Advances in Chemical Engineering and Science
Vol. 2 No. 2 (2012) , Article ID: 18893 , 16 pages DOI:10.4236/aces.2012.22026
Part 1: Synthesis and Evaluation of Novel Nano Scale Powdered Polyurethane Acrylate Binders
1Textile Research Division, National Research Centre, Cairo, Egypt
2Faculty of Science (Girls), Al-Azhar University, El-Nasser City, Cairo, Egypt
Email: melmolla@yahoo.com
Received September 22, 2011; revised October 27, 2011; accepted November 30, 2011
Keywords: Synthesis; Novel powdered; an Aqueous; Polyurethane; acrylate binders
ABSTRACT
Some new an aqueous polyurethane acrylate (waterborne binder) based on polyethylene glycol with different M. wt. mixed with polyol were carried out. Seven different polyurethane acrylate co-polymers were prepared aiming at substituting the two hydroxyl groups of polyethylene glycol (6000, 12,000 and 20,000 g/mol) and two primary hydroxyl groups of polyol through their reaction with corresponding calculated amounts of either isophorone diisoyanate or toluene diisocyanate and caped the remaining isocyanate group with either hydroxy ethyl acrylate or hydroxy propyl methacrylate to get on the polyurethane acrylate polymers [PUA]. From DSC measurement the result of Tg of synthesized PUA are in range from −8.78˚C to 36.4˚C. So they can classify as soft binders. The infrared spectra, rheological properties, viscosity measurement, the weight average molecular weight, of the synthesize binders were investigated.
1. Introduction
The binder is a film forming substance made up of long chain macromolecule which when applied to the textile, together with the pigment, produce three-dimensional linked network, during suitable fixation happened [1]. Binders employed in pigment printing of textile are polymer or copolymer of unsaturated monomer such as ethyl acrylate, butyl acrylate, styrene, acrylonitrile, vinyl acetate, butadiene, etc. [2].
The polymer composition being defined by application requirement however, pigment printing has few problems—relatively high temperature cure, stiff hand and poor crock fastness of printed goods, formaldehyde emission and clogging screen during the actual printing process. These disadvantages are related to binder used and must be taken into account. Thus to improve the quality of pigment printed goods the overall properties of the binder has to be improved. On the other hand, the chemistry of polyurethane (PU) formation is described in principally by the following reaction:
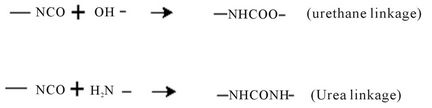
These reactions depend on the proportion of isocyanate and hydroxyl or amino groups [3]. Aliphatic isocyanate is represented by cycloaliphatic compounds, but simpler aliphatic compounds such as hexanethylene diisocyanate can also be used, the aromatic isocyanates can have more complicated structures. Hydroxyl-bearing materials, normal termed polyols, form the major part of polymer, polyesters, polyethers, and polycaprolactones are the three forms of commercial used polyols. Glycols usually used in the reaction include ethylene, propylene, diethyene, 1,4-butadienediol and 1,6-hexadi-enediol. Polyols based adipic acid and propylene or diethylene glycols are liquids, whereas those of the other three glycols are solids.
The chemical structure of polyol and isocyanate are important variables for polyurethane properties, other important variables include molecular weight, intermolecular force, and steric hinder. The molecular weight of polymers used in industry can be in the range of 600 to 8000 in case of oligomers. Oligomers have isocyanate fully reacted, but another functional groups (double bond or hydroxyl group) are left ready for further reaction in curing process [3].
The aim of this research is to synthesizing and evaluation of novel powdered waterborne polyurethane acrylate copolymer based on polyethylene glycol with different molecular weight and polyol mixture, with low viscosity for uses in pigment printing for all types of fabrics and colour fixation using UV light, microwaves and thermo fixation.
2. Experimental
2.1. Materials
Polyethylene glycol [(20,000 g/mol supplied by Sisco research laboratory pvt. ltd. India, PEG 12,000 g/mol, 6000 g/mol and dibutyl tin dilaurate (DBTDL) supplied by Fluka chemical [Co. Switzerland)], isophorone diisocyanate(IPDI) and toluene diisocyanate (TDI) supplied by Lypndell Chemical Co., Germany, hydroxy ethyl acrylate (HEA), hydroxyl propyl meth acrylate (HPMA) (Degussa, Germany) and polyol supplied by Sigma-Aldrich chemical Co., Germany were used.
2.2. Methods
Synthesis of Polyurethane Acrylate Co-Polymer
The reaction of polyethylene glycol with different molecular weight and polyol mixed with either isophorone diisocyanate and/or toluene diisocyante were conducting according to modification of procedure described elsewhere [4,5] as follows:
A calculated amount of polyethylene glycol (6000 g/mol, 12,000 g/mol and 20,000 g/mol (1:1) mol mixture were added as in a 70% acetone into a three necked flask equipped with stirrer, thermometer and reflux condenser under nitrogen atmosphere and left to about an hour to ensure complete mixing of the reaction.
A calculated amount of either IPDI and/or TDI containing 0.05 (w/w) Dibutyl tin dilaurate (DBTDL) as catalyst were slowly dropped into the reactor at 40˚C in case of IPDI and 30˚C in case of TDI for over an hour, the reaction mixture was stirred for additional 1 h. at 40˚C to ensure an acceptable reaction rate without gelation.
A calculated amount of either hydroxy ethyl acrylate (HEA) and/or hydroxy propyl methacrylate (HPMA) were gradually added to reaction mixture over an hour at 60˚C and the reaction mixture was stirred for another 2 hour continuously at 60˚C to cap the entire terminal NCO group.
The resulting product was a clear solution of polyurethane acrylate. It was divided into two parts; the first part was diluted by deionized water to obtain an-aqueous solution of resultant polyurethane acrylate, which was a clear solution with 50% (w/w), the reaction yield was evaluated. The part, which had left without dilution, had been solidified after cooling to obtain powdered polyurethane acrylate. The powdered polyurethane acrylate were broken up and ground down to very fine powder using GY-RO mill machine (mass ratio ball to material ratio 5:1). The function of ball mill is that use of highenergy milling to achieve particle size reduction. Particle sizes of resulting polyurethane acrylate had been characterized in two stages, one after the sample subjected to 5 days of milling and second after 15 days milling [6].
