Advances in Nanoparticles
Vol. 1 No. 3 (2012) , Article ID: 25169 , 10 pages DOI:10.4236/anp.2012.13007
Lipopolysaccharide Modified Liposomes for Amyotropic Lateral Sclerosis Therapy: Efficacy in SOD1 Mouse Model
1George M. Leader Family Laboratory, Department of Neurosurgery, Pennsylvania State University College of Medicine/Milton S. Hershey Medical Center, Hershey, USA
2Department of Neurology, Pennsylvania State University College of Medicine/Milton S. Hershey Medical Center, Hershey, USA
Email: jconnor@hmc.psu.edu
Received September 3, 2012; revised October 12, 2012; accepted November 4, 2012
Keywords: Amyotrophic Lateral Sclerosis; Minocycline; Liposomes; Microglia; TLR4; Lipopolysaccharides
ABSTRACT
Activation of microglia is a histological feature observed in neurodegenerative diseases like ALS. The oral administration of minocycline has been demonstrated to have minimal neuroprotection ability in the animal models and is also associated with inadvertent toxicity due to non-specific oral absorption of the drug. Nonetheless, the drug itself shows promise in a number of disease models suggesting it could be effective if delivered optimally. Thus, we utilized LPS modified liposomes to target TLR4 receptor on the microglia in SOD1G93A mice and compared its efficacy with nontargeted nanoliposomes. The in vitro results indicate that targeting the TLR4 receptor on microglia significantly increases (p < 0.01) the uptake of drug by 29% compared to non-targeted liposomes. In the SOD1G93A mouse model of ALS, targeted and non-targeted minocycline treatment significantly increased (p < 0.05) latency to endpoint stages compared to control mice. Targeting liposomes to microglia significantly delayed disease progression. Both targeted and non-targeted liposomes administration in SOD1 mice resulted in decreased TNF-α secretion in activated BV-2 microglial cells as compared to activated cells receiving no treatment. The non-targeted liposomes had a greater effect than the targeted liposomes in reducing the levels of TNF-α released by the BV-2 cells. In SOD1G93A mice, the nontargeted nanovesicles significantly increased the latency to rotarod failure and both targeted and non-targeted nanovesicles significantly delayed disease endpoints.
1. Introduction
Transgenic mice expressing mutant superoxide dismutase-1 (SOD1) are a model for studying amyotrophic lateral sclerosis (ALS). These animals experience progressive motor neuron degeneration, leading to leg weakness spreading to the upper limbs and bulbar muscles and eventually causing total paralysis and death [1,2] . On the cellular level, neuronal cell death is accompanied by inflammation and microgliosis, the combination of which has been shown to have a neurotoxic role in the pathogenesis of the disease [3,4] . Furthermore, activated microglia excretes several potentially neurotoxic molecules, including reactive oxygen species, proteases, and pro-inflammatory cytokines that amplify inflammation and harm other cells [2]. Recent studies have shown that diminishing expression of mutant SOD1 in microglia has little effect on the early disease phase but can sharply slow later disease progression. This suggests that onset and progression are represented by distinct disease phases that may represent SOD1 action within different CNS cell types [5,6] . This leads to a potential window for therapeutic intervention to target microglia activation, perhaps a different time points, to slow the kinetics of disease progression. Based on this concept, we developed a nanovesicle platform as a potential mechanism to enhance drug delivery to microglia in ALS patients. The drug we chose for delivery in this study, to establish proof of concept, was minocycline because it has recently been shown to have some positive effect in the SOD1 mouse model [7,8]
The delivery of anti-inflammatory compounds, such as minocycline, has been studied extensively in animal models of ALS as well as other neurodegenerative diseases where activation of microglia is part of the disease process. Although minocycline has been commercially available for nearly three decades, interest in its neuroprotective effects emanated from the finding that the drug significantly reduced infarct volume and neuronal cell loss after induced hypoxic injury in gerbils and rats [9,10] . Subsequent research with minocycline in various models of acute and chronic neurodegeneration corroborated its neuroprotective effect, and extended them to include traumatic brain and spinal cord injury [11,12] , multiple sclerosis [13] , Parkinson’s disease [14-16] , Huntington’s disease [17,18] , and ALS [7,19-23] . Despite promising results in the animal models of ALS, there has been a weak corollary of efficacy for minocycline in human clinical trials. Indeed, a recently published phase III trial of minocycline in patients with ALS reported a harmful effect [24] . Possible explanations for this effect are the inadvertent toxicity to motor neurons due to nonselective oral drug administration or the inability to deliver sufficient amounts of drug to where it is needed, both problems that may be circumvented via a targeted drug delivery system. As such, the hypothesis of this study is that nanovesicles carrying minocycline when infused intracerebroventricularly will delay disease onset and increase life span in mice carrying the SOD1G93A mutation by increasing site-specific drug action. The secondary hypothesis is that actively targeting microglia with minocycline encapsulated in nanovesicles will slow later disease progression as a function of attenuating neuroinflammation associated with microglia activation.
