Food and Nutrition Sciences
Vol. 3 No. 9 (2012) , Article ID: 22297 , 8 pages DOI:10.4236/fns.2012.39160
C16 Saturated Fatty Acid Induced Autophagy in A549 Cells through Topoisomerase I Inhibition
![]()
1Department of Oral Pathology, School of Dentistry, Chonnam National University, Gwangju, South Korea; 2Department of Oral & Maxillofacial Surgery, Chonnam National University Dental Hospital, Gwangju, South Korea; 3Department of Periodontology, School of Dentistry, Chonnam National University, Gwangju, South Korea.
Email: *js3894@chonnam.ac.kr
Received July 9th, 2012; revised August 9th, 2012; accepted August 16th, 2012
Keywords: Palmitic Acid (PA); Topoisomerase I; Monodansylcadaverine (MDC); 3-Methyladenine (3-MA); Autophagy
ABSTRACT
C16 saturated fatty acid (Palmitic acid) is one of the most common dietary fatty acids which played an important role in the cellular biological functions. Palmitic acid (PA) was tested for potential inhibition of DNA topoisomerase I (topo I) and it exhibited inhibitory activity in the nanomolar range. Treatment of lung adenocarcinoma cell line A549 with PA resulted in a decrease in cell viability in a concentration-dependent manner, and PA showed cytotoxicity with an IC50 value of 150 µM. DNA fragmentation assay and caspase activity indicated that PA does not induce apoptotic cell death in A549 cells. Finally, we found that PA was able to cause an increase in autophagic flux in a time-dependent manner, evidenced by the accumulation of LC3 through monodansylcadaverine (MDC) staining. More importantly, inhibition of autophagy by blocking autophagosome formation via the inhibition of type III Phosphatidylinositol 3-kinases (PI-3K), by 3-Methyladenine (3-MA) was able to effectively suppress PA-induced autophagy. We showed that inhibition of autophagy sensitized the cells signal to PA-induced apoptosis, suggesting the pro-survival function of autophagy induced by PA. Taken together, results from this study reveal that PA as a topo I inhibitor induced autophagic cell death in A549 cells.
1. Introduction
Natural resource products have been used as source of novel therapeutics for several years. Natural products have been selected during evolution to interact with biological targets and their high degree of chemical diversity make them attractive as lead structures for discovery of new drugs [1]. A number of plant-derived anticancer drugs have received FDA approval for marketing: Taxol, Vinblastine, Vincristine, Topotecan, Irinotecan, Etoposide and Teniposide [2]. Palmitic acid (PA) is one of the most common saturated fatty acid found in animals and plants. PA is a major component of the oil from palm trees (palm oil and palm kernel oil). It is also found in butter, cheese, milk and meat. Interestingly, during our investigation for a topoisomerase (Topo) I inhibitor, we found that PA shows strong inhibitory activity against Topo I. Topo I are DNA enzymes which are the regulators of the topology of the supercoiled DNA double helix during the transcripttion and replication of cellular genetic materials. There are two main types of topoisomerases, Topo I and II [3]. Topo I initiates the cleavage of a single strand of DNA molecule while Topo II cleaves both DNA strands. The complete or partial inhibition of this DNA replicative mechanism results in the accumulation and stability of cleavable complexes and subsequent death of the cell [3,4].
Cancer is the prominent cause of death in developed countries as well as the second leading cause of death in developing countries. The burden of cancer is increasing in economically developing countries as a result of population aging and an adoption of cancer-associated lifestyle choices including smoking, physical inactivity, and westernized diets. Lung cancer is the most common cause of cancer-related death worldwide [5]. Lung cancer survival rate is poor with only 15% of patients surviving 5 years after diagnosis. New chemotherapy drugs and radiotherapy have improved survival and quality of life of patients, but the overall effect in the last decade has been mainly on palliation rather than reduction in mortality. The antitumor effect of Topo I inhibitors suggests that the accumulation or prolongation of Topo I cleavable complexes results in irreversible DNA replication defects and subsequent cell cycle arrest followed by cell death [6]. Consequently, tumors that express significantly high levels of Topo I enzyme are presumed to be easy targets for Topo I inhibitors.
Topo I is a dual activity enzyme. First, it momentarily nicks double-stranded DNA which is responsible for relieving the torsional stress that occurs in DNA during transcription, replication, and chromatin condensation [7]. Second, it precisely phosphorylates arginine-serine-rich (SR proteins) essential splicing factors, is a regulator of splicing, and is potentially involved in pre-mRNA-splicing regulation [8]. The catalytic cycle of the enzyme consists of DNA cleavage to form a covalent enzyme-DNA intermediate, DNA relaxation, and finally, re-ligation of the phosphate backbone to restore the continuity of the DNA. DNA is the blueprint for humans and all other life forms, any damage to the DNA can result in cancer. Inhibitors of the topoisomerase enzymes have the ability to kill all cells undergoing DNA replication, reading of the DNA for protein production or experiencing repair of DNA damage. DNA, which is the genetic material, and the enzymes responsible for all the reactions in the body are major targets of interest for medical researchers looking for new cancer treatments. Therefore, topo I inhibitor is the novel target for anticancer drug development.