3. Measurement and Analysis
3.1. Infra Red Analysis (FT-IR)
The infra-red of the prepared polyurethane acrylate was measured using infra-red spectrophotometer, Perkin Elmer, system 2000FT-IR [7,8].
3.2. Rheological Properties
Rheological properties and apparent viscosity of the prepared aqueous polyurethane PUA’s solution of 10% (w/w) was measured using Brookfield model DV-111 programmable rheometer, USA at 25˚C and at different rate of shear.
3.3. Molecular Weight Measurement (Mw, Mn)
The molecular weight and molecular weight distributions of the polyurethane were obtained by Agilent Gel Permeation Chromatography (GPC), using a water peristaltic pump HPLC (Flow rate of DMF was 1 ml/min), and a water 410 refraction index detector [9,10].
3.4. Glass Transition Temperature (Tg)
The glass transition temperature Tg of the polyurethane acrylate was measured by Differential Scanning calorimetry shimadzu-50 (DSC) [10].
3.5. Transmission Electron Microscopy (TEM)
The samples size were measured by TEM Joel JEM 1230. ES 500 W model 782 Japan operation voltage 100 KV. The samples were investigated by prepared thin films from suspended solution of powders (by using hexane as suspended carrier) on cupper grad [9].
3.6. Scanning Electron Microscope (SEM)
Polyurethane acrylate’ samples mounted on aluminum stubs, and sputter coated with gold in a 150 Å sputter (Coated Edwards), and examined by Jeol (JXA-840A) Electron Probe Microanalysis (Japan),magnification range 35 - 10,000, accelerating voltage 19 kV [8,9].
4. Results and Discussion
Most acrylate are based on polyether, polyester, and epoxy resins, this is because the functionality of this condensation resins can be precisely controlled to 2 or 3 to prevent gelation and to keep low viscosity, diisocyanate react with either polyethylene glycol and/or polyol in the presence of catalyst (dibutyl tin dilaurate) to form isocyanate-terminated polyurethane prepolymer. Under controlled reaction condition and catalyst concentration one or two hydroxyl groups in the PEG and/or polyol react with the first isocyanate group of IPDI and/or TDI to form isocyanate-terminated polyurethane pre-polymer leaving the second isocyanate group intact for subsequent reaction with either hydroxyl ethyl acrylate or hydroxyl propyl meth acrylate to form polyurethane acrylate [4,5].
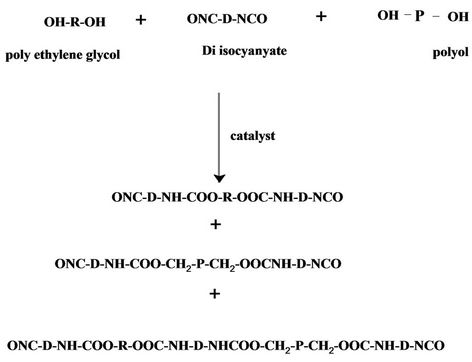
Urethane prepolymer where R =
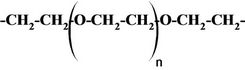
and D eiher
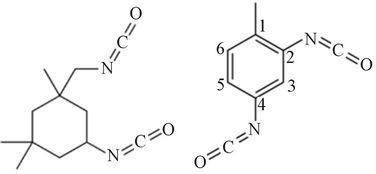
IPDI (1,3,3-trimethyl-cyclohexane) TDI (3, 5, 5-rimethyl-2-cyclohexen-1-one)
P = polyol Both IPDI and TDI is asymmetrical molecule, and thus have isocyanate groups with different reactivity. In IPDI, the secondary isocyanate group is more reactive than the primary isocyanate group [11], in TDI the 4-position is approximately four times more reactive than the 2-posion. 2, 6-TDI is a symmetrical molecule and thus has two isocyanate groups of similar reactivity, similar to the 2- position on 2, 4-TDI. However, since both isocyanate groups are attached to the same aromatic ring, reaction of one isocyanate group will cause a change in the reactivity of the second isocyanate group.
After that, the urethane prepolymer was reacted with either hydroxy ethyl acrylate or hydroxyl propyl methacrylate to give polyurethane acrylate as follows:
Polyurethane acrylate

Polyurethane acrylate Aromatic diisocyanates are used for the manufacture of the so-called aromatic urethane acrylates (e.g. Toluene diisocyanate). The incorporation of an aromatic diisocyanate makes the urethane acrylate harder, gives it a better scratch resistance. Aromatic urethane acrylates are also significantly lower cost than aliphatic urethane acrylate. The major drawback of aromatic urethane acrylates is that they tend to yellow and therefore they are less appropriate for long lasting applications on white or light colored substrates.
Aliphatic urethane acrylates, which based on aliphatic isocyanate (e.g. Isophorone diisocyanate) are slightly more flexible than aromatic urethane acrylates with the same functionality. The main advantage of aliphatic urethane acrylates is the fact that they are virtually non-elwing and therefore can be used for long lasting applications, on white or light colored substrates. Improvement of flexibility of acrylated urethane may be achieved by chain extension using long chain diol, and polyol to increase the cross-linking producing higher molecule weight isocyanate functional prepolymer, which is subsequently caped, by hydroxyl acrylate monomer.
Seven different polyurethane acrylate polymers were prepared aiming at substituting the two hydroxyl groups of polyethylene glycol (6000, 12,000 and 20,000 g/mol) and two primary hydroxyl groups of polyol through its reaction with corresponding calculated amounts of either isophorone diisocyanate or toluene diisocyanate and caped the remaining isocyanate groups with either hydroxy ethyl acrylate or hydroxy propyl methacrylate to get on the polyurethane acrylate polymers [PUA].