2. Experimental Methods
2.1. Materials
1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), Cholesterol, 2-distearoyl-sn-glycero-3-phosphoethanolamine N-[carboxy (polyethylene-glycol)2000(ammonium salt) (DSPE-PEG2000) and rhodamine-labeled phospholipids [L-a-phosphatidylethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) were purchased from Avanti Polar Lipids (Alabaster, AL). Lipopolysaccharide was from the source E. Coli 0128:B12 Sigma (St. Louis, Missouri). Minocyline was from Sigma Chemicals (St. Louis, Missouri). Mouse microglia-derived BV-2 cell line was a gift sample from Dr. Steven Levison (New Jersey Medical School). All cell culture materials were purchased from Invitrogen (Carlsbad, CA). The IFN-γ was purchased from Sigma, St. Louis, MO. Standard mouse ELISA kit was from R & D Systems (Minneapolis, MN). BioRad DC Protein Assay kit was purchased from Bio-Rad Laboratories (Hercules, CA). Transgenic mice with mutation B6SJL-Tg(SOD1-G93A)1Gur/J were purchased from Jackson Laboratory (Bar Harbor, ME). Osmotic pumps used for this study was from Alzet Inc. (Cupertino, CA).
2.2. Preparation and Characterization of Liposomes
Sterically stable liposomes were formulated using the lipids DPPC, DSPE-PEG-2000 and cholesterol in a molar ratio 10:2.5:1.5 respectively. To study cellular uptake, the liposomes were labeled with rhodamine by adding 1% (molar ratio) of rhodamine-labeled phospholipids [L-α-phosphatidylethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) to the liposomes formulation. The lipid constituents were dissolved in methanol/tbutanol (1:1 v/v) mixture in a round bottomed flask. A 600 μl saturated solution (chloroform:methanol, 1:1 v/v) of lipopolysaccharide (source from E.Coli 0128:B12) was then added to the lipid mixture for synthesis of the targeted nanovesicles. The non-targeted nanovesicles were formulated separately in the same manner, but without the addition of LPS. The lipid mixture was subsequently rotary evaporated (Buchi rotary evaporator, Germany) at 55˚C to obtain a thin lipid film, which was further dried by passing nitrogen and then placed in a desiccator overnight for complete drying. For minocycline encapsulation, the lipid film was hydrated in 1.0 ml of 10 mg/ml concentration of minocyline (Sigma, St. Louis, Missouri, Cat No. M9511) and sterile phosphate buffered saline (PBS) mixture and then reconstituted in a bath-type sonicator ( Branson Model 3510 labtop sonicator, Bransonic Ultrasonic Corporation, CT ) for 5 minutes. The appropriate minocycline concentration was determined by the solubility limitations of minocycline as well as from previous in vivo studies [7,20,25] . Unilammelar nanovesicles were obtained by extruding the liposomes though a two-stacked polycarbonate membrane with pore size of 0.1 µm for 10 times, followed by a 0.05 µm polycarbonate membrane for 10 times using a nitrogen pressure-operated extruder (Lipex extruder, Northern Lipids, Inc., Vancouver, British Columbia, Canada). All extrusions were