastatin Reduces Severity of L-DOPA-Induced Abnormal Involuntary Movements in Experimental Parkinson’s Disease,” Journal of Neuroscience, Vol. 28, No. 17, 2008, pp. 4311-4316. doi:10.1523/JNEUROSCI.4720-07.2008
- H. Kita and S. T. Kitai, “Glutamate Decarboxylase Immunoreactive Neurons in Rat Neostriatum: Their Morphological Types and Populations,” Brain Research, Vol. 447, No. 2, 1988, pp. 346-352. doi:10.1016/0006-8993(88)91138-9
- T. Gonzalez-Hernandez and M. Rodriguez, “Compartmental Organization and Chemical Profile of Dopaminergic and GABAergic Neurons in the Substantia Nigra of the Rat,” Journal of Computational Neurology, Vol. 421, No. 1, 2000, pp. 107-135.
- E. Santini, E. Valjent and G. Fisone, “Parkinson’s Disease: Levodopa-Induced Dyskinesia and Signal Transduction,” FEBS Journal, Vol. 275, No. 7, 2008, pp. 1392- 1399. doi:10.1111/j.1742-4658.2008.06296.x
- J. Aceves, B. Floran, D. Martinez-Fong, A. Sierra, S. Hernandez and S. Mariscal, “L-Dopa Stimulates the Release of [3H]Gamma-Aminobutyric Acid in the Basal Ganglia of 6-Hydroxydopamine Lesioned Rats,” Neuroscience Letters, Vol. 121, No. 1-2, 1991, pp. 223-226. doi:10.1016/0304-3940(91)90690-U
- P. Barroso-Chinea and E. Bezard, “Basal Ganglia Circuits Underlying the Pathophysiology of Levodopa-Induced Dyskinesia,” Frontiers in Neuroanatomy, Vol. 4, 2010, p. 131.
- K. K. Yung, J. P. Bolam, A. D. Smith, S. M. Hersch, B. J. Ciliax and A. I. Levey, “Immunocytochemical Localization of D1 and D2 Dopamine Receptors in the Basal Ganglia of the Rat: Light and Electron Microscopy,” Neuroscience, Vol. 65, No. 3, 1995, pp. 709-730. doi:10.1016/0306-4522(94)00536-E
- E. Valjent, V. Pascoli, P. Svenningsson, S. Paul, H. Enslen, J. C. Corvol, A. Stipanovich, J. Caboche, P. J. Lombroso, A. C. Nairn, P. Greengard, D. Herve and J. A. Girault, “Regulation of a Protein Phosphatase Cascade Allows Convergent Dopamine and Glutamate Signals to Activate ERK in the Striatum,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 102, No. 2, 2005, pp. 491-496. doi:10.1073/pnas.0408305102
- H. Ikeda, A. Kotani, N. Koshikawa and A. R. Cools, “Differential Role of GABAA and GABAB Receptors in Two Distinct Output Stations of the Rat Striatum: Studies on the Substantia Nigra Pars Reticulata and the Globus Pallidus,” Neuroscience, Vol. 167, No. 1, 2010, pp. 31-39. doi:10.1016/j.neuroscience.2010.01.054
- G. L. Snyder, G. Fisone and P. Greengard, “Phosphorylation of DARPP-32 Is Regulated by GABA in Rat Striatum and Substantia Nigra,” Journal of Neurochemistry, Vol. 63, No. 5, 1994, pp. 1766-1771. doi:10.1046/j.1471-4159.1994.63051766.x
- C. C. Ouimet, P. E. Miller, H. C. Hemmings Jr., S. I. Walaas and P. Greengard, “DARPP-32, a Dopamineand Adenosine 3’:5’-Monophosphate-Regulated Phosphoprotein Enriched in Dopamine-Innervated Brain Regions. III. Immunocytochemical Localization,” Journal of Neuroscience, Vol. 4, No. 1, 1984, pp. 111-124.