Characterization of synthesized PUA is shown in Figures 1-9 which represent the infra-red spectra. The formation of polyurethane acrylate was evidenced by the emergence of absorption bands at around 840 cm−1 cor-
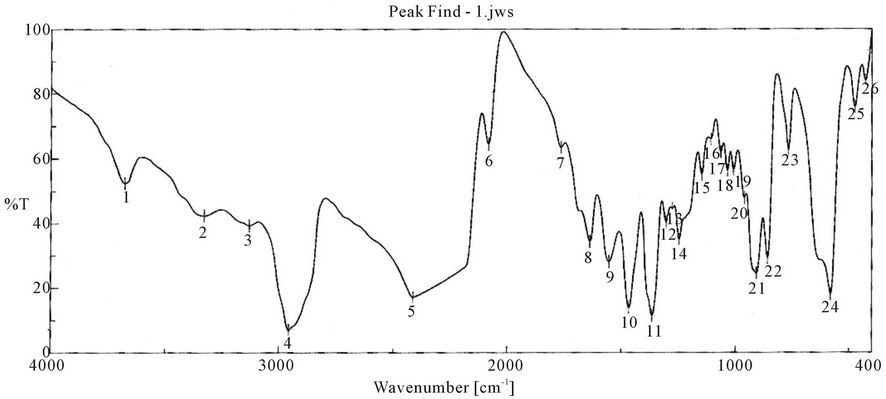
Figure 1. FTIR spectra of isophorone diisocyanate.
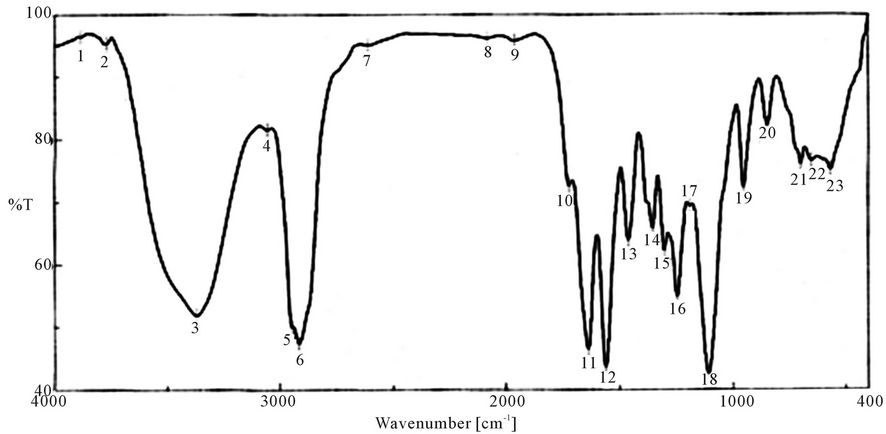
Figure 2. FTIR spectra of PUA* 1 (PEG 6000 g/mol + Polyol + IPDI + HE. *PUA 1 is Poly ethylene glycol 6000 + polyol + iso phoron di isocyanet + hydroxyl ethyl acrylate (di butyl tin di laurite used as catalyst).
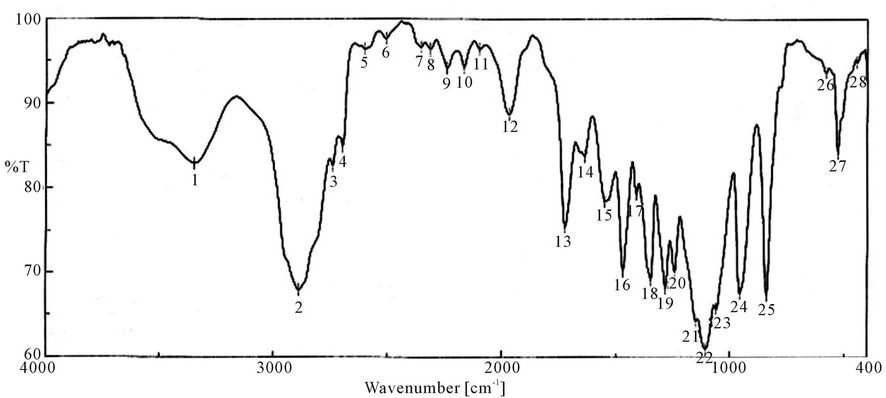
Figure 3. FTIR spectra of PUA* 2 (PEG 12,000 g/mol + Polyol + IPDI + HEA). *PUA 2 is Poly ethylene glycol 12,000 + polyol + iso phoron di isocyanet + hydroxyl ethyl acrylate (di butyl tin di laurite used as catalyst).
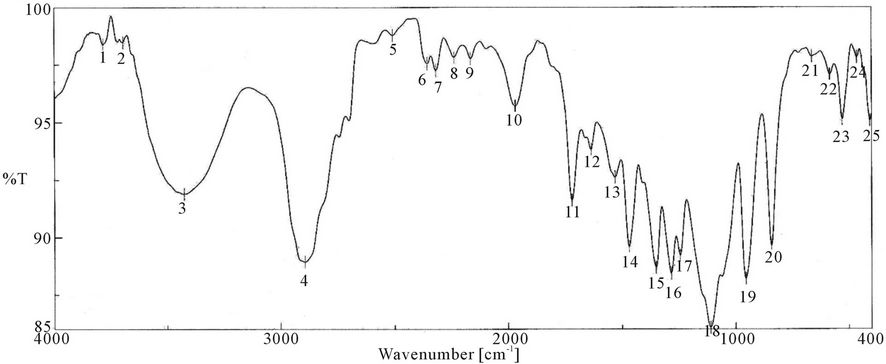
Figure 4. FTIR spectra of PUA* 3 (PEG 20,000 g/mol + Polyol + IPDI + HEA). *PUA 3 is Poly ethylene glycol 20,000 + polyol + iso phoron di isocyanet + hydroxyl ethyl acrylate (di butyl tin di laurite used as catalyst).
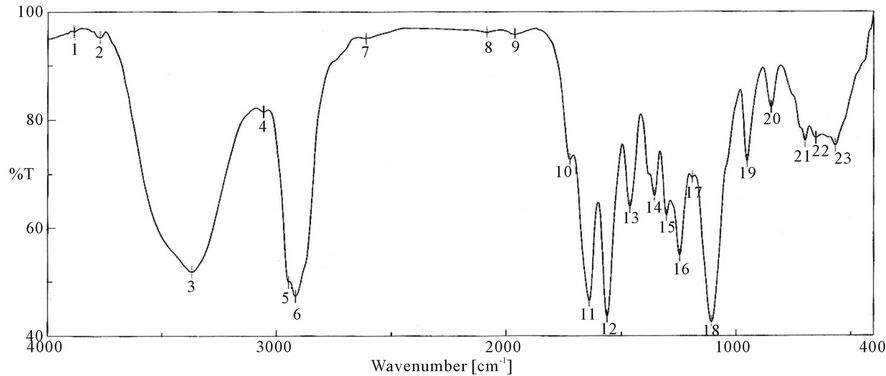
Figure 5. FTIR spectra of PUA* 4 (PEG 20,000 g/mol + Polyol + IPDI + HPMA). PUA 4 is polyethylene glycol 20,000 + polyol + isophoron di isocanet + hydroxyl propyl metha acrylate (di butyl tin di laurite used as catalyst).