performed at an operating pressure of 300 p.s.i. The unencapsulated minocycline from the liposomes were removed by dialyzing them against phosphate buffered saline. Liposome size and polydispersity index (PDI) were determined by dynamic light scattering, using a Malvern Zetasizer Nano apparatus (Malvern Instruments, Worcestershire, England). Zeta potential of the liposomes was measured via phase analysis light scattering utilizing a Zeta PALS instrument (Brookhaven Instruments, Holtsville, NY) in a 100 mM NaCl solution. The final minocycline concentration in targeted and non-targeted nanovesicles was analyzed by solubilizing an aliquot of nanovesicle in methanol and sonicating them at room temperature for 5 min. This solution of solubilized nanovesicles and minocycline was then injected into a HPLC for quantification of the minocycline (Beckman Coulter, Fullerton, CA) fitted with C18 column. The targeted and non-targeted nanovesicles were diluted in PBS to maintain an equal concentration of the liposomes. The LPS content of the LPS modified minocycline liposomes and unmodified liposomes were analyzed using Toxin sensor chromogenic LAL endotoxin Assay kit (Genscript, Inc.). The schematic picture of the steps involved in the liposome preparation can be found in Figure 1.
Lipopolysaccharide (LPS) was chosen as the targeting moiety for conjugation to the liposomes because of its high affinity for surface markers on microglia [26,27] . The signal-transducing receptor for LPS is Toll-Like Receptor 4 (TLR4), which normally is not expressed on cortical neurons, astrocytes, or oligodendrocytes [28-30] . Binding of this complex initiates an intracellular signaling pathway of the innate immune system, which includes both the neuroprotective and neurotoxic effects of microglia as described above.
2.3. In Vitro Experiments
2.3.1. Cell Culture
The murine BV-2 cell line was originally generated by Blasi et al [31] and exhibits phenotypic and functional properties of reactive microglial cells. Cells were maintained in Dulbecco’s Modified Eagle Medium supplemented with 10% heat inactivated Fetal Bovine Serum and 1% Penicillin-Streptomycin at 37˚C in a 5% CO2 atmosphere.
2.3.2. Cell Uptake Study
An uptake study with BV-2 cells was performed to inves tigate the ability of the cells to internalize the liposomes. For this experiment, targeted liposomes labeled with rhodamine lipid as mentioned in the methods section was utilized. BV-2 cells were cultivated on a chamber slide for 24 hours at a starting density of 10,000 cells each. The rhodamine-labeled liposomes were added to the cells at 200 μmol/ml and incubated at 37˚C for 120 minutes. Media was removed from the chambers and the slide fixed with 4% paraformaldehyde. The cells were stained with 4,6-diamidino-2-phenylindole (DAPI) for visualizetion of the nuclei, and then washed three times with PBS to end exposure to adherent liposomes. Slides were then viewed under a fluorescent microscope and images were captured using a digital camera.