There are two types of programmed cell death that have been well defined in both normal cells as well as cancer cells. Type I programmed cell death (apoptosis) has been studied extensively at the morphological and biochemical level. Type II programmed cell death (autophagy) is involved in the formation of autophagosome, which then fuses with lysosomes to form single-membrane autophagolysosomes, followed by degradation through liposomal hydrolases. Autophagy is an evolutionarily conserved process in which long-lived proteins and intact organelles are engulfed within double-membraned vacuoles (autophagosomes) and then degraded after the vacuoles fuse with lysosomes (autophagolysosomes or autolysosomes). At the end of the process, the degradation products are released into the cytosol and reutilized for metabolism. The molecular machinery of autophagy is tightly regulated by diverse signaling pathways that are activated in response to various stimuli. Under physiological conditions, autophagy occurs at a basal level to maintain homeostasis. However, autophagy can result in cell death if it is massively activated under certain stresses [9]. The autophagy pathway is controlled by autophagy-related protein Atg which is involved in autophagosome formation [10]. MAP 1 (microtubule-associated protein 1) LC 3 (light chain 3), the mammalian homologue of yeast Atg8, plays a role in activation of LC3 I which is then converted to LC3 II by protease and is targeted to the autophagosome membrane. Hence, LC3 II has been used as an autophagosome marker [11].
In this study, we aimed to investigate whether PA induces DNA damage by interacting with Topo I enzyme followed by autophagy and the biological significance of autophagy. We found that PA induced autophagy. Previously it was reported that the UV irradiation induced onrate autophagic flux and it was observed that autophagy acted as a cytoprotective mechanism in both the A549 and H1299 cell lines because knockdown of the two autophagy proteins, Beclin-1 and ATG5, enhanced the UV-induced cell death [12]. In our study, autophagy was inhibited by 3-Methyladenine (3-MA) by blocking autophagosome formation and increasing apoptosis, which suggests that autophagy acts as cytoprotective mechanism against DNA damage in A549 cells. During our investigation we found that PA induced cell death in A549 cells by activation of autophagy.
2. Materials and Methods
2.1. Cell Culture and Chemicals
The lung cancer cell line A549 was obtained from the American Type Culture Collection. Cells were cultured at 37˚C in RPMI 1640 medium containing 5% heat-inactivated fetal bovine serum (FBS) and 1% antibiotic (WelGENE Inc., Daegu, South Korea) in a 5% CO2 incubator. Cells were maintained and subcultured. 3-Methyladenine (3-MA), fatty acid free bovine serum albumin (BSA), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), monodansylcadaverine (MDC), 4,6-Diamidino-2-phenylindole (DAPI) and lysis buffer were obtained from Sigma Chemicals Co. (St. Louis, MO, USA). Other laboratory grade chemicals and reagents were purchased from Sigma Chemicals Co.
2.2. Cytotoxicity Assay
Cell viability was analyzed by reduction of MTT as previously described [13] with a slight modification. Cells were seeded at 1 × 105 cells/ well in 100 μl medium on 96-well plates and cultured at 37˚C in a 5% CO2 incubator. Twenty-four hours after exposure different concentration (5000 µM to 39 µM) of PA, media were removed and cells were incubated in PBS containing 20 μl MTT at 37˚C for 3 h. The formazan product was solubilized by the addition of 50 μl of dimethyl sulfoxide (DMSO; Calbiochem Bio-Mol, La Jolla, CA, USA). Optical density was measured at 570 nm using a microplate reader (BioRad®, Hercules, CA, USA).
2.3. Caspase Activity Assay
Caspase activities were determined using Caspase-3, -8 and -9 assay kits (Clontech, Terra Bella Ave, USA) according to the manufacturer’s protocol. Protein concentration was measured using BSA Protein assay kit (Pierce Biotechnology®, Rock-Ford, USA). Caspase activity was measured using a microplate reader (Bio-Rad, Hercules, CA, USA) at 405 nm.
2.4. DNA Topoisomerase I Activity Assay
DNA topoisomerase I assay was performed using DNA topoisomerase assay kit (TopoGEN, Port Orange, FL) according to the manufacturer’s protocol. The reaction mixture was prepared by adding pRYG DNA, topoisomerase enzyme (Takara®, Shiga, Japan) and different concentrations of PA. After incubation at 37˚C for one hour, each sample was loaded into 1.5% agarose (Bioneer, Corp., Daejeon, South Korea) gel and electrophoresis was carried out.