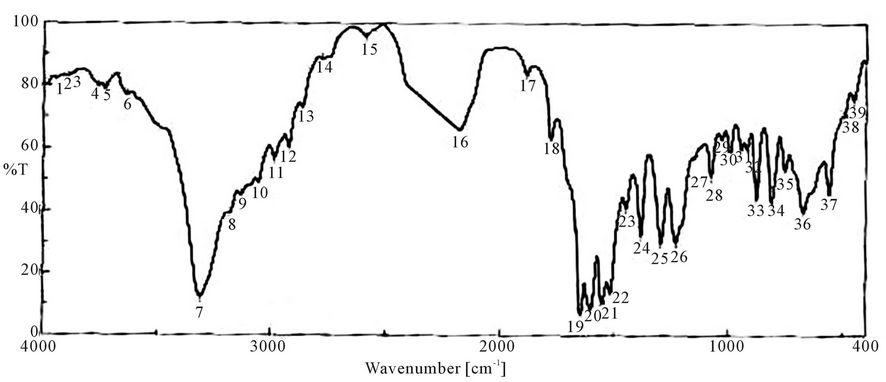
Figure 6. FTIR spectra of toluene diisocyanate.
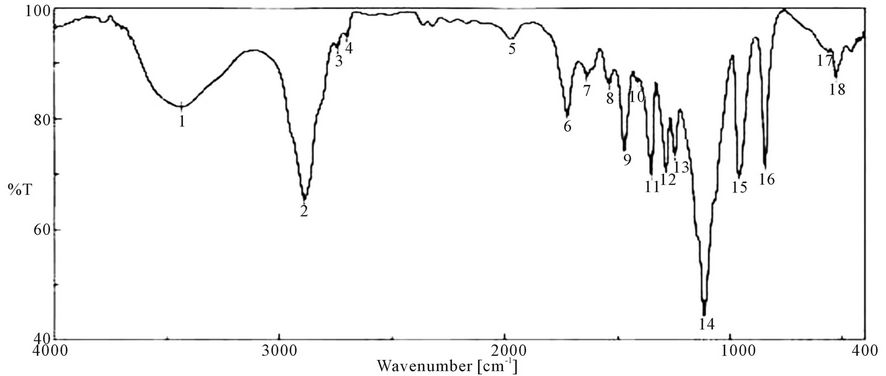
Figure 7. FTIR spectra of PUA* 5 (PEG 20,000 g/mol + Polyol + TDI + HEA). *PUA 5 is polyethylene glycol 20,000 + polyol + isophoron di isocanet + hydroxyl ethyl acrylate (di butyl tin di laurite used as catalyst).
Figure 8. FTIR spectra of PUA 6 (PEG 20,000 g/mol + Polyol + TDI + HPMA). *PUA 6 is polyethylene glycol 20,000 + polyol + isophoron di isocanet + hydroxyl propyl metha acrylate (di butyl tin di laurite used as catalyst).
Figure 9. FTIR spectra of PUA 7 (PEG 6000 g/mol + Polyol + TDI + HPMA). *PUA 7 is polyethylene glycol 6000 + polyol + isophoron di isocanet + hydroxyl propyl metha acrylate (di butyl tin di laurite used as catalyst).
responding to δ: C-H, 958 cm−1 δ:=C-H , strong absorption bands at around 1100 cm−1 (C-O-C), 1347 cm−1 st C-N ,1470 cm−1 δ: CH2, 1543 cm−1 δ: N-H, 1725 st C=O, 1986 cm−1 ast C=C (give three band which mean the presence of conjugated double bond aromatic ring and two in case of alicyclic ring) 2888 cm−1 st CH2, CH3,CH and broad band at around 3346 cm−1 originated from the hydrogen-bond OH group which is free and doesn’t take part in whole reaction.
It is clear from the spectra Figures 4-7 and 9-11 that no absorption band was observed at approximately 2100 - 2270 cm-1 which corresponding to N=C=O group as shown in Figures 3 and 8 and this indicates that the entire amount of either IPDI or TDI were consumed during the reaction and the final product is free from isocyanates.
All this confirm the occurrence of the addition reaction to obtain the aqueous polyurethane acrylate of PUA polymers.
4.1. Rheological Properties of Prepared Polyurethane Acrylate
The rheological properties and viscosity of PUA based on mixture of PEG of different molecular weight and polyol using 10% concentration had measured using Brookfield model DV-Ш, programmable Rheometer, at different rate of shear. Figure 10 shows the rheological properties of prepared polyurethane acrylate based on PEG with different M.wt and mixed with polyol. It is clear from these rheograms that all samples are characterized by non-Newtonian pseudo plastic behavior, where up and down curves is coincident. It is also clear from the same Figure 10 that, the location of the rheogram and its slope seems to be dependent on the type of PUA used.
The rheogram curve shifted far from the axis of the rate of shear indicating an increase in the apparent viscosity. It can be noticed that for aromatic polyurethane acrylate (polyurethane acrylate 5, 6, 7) from Figure 10, the PUA 5 has a rheogram curve shifted far from the axis of the rate of shear compared with the other polyurethane acrylate i.e. PUA 6 & 7 indicating an increase in the apparent viscosity and this is may be due to the difference in the M.wt, configuration& constituent. The apparent viscosity increased with increasing the molecular weight of the PEG used, as shown in Table 1, for example at a rate of shear 46.5 sec−1 the apparent viscosity of PUA 1 is 2.6 centipoises compared with 10 centipoises in case of PUA 4. The increase in apparent viscosity in PUA 4 may be due to the increase of PEG molecular weight from 6000 g/mol to 20,000 g/mol respectively and M.wt. of HPMA than that of HEA.
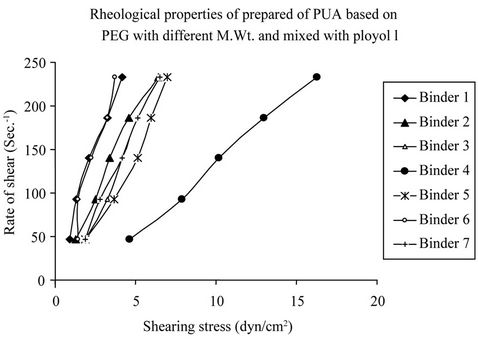
Figure 10. Rhograms of prepared polyurethane acrylate.