- Y. Ding, L. Won, J. P. Britt, S. A. Lim, D. S. McGehee and U. J. Kang, “Enhanced Striatal Cholinergic Neuronal Activity Mediates L-DOPA-Induced Dyskinesia in Parkinsonian Mice,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 108, No. 2, 2011, pp. 840-845. doi:10.1073/pnas.1006511108
- E. Santini, M. Feyder, G. Gangarossa, H. S. Bateup, P. Greengard and G. Fisone, “Dopamineand cAMP-Regulated Phosphoprotein of 32-kDa (DARPP-32)-Dependent Activation of ERK and Mammalian Target of Rapamycin Complex 1 (TORC1) Signaling in Experimental Parkinsonism,” Journal of Biological Chemistry, Vol. 287, 2012, pp. 27806-27812. doi:10.1074/jbc.M112.388413
- R. A. Hauser, J. P. Hubble and D. D. Truong, “Randomized Trial of the Adenosine A2A Receptor Antagonist Istradefylline in Advanced PD,” Neurology, Vol. 61, No. 3, 2003, pp. 297-303. doi:10.1212/01.WNL.0000081227.84197.0B
- H. Kase, S. Aoyama, M. Ichimura, K. Ikeda, A. Ishii, T. Kanda, K. Koga, N. Koike, M. Kurokawa, Y. Kuwana, A. Mori, J. Nakamura, H. Nonaka, M. Ochi, M. Saki, J. Shimada, T. Shindou, S. Shiozaki, F. Suzuki, M. Takeda, K. Yanagawa, P. J. Richardson, P. Jenner, P. Bedard, E. Borrelli, R. A. Hauser and T. N. Chase, “Progress in Pursuit of Therapeutic A2A Antagonists: The Adenosine A2A Receptor Selective Antagonist KW6002: Research and Development toward a Novel Nondopaminergic Therapy for Parkinson’s Disease,” Neurology, Vol. 61, Suppl 6, 2003, pp. S97-S100. doi:10.1212/01.WNL.0000095219.22086.31
- P. Jenner, “Istradefylline, a Novel Adenosine A2A Receptor Antagonist, for the Treatment of Parkinson’s Disease,” Expert Opinion on Investigational Drugs, Vol. 14, No. 6, 2005, pp. 729-738. doi:10.1517/13543784.14.6.729
- A. Pinna, J. Wardas, N. Simola and M. Morelli, “New Therapies for the Treatment of Parkinson’s Disease: Adenosine A2A Receptor Antagonists,” Life Sciences, Vol. 77, No. 26, 2005, pp. 3259-3267. doi:10.1016/j.lfs.2005.04.029
- M. Ochi, S. Shiozaki and H. Kase, “Adenosine A2A Receptor-Mediated Modulation of GABA and Glutamate Release in the Output Regions of the Basal Ganglia in a Rodent Model of Parkinson’s Disease,” Neuroscience, Vol. 127, No. 1, 2004, pp. 223-231. doi:10.1016/j.neuroscience.2004.04.050
- N. Yamamoto, R. C. Pierce and J. J. Soghomonian, “Subchronic Administration of L-DOPA to Adult Rats with a Unilateral 6-Hydroxydopamine Lesion of Dopamine Neurons Results in a Sensitization of Enhanced GABA Release in the Substantia Nigra, Pars Reticulata,” Brain Research, Vol. 1123, No. 1, 2006, pp. 196-200. doi:10.1016/j.brainres.2006.09.027
- P. Huot, T. H. Johnston, J. B. Koprich, S. H. Fox and J. M. Brotchie, “The Pharmacology of L-DOPA-Induced Dyskinesia in Parkinson’s Disease,” Pharmacological Reviews, Vol. 65, No. 1, 2013, pp. 171-222.
- T. Wichmann, M. A. Kliem and M. R. DeLong, “Antiparkinsonian and Behavioral Effects of Inactivation of the Substantia Nigra Pars Reticulata in Hemiparkinsonian Primates,” Experimental Neurology, Vol. 167, No. 2, 2001, pp. 410-424. doi:10.1006/exnr.2000.7572
- B. B. Carlson, S. Behrstock, A. J. Tobin and J. D. Salamone, “Brain Implantations of Engineered GABA-Releasing Cells Suppress Tremor in an Animal Model of Parkinsonism,” Neuroscience, Vol. 119, No. 4, 2003, pp. 927-932. doi:10.1016/S0306-4522(03)00218-5
- C. C. Tenn and L. P. Niles, “Mechanisms Underlying the Antidopaminergic Effect of Clonazepam and Melatonin in Striatum,” Neuropharmacology, Vol. 36, No. 11-12, 1997, pp. 1659-1663. doi:10.1016/S0028-3908(97)00165-2
- C. Winkler, C. Bentlage, G. Nikkhah, M. Samii and A. Bjorklund, “Intranigral Transplants of GABA-Rich Striatal Tissue Induce Behavioral Recovery in the Rat Parkinson Model and Promote the Effects Obtained by Intrastriatal Dopaminergic Transplants,” Experimental Neurology, Vol. 155, No. 2, 1999, pp. 165-186. doi:10.1006/exnr.1998.6916
NOTES
*Corresponding author.