2.5. Determination of Apoptosis Using Agarose Gel
A549 cells were seeded in 60 mm disc plates and treated with PA for 0, 6, 12 and 24 hours respectively and then washed with phosphate buffered saline (PBS). DNA was extracted using the Wizard genomic DNA purification kit (Promega®, Madison, USA) according to the manufacturer’s instructions. The extracted DNA was electrophoresed on a 1.5% agarose gel containing ethidium bromide.
2.6. DAPI and Monodansylcadaverine (MDC) Staining
A549 cells were grown on coverslips at a density of 1 × 105 cells/cm2 overnight at 37˚C in a 5% CO2 incubator. The following day, A549 cells were treated with PA (150 µM) for 0, 1, 6, 12 and 24 hours and washed with PBS.
Cells were treated with 50 µM of MDC for 5 minutes. Then, cells were washed with PBS followed by staining with DAPI (to a final concentration of 6 µM in PBS) for 5 minutes. Finally, cells were washed with PBS and fixed in 3% formalin solution for 30 minutes. Fluorescence intensity was monitored using a confocal microscope (Carl Zeiss, Oberkochen, Germany) and images were captured.
2.7. Immunoblot Analysis
Cells were washed and lysed using 10 µl/ml of lysis buffer containing protease inhibitor cocktail (Thermo Scientific, Rockford, IL, USA). The lysed cells were centrifuged and the protein concentration was quantified. Equal amounts of protein were separated on sodium dodecyl sulfate polyacrylamide gel (SDS-PAGE) electrophoresis (10% - 15%) and then transferred onto polyvinylidene difluoride (PVDF) membrane and analyzed by immunoblotting using corresponding antibodies. LC3 primary antibody was purchased from Cell Signaling Technology (MA, USA). Bcl2, Bax, GAPDH and β-actin were purchased from Santa Cruz Biotec. (Delaware Avenue, Santa Cruz, CA, USA). Immobilized proteins were incubated with goat anti-mouse IgG and goat anit-rabbit IgG (Santa Cruz Biotec.) and signals were detected using enhanced chemiluminescence (ECL) detection kit (Santa Cruz Biotec.).
2.8. Statistical Analysis
All the experiments were repeated at least 3 times and the significance of difference between treatment and control was analyzed by t-test. Values are expressed as the mean ± SD.
3. Results and Discussion
3.1. PA as a Topoisomerase I Inhibitor and Its Cytotoxicity in A549 Cell Line
Samples of PA were prepared in ethanol and inhibitory activity of PA was checked in the agarose gel as described in the “Materials and Methods” Section.
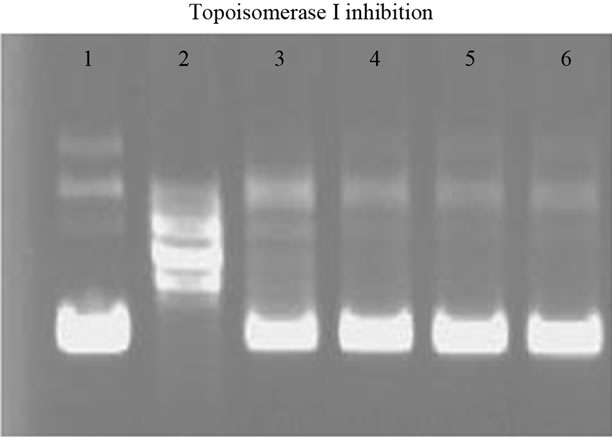
The result showed that there was complete inhibition of Topo I in the presence of PA as shown in Figure 1(a). We further checked the cytotoxicity of PA in A549 cells. Samples of PA were prepared as previously described [14] with a slight modification. Cytotoxicity IC50 of PA was found to be around 150 µM as presented in Figure 1(b). Hence, PA induced cell death in A549 cells and further experiments were performed using the above mentioned concentration of PA.
Excessive intake of fatty acids causes lipotoxicity [15]. Lipotoxicity refers to the cytotoxic effects of excess fat accumulation in cells and has been implicated as one of the contributing factors to diseases like obesity, diabetes and non-alcoholic fatty liver [16-18]. Previously it was reported that PA has been identified as a novel CD4 fusion inhibitor that blocks HIV entry and infection [19,20], as an antibacterial compound [21]. PA also shows selective cytotoxicity to human leukemic cells, but no cytotoxicity to normal HDF cells [22]. The present study showed that PA inhibits topo I at nanomolar concentrations and its cytotoxic effect was observed in human lung adenocarcinoma cell line A549.