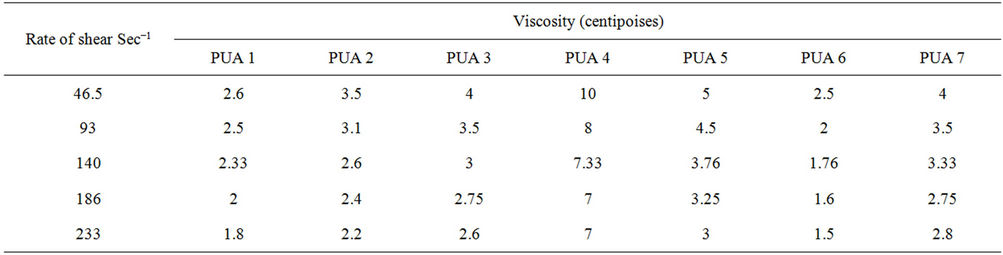
Table 1. The apparent viscosity of synthesized PUA polymers.
4.2. Effect of Structure of Polyurethane Acrylate Polymers on It’s the Glass Transition Temperature
The Tg is the temperature when a rigid amorphous thermoplastic glass polymer becomes elastomeric, and its stiffness drops steeply. These transitions appear as a step in the baseline of the recorded DSC signals. There is a relation between the locked-in configuration of polymer backbone chains and its Tg [12].
It is most easily appreciated by examining the effect of different backbone configurations of prepared polyurethane acrylate on the glass-transition temperature. The DSC signals of PUA based on PEG with different M∙wt mixed with polyol are measured and the curves are given in Figures 11-17.
The curves had one endothermic slope and one sharp endothermic peak. The onset point of endothermic peak showed that, cross-linking reaction happened at around 50˚C, providing the information that the printed fabrics should be subjected to heat treatment over 50˚C for strong pigment—fixation. From the data of DSC, it is noticed that Tg was affected by molecular weight of prepared polyurethane. As the molecular weight decreases, crosslink density increases and the Tg increases for polymers having similar chemical structures. This can be seen with polyurethane acrylate1, 2, 3 (10.6˚C, 6.66˚C, −8.78˚C respectively), that are very similar in backbone, but vary in molecular weight (PEG 6000, 12,000 and 20,000 g/mol respectively) [13]. It is also clear that Tg of PUA 4 (36.4˚C) is higher than those of later mentioned polyurethane acrylate, this may be attributed to the chain length and structure configuration (stuff CH3 group) which results in the difficulty of mobility of polymer chains and also the chain rigidity is increased.
In case of aromatic polyurethane acrylate (polyurethane acrylate 5, 6, 7), the benzene rings are trapped within the backbone which results in increasing the steric hindrance, and hence the Tg. PUA 6 shows irregular behavior as it has lower Tg than PUA 5. PUA 7 has higher Tg, this may be due to the lowest molecular weight of PEG (6000 g/mol) and to the longer chain length of HPMA and the existing of substitution group (CH3 group). The values of Tg of PUA 5, 6, 7 are 24.5˚C, 17.8˚C, 28.4˚C respectively.
4.3. Effect of Structure of Polyurethane Acrylate on the Molecular Weight Distribution and Poly Dispersity Index
Polymer molecular weight is important because it determines many physical properties. The number average molecular weight is a way of determining the molecular weight of a polymer [13]. Moreover, it considers a property, which is only sensitive to the number of molecules present—a property that is not influenced by the size of any particle in the mixture. The weight average molecular weight is a way of describing the molecular weight of a polymer and it considers of a polymer property which depends not just on the number of polymer molecules but on the size or weight of each polymer molecule [13].
In organic chemistry, the polydispersity index PDI is a measure of the distribution of molecular mass in a given sample and it is the ratio of weight average to the number average [14]. Ideal step-growth polymerization gives polymer with poly disperisty of 2. Ideal living polymerization results in polydisperisty of 1 [14].
The data obtained from GPC analysis of prepared polyurethane acrylate were hown in Table 2. The values of weight average molecular weight (Mw) for polyurethane acrylate 1, 2, 3 and 4 increases by increasing the molecular weight of polyethylene glycol (PEG 6000 g/mol, 12,000 g/mol and 20,000 g/mol respectively) used in preparation of the PUA polymers. The rate of increasing in value of Mw in case of PUA4 is faster than the others, which may be due to the high molecular weight of acrylate portion (HPMA). While the higher value of Mw of PUA 5 than that of PUA 6(both based on PEG 20,000 g/mol), may be due to ability of hydroxy propyl meth acrylate to extend polyurethane chain. In case of PUA 7, the decreases in Mw may be due to the drop in molecular weight of PEG from 20,000 g/mol to 6000 g/mol.
On the other hand the values of poly dispersity index PDI (Mw/Mn) for the first four polyurethane acrylate
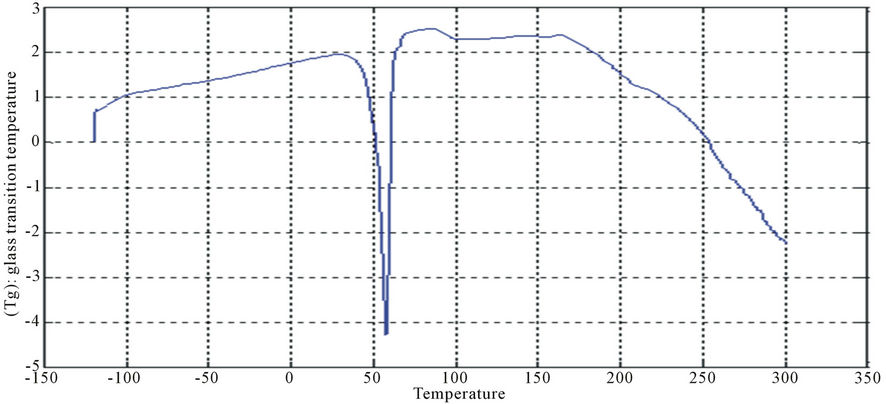
Figure 11. DSC curve of PUA 1.
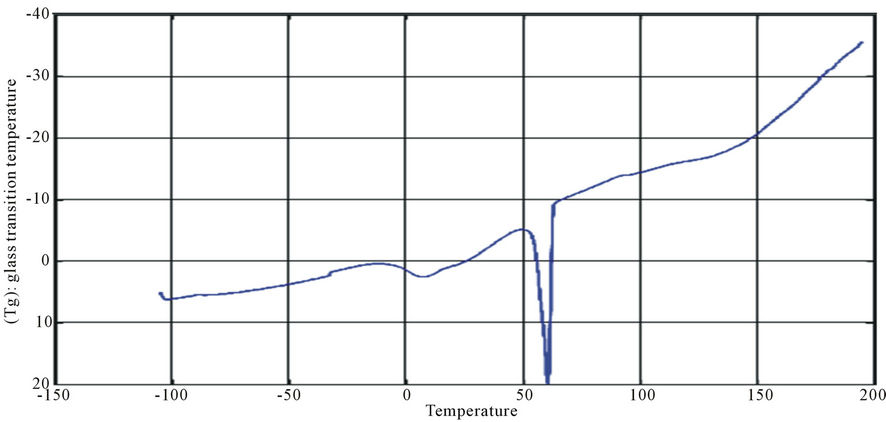
Figure 12. DSC curve of PUA 2.
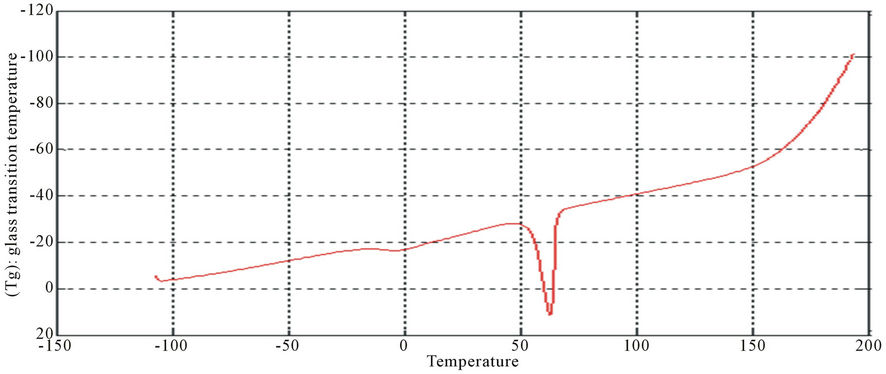
Figure 13. DSC curve of PUA 3.
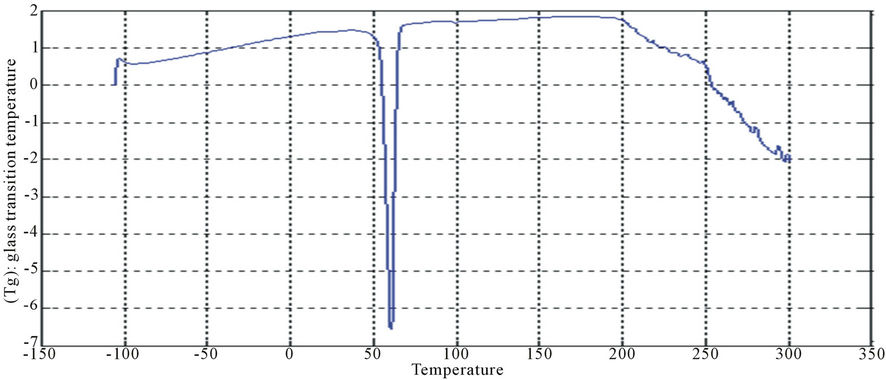
Figure 14. DSC curve of PUA 4.
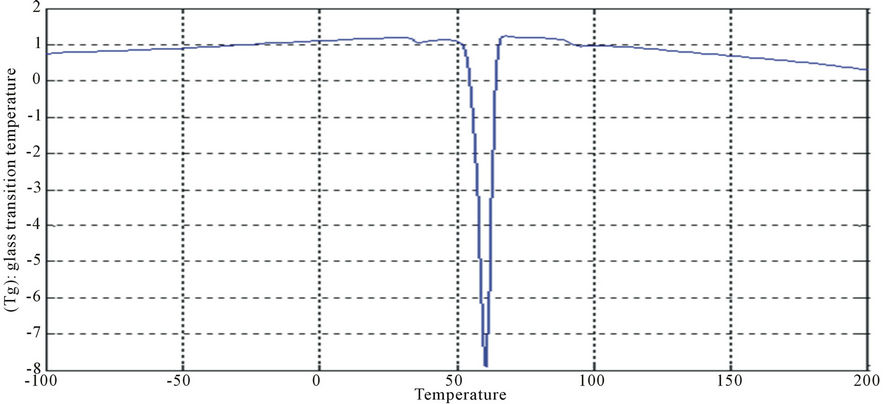
Figure 15. DSC curves of PUA 5.
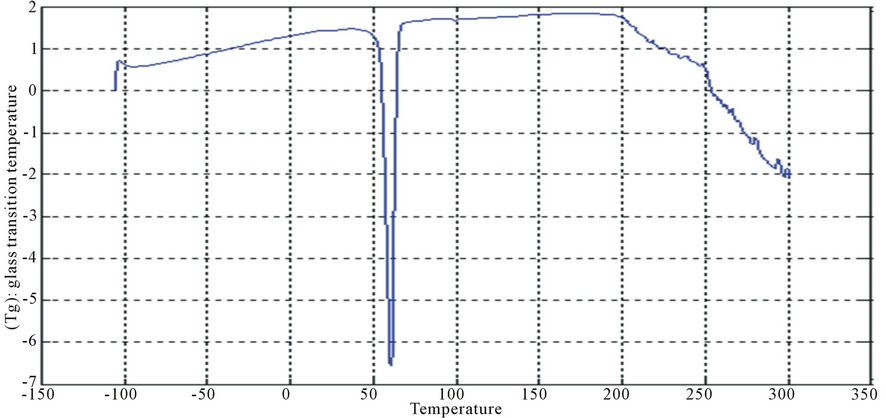
Figure 16. DSC curve of PUA 6.