3.2. Determination of Apoptosis Using Agarose Gel and Caspase Activity
MTT assay showed cell death in A549 cells in the presence of PA. We tried to identify the type of programmed cell death (either apoptosis or autophagy) which occurred during experimental treatment with PA. Firstly, DNA fragmentation assay was performed in agarose gel electrophoresis as presented in Figure 2(a). This figure confirmed that cell death was not induced via apoptosis. Secondly, the caspase activities (caspase-3, -8 and -9) which are known to be the markers of type I programmed cell death (apoptosis) were measured as shown in Figures 2(b)-(d), respectively. Data of 24 hours treatment with PA showed that there were no changes in caspase-3 and caspase-9
 (a)
(a)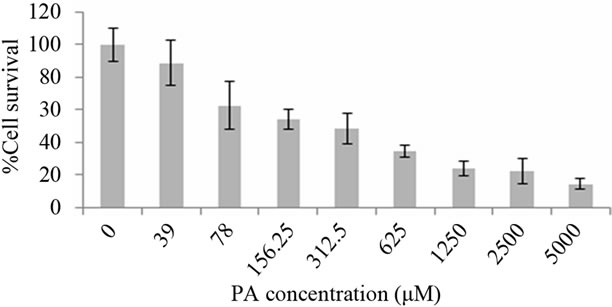 (b)
(b)
Figure 1. Inhibitory activity of PA on human DNA topoisomerase I (a) Lane 1, 0.1 µg of supercoiled DNA without the added enzyme; lane 2, supercoiled DNA with 1 U DNA topoisomerase I; lane 3 - 6 (supercoiled DNA, 1 U DNA topoisomerase I with PA concentration of 0.004, 0.04, 0.4 and 4 µM, respectively). Cytotoxicity effect of PA in A549 cells at different concentrations (µM) after treatment for 24 hours is presented in Figure 1(b).
activities but there was a slight increase in caspase-8 activity, which confirmed that cell death did not occur via apoptosis as previously described and that caspase-8 is not required for apoptosis [23]. Finally, we checked the caspase activities (caspase-3, -8 and -9) in the presence of autophagy inhibitor 3-MA, but we did not find any changes in caspase activity with respect to control. When, PA and 3-MA was treated together in the A549 cells, caspase activity was increased by 30% as shown in Figures 2(b)-(d), respectively. Hence, we further checked the type of cell death by analyzing MDC staining and the expression level of autophagy marker LC3.
Biochemically, apoptosis includes DNA fragmentation and caspase cascade activation. Previously, it was reported that PA induced apoptosis in a human granulosa cell line and this was confirmed by DNA fragmentation [24], but in our study, DNA fragmentation was not observed in A549 cells. This study showed that treatment of cells with PA did not induce caspase-3 and -9 activity MDC, a fluorescent compound has been proposed as a tracer for autophagic vacuoles under in vivo conditions and is not present in the early and late endosome [25]. MDC positive structures accumulate predominantly at perinuclear regions whereas autophagosomes are detected almost evenly throughout the cytoplasm. MDC dots co-localize well with staining for late-endosomal and lysosomal markers [26]. Thus, formation of autophagosomes is the key step in autophagy and this was determined by MDC staining which gives a green although it induced caspase-8 activity. Actually caspase-8 is not required for apoptosis but it can accelerate apoptosis [23]. These data suggested that PA-treated cells did not show type I programmed cell death.
3.3. MDC Staining Confirmed Autophagosome Formation by PA in A549 Cells
Florescence while DAPI staining of nuclei shows a blue color as shown in Figure 3. At 0 and 6 hours of exposure to experimental treatment, the DAPI staining was clear and uniform but the MDC staining was not clear. At 12 hours of treatment with PA, MDC staining for autophagosomes was not clear, but at 24 hours of treatment with PA, autophagosomes were clearly stained with MDC. At the same time, DAPI staining showed that there were nuclei fragmentations suggesting autophagic cell death.
Autophagy is controlled by a group of autophagy-related genes (ATG genes) that control several sequential steps of the autophagic process, including induction, autophagosome nucleation and elongation, autophagosome docking and fusion with the lysosome, and finally autophagic body breakdown and release of the contents into the cytosol [27]. MDC positive structures accumulate primarily at perinuclear regions whereas autophagosomes are mottled almost evenly throughout the cytoplasm [26]. Autophagosome formation, which is key step in autophagy, was detected by MDC staining.
3.4. PA Induced Cell Death via Autophagy
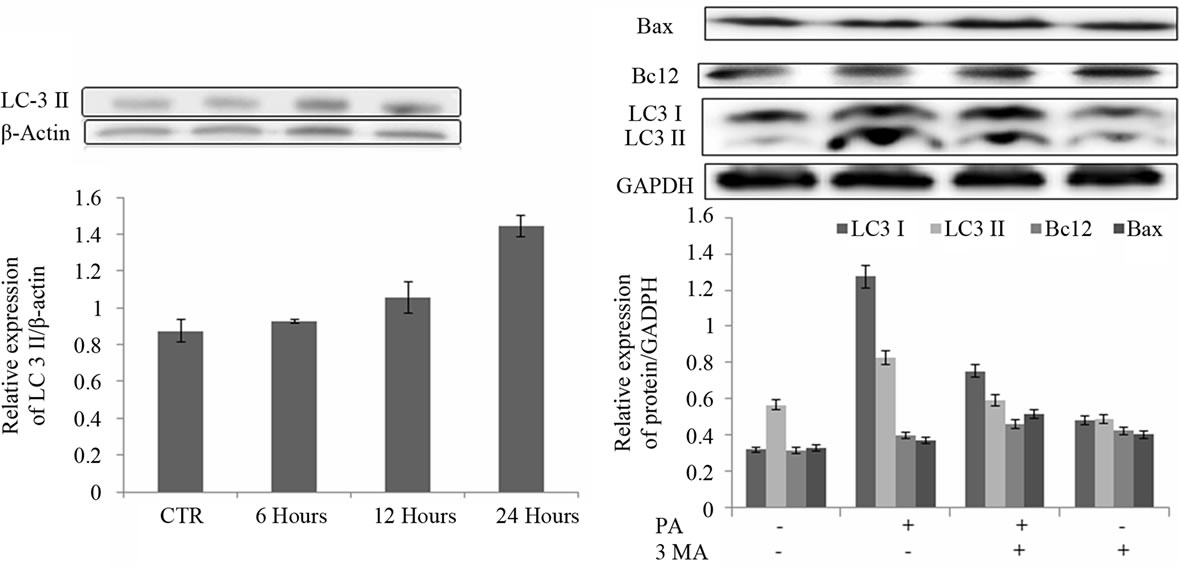
Many studies suggested that LC3 has been used as a marker for autophagy. Hence, A549 cells were treated with PA (150 µM) and formation of LC3 was checked by immunoblot analysis. The data showed that LC3 formation increased in a time-dependent manner as shown in Figure 4(a). Further, we investigated the role of autophagy inhibitor (3-MA) in the formation of LC3. Figure 4(b) demonstrates that after the A549 cell line was treated with PA for 24 hours, the conversion of LC3 I to LC3 II was increased with respect to control. When A549 cells were treated with the combination of PA and 3-MA, formation of LC3 II was decreased than that in A549 cells treated with PA alone suggesting the inhibition of autophagosome formation in A549 cells. When the A549 cell line was treated with 3-MA, immunoblot analysis of LC 3I and LC 3 II showed effective blockage of LC3
 (a)
(a)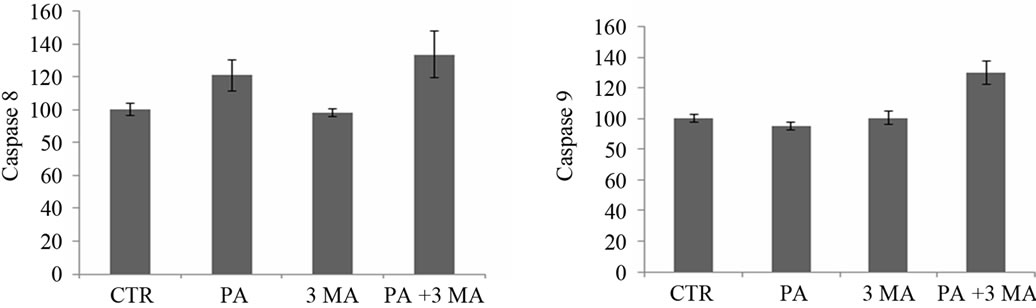 (b)
(b)
Figure 2. Determination of apoptosis in A549 cells by 1.5% agarose gel in the presence of 150 µM of PA at different time intervals (a) Caspase-3, -8 and -9 activities were observed in the presence of CTR (control), PA, 3-MA and with the combination of PA (150 µM) and 3-MA (5 mM) after treatment for 24 hours ((b), (c), (d) respectively).