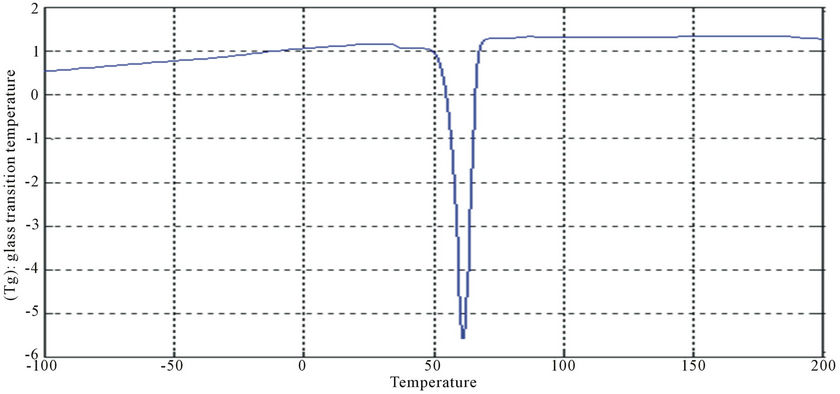
Figure 17. DSC curve of PUA 7.
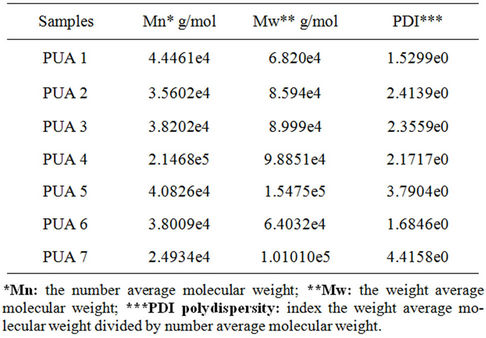
Table 2. GPC data from analysis of prepared PUA.
show normal values which indicate that the polymerization process occurred in homogenous order reactions condition (For typical step polymerization, most probable values of the PDI are around 2), while PDI values for prepared polyurethane acrylate 5, 6, 7 are irregular, which may be due to the presence of aromatic ring.
4.4. Effect of High Energy Electric Milling on the Spheres Diameters of Synthesized Powered Polyurethane Acrylate Polymers
In general, there are two approaches to nanoparticle production that are commonly referred to as “top-down” and “bottom-up”. “Top-down” as mentioned before nanoparticles are generated from the size reduction of bulk materials. They generally rely on physical, the combination of physical and chemical, electrical or thermal processes for their production. Such methods include high-energy milling, mechano-chemical processing, electro-explosion, laser ablation, sputtering and vapor condensation [15]. Synthesized polyurethane acrylate was subjected to size reduction (top-down) using GY-RO high-energy mill machine (mass ratio ball to material 5:1) for different periods (5, 15 days) [16].
It is noticed that after 5 days of milling the spheres diameters almost are in range of 500 - 100 nm, while after subjecting to 15 days of milling the spheres diameters are highly reduced to become below 100 nm The results of TEM of the prepared polyurethane acrylate (2, 3, 4, 5, 7) are shown in Figures 18-22, it was clear that spheres diameters of prepared polyurethane acrylate are in range of 324 nm to 46 nm when subjected to mill for about 5 days, while spheres diameters of polyurethane acrylate subjected to mill for 15 days are below 60 nm and in range at 58 - 20 nm i.e. in nano scale. Polyurethane acrylate 1, 6 are not subjected to milling process because they are stick in natures, and could not be completely solidified.
4.5. Scanning Electron Microscope
The surface morphology of prepared polyurethane acrylate of PUA based on PEG with different molecular weight mixed with polyol was observed using scanning electron microscope (SEM) [16]. Figures 23-29 show SEM of prepared polyurethane acrylate before and after milling. It is clear that the surface of prepared polyurethane acrylate polymers based on IPDI exhibits almost spherical shape (beads) except PUA 1 which is has sticky liquid form and couldn’t be completely solidified, this may be attributed to the low molecule weight of the PEG (6000 g/mol) used in its synthesis (the low in degree of polymerization results in decrease in viscosity, and polymer change to liquid form as that decrease is increase). SEM of polyurethane acrylate polymers that were based on TDI were appears as heterogeneous shape.
For polyurethane acrylate 2, 3, 4, 5 we notice that the
 (a) (b)
(a) (b)
Figure 18. TEM of PUA 2 (a) after 5 days of milling and (b) after 15 days. (a) 6 kx; (b) 6 kx.
 (a) (b)
(a) (b)
Figure 19. TEM of PUA 3 (a) after 5 days of milling and (b) after 15 days of milling. (a) 6 kx; (b) 6 kx.
 (a) (b)
(a) (b)
Figure 20. TEM of PUA 4 (a) after 5 days of milling and (b) after 15 days of milling. (a) 6 kx; (b) 10 kx.
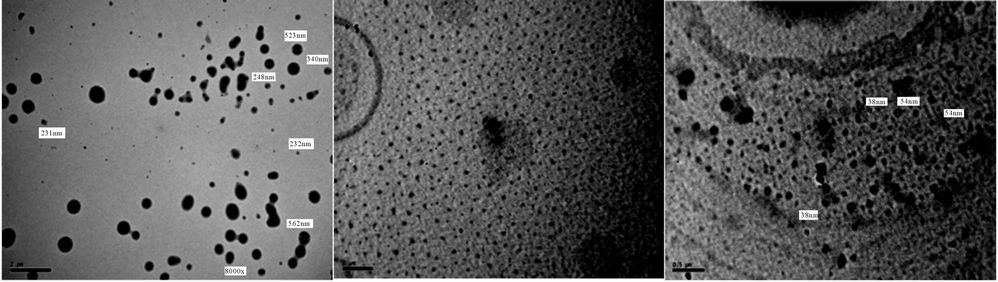 (a) (b) (c)
(a) (b) (c)
Figure 21. TEM of PUA 5 after (a) 5 days of milling and (b) & (c) after 15 days of milling. (a) 8 kx; (b) 10 kx; (a) 6 kx; (b) 25 kx.
 (a) (b)
(a) (b)
Figure 22. TEM of PUA 7 (a) after 5 days of milling and (b) after 15 days of milling. (a) 4 kx; (b) 6 kx.

Figure 23. SEM of PUA 1.
polyurethane acrylate appear as beads shape very close to polystyrene beads shape, those beads had been disintegrated away from each other after subjected to GY-RO mill machine. Samples based on TDI appear as porous surface and this may be due to ability of polyurethane to form foam, which leads to the possibility of creating of such pores in the polymers.
However, after milling (5 days) and grinding, this phenomenon of porosity was disappearing, and the shape was changed to be similar to beads shape.
4.6. Textile Pigment Applications
The application of these types of binders will be discuss in the second part.
5. Conclusion
Seven different polyurethane acrylate co-polymers were prepared aiming at substituting the two hydroxyl groups of polyethylene glycol (6000, 12,000 and 20,000 g/mol) and two primary hydroxyl groups of polyol through their reaction with corresponding calculated amounts of either isophorone diisoyanate or toluene diisocyanate and caped
 (a) (b)
(a) (b)
Figure 24. SEM of PUA 2 (a) before milling (b) after milling.
 (a) (b)
(a) (b)
Figure 25. SEM of PUA 3 (a) before milling (b) after milling.
 (a) (b)
(a) (b)
Figure 26. SEM PUA 4 (a) before milling (b) after milling.
 (a)
(a) (b)
(b)
Figure 27. SEM PUA 5 (a) before milling (b) after milling.

Figure 28. SEM of PUA 6.
 (a)
(a) (b)
(b)
Figure 29. SEM PUA 7 before milling (a) after milling (b).
the remaining isocyanate group with either hydroxy ethyl acrylate or hydroxy propyl meth acrylate to get on the polyurethane acrylate polymers [PUA]. The infra red (FTIR) spectra of the new polyurethane acrylate confirm the occurrence of the addition reactions in the process of the synthesis of polyurethane acrylate polymer. Also from FT-IR spectra of the synthesized PUA polymer, it clear that there are fully reacted isocyanate groups, and other functional groups (double bond) are left ready for further reaction in fixation process. The Tg measurement of synthesized PUA are in range from −8.78˚C to 36.4˚C so they can classify as soft binders. The weight average molecular weight of the synthesized PUA polymer shows that, the values are in range of 9.8851e4 to 1.01010e5. The spheres diameters of prepared polyurethane acrylate are in range of 324 nm to 46 nm when subjected to mill for about 5 days, while spheres diameters of polyurethane acrylate subjected to mill for 15 days are below 60 nm and in range at 58 - 20 nm i.e. in nano scale. The surface of all prepared polyurethane acrylate exhibits spherical shape in-group of aliphatic polyurethane acrylate, except PUA 1. Aromatic polyurethane acrylate appears as porous surface and this may be due to ability of polyurethane to form foam. After milling and grinding, this phenomenon of porosity was disappearing, and the shape was changed to be similar to beads shape.
REFERENCES
- V. Giesen and R. Eisenlohr, “Pigment Printing,” Review of Progress in Coloration and Related Topics, Vol. 24, 1994, p. 24.
- P. K. Shah and O. H. Westlake, “Formaldehyde Free Print Binder,” US Patent No. 5969018, 1999.
- J. Wypych, “Polymer Modified Textile Materials,” 1st Edition, Wiley-Inter Science, Hoboken, 1988, pp. 2-6.
- M. M. El-Molla, “Synthesis and Characterization of Aqueous UV-Curable Binder for Ink Preparation in Ink Jet Printing and Pigment Dyeing of Fabrics,” Indian Journal of Fiber & Textile Research, Vol. 32, No. 1, 2007, pp. 105-113.
- M. M El-Molla, “Synthesis of Polyurethane Acrylate Oligomers as Aqueous UV-Curable Binder for Inks of Ink Jet in Textile Printing and Pigment Printing,” Dyes and Pigment, Vol. 47, 2007, pp. 371-379.
- K. Arshak and I. Gaidan, “NiO/Fe2O3 Polymer Thick Films as Room Temperature Gas Sensors,” Thin Solid Films, Vol. 495, No. 1-2, 2006, pp. 286-291. doi:10.1016/j.tsf.2005.08.298
- W. F. Reed, “Miniature, Submersible, Versatile, Light Scattering Probe for Absolute Equilibrium and Non-Equilibrium Characterization of Macromolecular and Colloidal Solutions,” US Patent No. 6052184, 2000.
- W. F. Reed, “Automatic Mixing and Dilution Methods for Online Characterization of Equilibrium and Non-Equilibrium Properties of Solutions Containing Polymers and/or Colloids,” US Patent No. 6653150, 2003.
- A. M. Alb, M. F. Drenski and W. F. Reed, “Perspective Automatic Continuous Online Monitoring of Polymerization Reactions (ACOMP),” Polymer International, Vol. 57, No. 3, 2008, pp. 390-396. doi:10.1002/pi.2367
- J. Brandrup, E. H. Immergut and E. A. Grulke, “Polymer Handbook,” 4th Edition, John Wiley & Sons Ltd., Hoboken, 1999.
- D. Randall and S. Lee, “The Polyurethanes Book,” John Wiley & Sons Ltd., New York, 2002, pp. 1-477.
- T. Hatakeyama and F. X. Quinn, “Thermal Analysis Fundamentals and Application to Polymer Science,” 2nd Edition, John Wiley & Sons Ltd., West Sussex, 1999, pp. 10-21.
- Y. T. Choi, M. S. El-Aasser, E. D. Sudol and J. W. Vanderhoff, “Polymerization of Styrene Miniemulsions,” Journal of Polymer Science: Polymer Chemistry Edition, Vol. 23, No. 12, 1985, pp. 2973-2987. doi:10.1002/pol.1985.170231206
- M. Antonietti and K. Landfester, “Polyreactions in Miniemulsions,” Progress in Polymer Science, Vol. 27, No. 4, 2002, pp. 689-757. doi:10.1016/S0079-6700(01)00051-X
- C. H. Xue, M. M. Shi, H. Z. Chen and M. Wang, “Preparation and Application of Nanoscale Microemulsion as Binder for Fabric Inkjet Printing,” Colloids and Surfaces A: Physicochemical and Engineering Aspects, Vol. 287, No. 1-3, 2006, pp. 147-152. doi:10.1016/j.colsurfa.2006.03.046
- R. Zhao, L. C. Waclsworth, D. Z. Hang and C. Sun, “Plasma Treatment Effect on Dyeing Properties and Surface Characteristic of PAT,” Proceedings of the ATCC 2003, Manassas, February 2003, pp. 21-27.