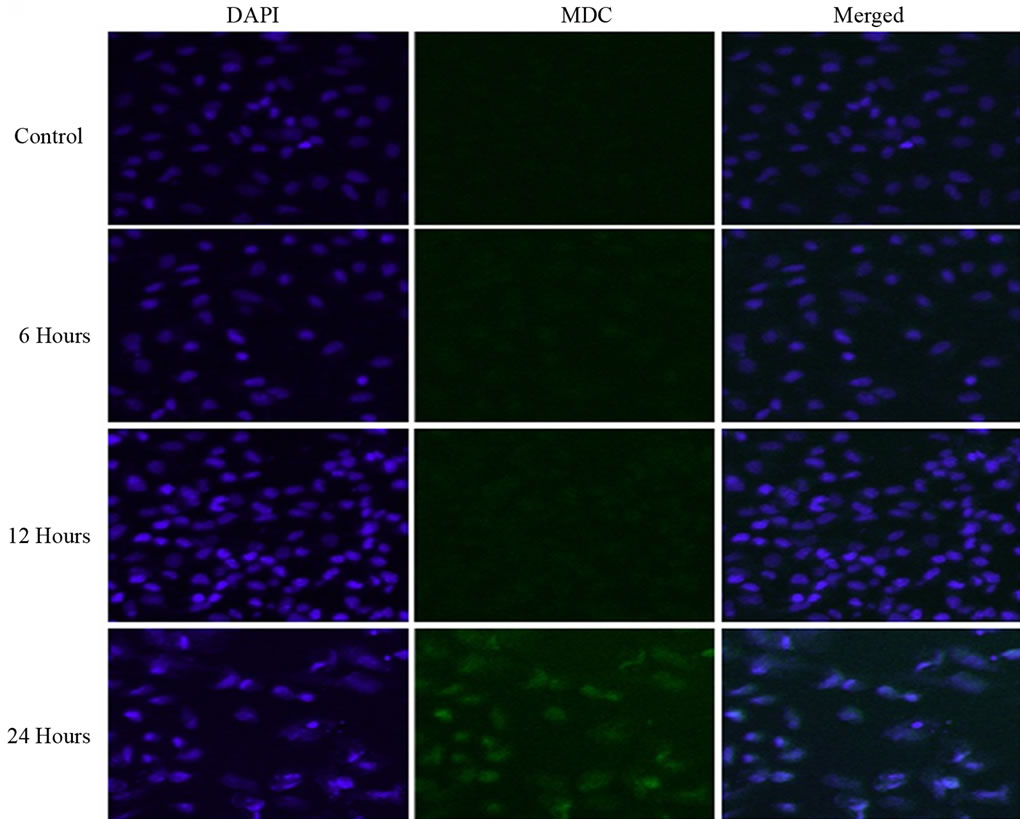
Figure 3. DAPI staining of nuclei showed a blue color and MDC staining of autophagosomes showed a green fluorescence. Treatment with 150 of µM PA caused induction of autophagy in a time-dependent manner as described in the “Materials and Methods” section and images were captured by a confocal microscope (×200).
 (a) (b)
(a) (b)
Figure 4. LC3 II expression after treatment with PA (150 µM) for different time intervals (a) Expression levels of LC3 I, LC3 II, Bcl2 and Bax after treatment for 24 hours with PA (150 µM), 3-MA (5 mM) and with the combination of PA and 3-MA (b).
formation. These data suggested that 3-MA effectively blocked autophagosome formation in A549 cells. Further, we investigated Bcl2 and Bax expression levels in the same treatment condition using PA, 3-MA and the combination of both PA and 3-MA as shown in Figure 4(b). Western blot result showed that treatment with PA or 3- MA and combined treatment with both drugs induced Bax and Bcl2 expression with respect to control.
3-MA, an inhibitor of type III Phosphatidylinositol 3- kinases (PI-K3) is known to be a selective and potent inhibitor which inhibits autophagy-dependent protein degradation and suppresses the formation of autophagosomes; cells were treated with the combination of PA and 3-MA and caspase cascade activities were measured. Caspase-3, -8 and -9 activities were increased by around 30% which suggested the induction of apoptosis. Yen R., et al. and Qiao C., et al. also reported that apoptosis increased in NB4 cells and HeLa cells after treatment with the combination of drug and 3-MA [28,29], suggesting that the pro-survival function of autophagy was induced by PA. PA induced autophagy in a time-dependent manner and maximum amount of LC3 formation was observed at 24 hours in A549 cell line. In the A549 cells treated with 3-MA, the expression level of LC was decreased with respect to those in the A549 cells treated with PA alone and with the combination of PA and 3-MA. Bax and Bcl2 level was increased in the presence of PA or 3-MA and the combination of both PA and 3-MA. BCL-2 family proteins at the endoplasmic reticulum (ER) also regulate autophagy, a survival pathway that limits metabolic stress, genomic instability and tumorigenesis [30]. Taken together, PA induced autophagy in A549 cell line.
4. Conclusions
Lipids are an important part of our diet: they serve as a potential source of energy and fuel for our cells, and some types of fats play an important role in maintaining a healthy heart, blood vessels and brain. One type of fatty acid which is present in commonly consumed foods is PA, a type of saturated fatty acid. PA exhibits DNA topo.
I inhibitory activity, which apparently promotes the formation of supercoiled DNA. Inhibitors of the topoisomerase enzymes have the ability to kill all cells undergoing DNA replication, reading of the DNA for protein production or experiencing repair of DNA damage. Since cancer cells divide much more rapidly than normal cells, the cancer cells will be killed by the topoisomerase inhibitors, although to some extent, normal cells with topoisomerase activity will be killed [31].
DNA, which is the genetic material, and the enzymes responsible for all the reactions in the body are major targets of interest for medical researchers looking for new cancer treatments. Apoptosis has been widely studied as a cellular response to DNA damage, although this report suggests that autophagy also plays an important role in determining cell fate. Rodriguez-Rocha H., et al. suggested that autophagy as an integral part of DNA damage response [32] and DNA damage by PA (topo I inhibitor) induced autophagy. Herein, we reported for the first time that PA as a topo I inhibitor induced autophagy in A549 cells and it can be a potential drug in cancer treatment.
5. Acknowledgements
This work was supported by the Korea Research Foundation Grant funded by the Korean Government (MOEHRD) (KRF-2009-0069839).
REFERENCES
- F. E. Koehn and G. T. Carter, “The Evolving Role of Natural Products in Drug Discovery,” Nature Reviews Drug Discovery, Vol. 4, No. 3, 2005, pp. 206-220. doi:10.1038/nrd1657
- V. Srivastava, A. S. Negi, J. K. Kumar, M. M. Gupta and S. P. Khanuja, “Plant-Based Anticancer Molecules: A Chemical and Biological Profile of Some Important Leads,” Bioorganic & Medicinal Chemistry, Vol. 13, No. 21, 2005, pp. 5892-5908. doi:10.1016/j.bmc.2005.05.066
- A. Y. Chen and L. F. Liu, “DNA Topoisomerases: Essential Enzymes and Lethal Targets,” Annual Review of Pharmacology and Toxicology, Vol. 34, 1994, pp. 191- 218. doi:10.1146/annurev.pa.34.040194.001203
- G. J. Creemers, B. Lund and J. Verweij, “Topoisomerase I Inhibitors: Topotecan and Irenotecan,” Cancer Treatment Reviews, Vol. 20, No. 1, 1994, pp. 73-96. doi:10.1016/0305-7372(94)90011-6
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, “Global Cancer Statistics,” CA: Cancer Journal for Clinicians, Vol. 61, No. 2, 2011, pp. 69-90. doi:10.3322/caac.20107
- Y. Pommier, “Eukaryotic DNA Topoisomerase I: Genome Gatekeeper and Its Intruders, Camptothecins,” Semin Oncology, Vol. 23, No. 1, 1996, pp. 3-10.
- J. B. Leppard and J. J. Champoux, “Human DNA Topoisomerase I: Relaxation, Roles, and Damage Control,” Chromosoma, Vol. 114, No. 2, 2005, pp. 75-85. doi:10.1007/s00412-005-0345-5
- J. Soret, M. Gabut, C. Dupon, G. Kohlhagen, J. Stevenin, Y. Pommier and J. Tazi, “Altered Serine/Arginine-Rich Protein Phosphorylation and Exonic Enhancer-Dependent Splicing in Mammalian Cells Lacking Topoisomerase I,” Cancer Research, Vol. 63, No. 23, 2003, pp. 8203-8211.
- B. Levine and J. Yuan, “Autophagy in Cell Death: An Innocent Convict?” Journal of Clinical Investigation, Vol. 115, No. 10, 2005, pp. 2679-2688. doi:10.1172/JCI26390
- D. J. Klionsky, “The Molecular Machinery of Autophagy: Unanswered Questions,” Journal of Cell Science, Vol. 118, No. 1, 2005, pp. 7-18. doi:10.1242/jcs.01620
- Y. Kabeya, N. Mizushima, T. Ueno, A. Yamamoto, T. Kirisako, T. Noda, E. Kominami, Y. Ohsumi and T. Yoshimori, “LC3, a Mammalian Homologue of Yeast Apg8p, Is Localized in Autophagosome Membranes after Processing,” EMBO Journal, Vol. 19, No. 21, 2000, pp. 5720-5728. doi:10.1093/emboj/19.21.5720
- W. P. Roos and B. Kaina, “DNA Damage-Induced Cell Death by Apoptosis,” Trends in Molecular Medicine, Vol. 12, No. 9, 2006, pp. 440-450. doi:10.1016/j.molmed.2006.07.007
- T. Mosmann, “Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays,” Journal of Immunological Methods, Vol. 65, No. 1-2, 1983, pp. 55-63. doi:10.1016/0022-1759(83)90303-4
- D. Dyntar, M. Eppenberger-Eberhardt, K. Maedler, M. Pruschy, H. M. Eppenberger, G. A. Spinas and M. Y. Donath, “Glucose and Palmitic Acid Induce Degeneration of Myofibrils and Modulate Apoptosis in Rat Adult Cardiomyocytes,” Diabetes, Vol. 50, No. 9, 2001, pp. 2105- 2113. doi:10.2337/diabetes.50.9.2105
- R. H. Unger and Y. T. Zhou, “Lipotoxicity of Beta-Cells in Obesity and in Other Causes of Fatty Acid Spillover,” Diabetes, Vol. 50, Suppl. 1, 2001, pp. S118-S121.
- P. Almeda-Valdes, D. Cuevas-Ramos and C. A. AguilarSalinas, “Metabolic Syndrome and Non-Alcoholic Fatty Liver Disease,” Annals of Hepatology, Vol. 8, Suppl. 1, 2009, pp. S18-S24.
- R. T. Brookheart, C. I. Michel and J. E. Schaffer, “As a Matter of Fat,” Cell Metabolism, Vol. 10, No. 1, 2009, pp. 9-12. doi:10.1016/j.cmet.2009.03.011
- J. A. Chavez and S. A. Summers, “Lipid Oversupply, Selective Insulin Resistance, and Lipotoxicity: Molecular Mechanisms,” Biochimica et Biophysica Acta, Vol. 1801, No. 3, 2010, pp. 252-265. doi:10.1016/j.bbalip.2009.09.015
- D. Y. Lee, X. Lin, E. E. Paskaleva, Y. Liu, S. S. Puttamadappa, C. Thornber, J. R. Drake, M. Habulin, A. Shekhtman and M. Canki, “Palmitic Acid Is a Novel CD4 Fusion Inhibitor That Blocks HIV Entry and Infection,” AIDS Research and Human Retroviruses, Vol. 25, No. 12, 2009, pp. 1231-1241. doi:10.1089/aid.2009.0019
- X. Lin, E. E. Paskaleva, W. Chang, A. Shekhtman and M. Canki, “Inhibition of HIV-1 Infection in ex Vivo Cervical Tissue Model of Human Vagina by Palmitic Acid; Implications for a Microbicide Development,” PLoS ONE, Vol. 6, No. 9, 2011, Article ID: e24803. doi:10.1371/journal.pone.0024803
- B. T. Yff, K. L. Lindsey, M. B. Taylor, D. G. Erasmus and A. K. Jager, “The Pharmacological Screening of Pentanisia Prunelloides and the Isolation of the Antibacterial Compound Palmitic Acid,” Journal of Ethnopharmacology, Vol. 79, No. 1, 2002, pp. 101-107. doi:10.1016/S0378-8741(01)00380-4
- H. Harada, U. Yamashita, H. Kurihara, E. Fukushi, J. Kawabata and Y. Kamei, “Antitumor Activity of Palmitic Acid Found as a Selective Cytotoxic Substance in a Marine Red Alga,” Anticancer Research, Vol. 22, No. 5, 2002, pp. 2587-2590.
- M. Kruidering and G. I. Evan, “Caspase-8 in Apoptosis: The Beginning of “The End”?” IUBMB Life, Vol. 50, No. 2, 2000, pp. 85-90. doi:10.1080/713803693
- Y. M. Mu, T. Yanase, Y. Nishi, A. Tanaka, M. Saito, C. H. Jin, C. Mukasa, T. Okabe, M. Nomura, K. Goto and H. Nawata, “Saturated FFAs, Palmitic Acid and Stearic Acid, Induce Apoptosis in Human Granulosa Cells,” Endocrinology, Vol. 142, No. 8, 2001, pp. 3590-3597. doi:10.1210/en.142.8.3590
- A. Biederbick, H. F. Kern and H. P. Elsasser, “Monodansylcadaverine (MDC) Is a Specific in Vivo Marker for Autophagic Vacuoles,” European Journal of Cell Biology, Vol. 66, No. 1, 1995, pp. 3-14.
- D. B. Munafo and M. I. Colombo, “A Novel Assay to Study Autophagy: Regulation of Autophagosome Vacuole Size by Amino Acid Deprivation,” Journal of Cell Science, Vol. 114, No. 20, 2001, pp. 3619-3629.
- C. He and D. J. Klionsky, “Regulation Mechanisms and Signaling Pathways of Autophagy,” Annual Review of Genetics, Vol. 43, 2009, pp. 67-93.
- Y. Ren, F. Huang, Y. Liu, Y. Yang, Q. Jiang and C. Xu, “Autophagy Inhibition through PI3K/Akt Increases Apoptosis by Sodium Selenite in NB4 Cells,” BMB Reports, Vol. 42, No. 9, 2009, pp. 599-604. doi:10.5483/BMBRep.2009.42.9.599
- Q. Cui, S. Tashiro, S. Onodera and T. Ikejima, “Augmentation of Oridonin-Induced Apoptosis Observed with Reduced Autophagy,” Journal of Pharmacological Sciences, Vol. 101, No. 3, 2006, pp. 230-239. doi:10.1254/jphs.FPJ06003X
- H. M. Heath-Engel, N. C. Chang and G. C. Shore, “The Endoplasmic Reticulum in Apoptosis and Autophagy: Role of the BCL-2 Protein Family,” Oncogene, Vol. 27, No. 50, 2008, pp. 6419-6433. doi:10.1038/onc.2008.309
- C. Bailly, “Topoisomerase I Poisons and Suppressors as Anticancer Drugs,” Current Medicinal Chemistry, Vol. 7, No. 1, 2000, pp. 39-58.
- H. Rodriguez-Rocha, A. Garcia-Garcia, M. I. Panayiotidis and R. Franco, “DNA Damage and Autophagy,” Mutation Research, Vol. 711, No. 1-2, 2011, pp. 158-166. doi:10.1016/j.mrfmmm.2011.03.007
NOTES
*Corresponding author.