2.3.3. Flow Cytometry Assay
Flow cytometry was used to measure total intracellular liposome fluorescence, which is an indirect measure of intracellular drug content. Fresh batches of targeted and non-targeted liposomes were synthesized by including rhodamine phospholipid at 1 molar percent in the liposome preparation method described above. BV-2 cells (1 × 106) were exposed to 200 μmol/ml media of either targeted or non-targeted liposomes or the equivalent volume of PBS and incubated at 37˚C for 120 minutes. The cells were then centrifuged (5 minutes, 1000 rpm) and washed with PBS twice to remove any non-internalized and unbound liposomes. Following the last centrifugation, cells were re-suspended in 1 ml PBS for flow cytometry assay. The intracellular accumulation of rhodaminelabeled liposomes was evaluated using a fluorescence activated cell sorting analyzer (FACSII; Becton Dickinson, Sparks, MD). A single 15-mW argon ion laser beam (570 nm) was used to excite the fluorescence of rhodamine with a total of 10,000 cells analyzed for each inten-
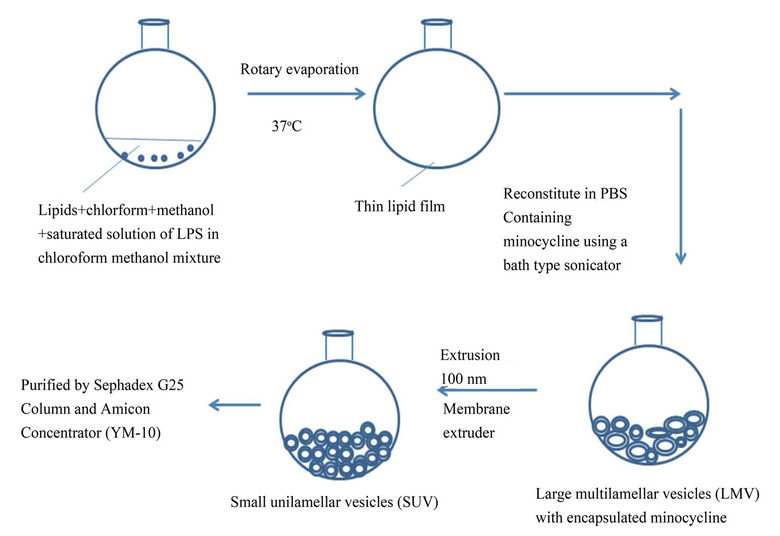
Figure 1. Schematic representation of the steps involved in the LPS modified minocycline liposomes.
sity histogram. The flow cytometry experiment was repeated three times for each treatment condition.
2.3.4. Cytokine Enzyme-Linked Immunosorbent Assays (ELISA)
BV-2 cells were grown to confluency in 24-well cell culture plates (BD Biosciences, San Jose, CA) with 0.5 ml media. The cells were activated with 10 ng/ml IFN-γ and simultaneously treated with 40 μM minocycline (Sigma, St. Louis, MO) contained in targeted liposomes or non-targeted liposomes, or as free drug. Cells activated with IFN-γ and receiving no treatment served as positive controls for activation, while BV-2 cells receiving media alone served as true controls. The concentration of minocycline was chosen based on previous in vitro studies [32,33] , as well as our own studies involving graded minocycline concentrations (data not shown). After 24-hour incubation at 37˚C, the supernatant was collected for each condition and TNF-α was measured using a standard mouse ELISA kit according to the manufacturer’s protocol. Plates were then washed with Hank’s Balanced Salt Solution and the cells lysed with RIPA buffer for protein quantification with a Bio-Rad DC Protein Assay kit.
2.4. In Vivo Experiments
2.4.1. Animals
Transgenic mice with mutation B6SJL-Tg(SOD1- G93A) 1Gur/J overexpress mutant human SOD1G93A and develop ALS symptoms (age-related spinal neuron degeneration, muscle atrophy, and shortened lifespan) at approximately 90 days of age. Treatment with minocycline containing liposomes began at 70 days, wherein the animals were fitted with an intracerebroventricular catheter and osmotic pump to continuously deliver minocycline to the lateral ventricle. The experiments were conducted in accordance with Institutional Animal Care and Use Committee (IACUC) guidelines of Pennsylvania State University. All the studies were performed using the first generation mSOD1 transgenic mice. A total of 7 mice per group were used in the control and targeted nanoliposome groups and 6 mice were used in the non-targeted liposome group.
2.4.2. Implant of Osmotic Pump
An osmotic pump was used to continuously deliver liposomes at a rate of 0.25 μl per hour for 28 days. The final concentration of minocycline for delivery was 2 mg/200 µl. Mice were implanted with the pump under general anesthesia (70 mg/kg ketamine and 14 mg/kg xylazine, injected intramuscularly), wherein the animal’s scalp was shaved and cleaned and a burr hole (approximately the size of a 25-gauge needle) made at +1.0 mm lateral and
–0.46 mm caudal to Bregma and a cannula (depth of 2.0 mm) was fixed to the animal’s skull with cyanoacrylate adhesive. Cannula placement was confirmed following disease endpoint on brains perfused and fixed in 4% paraformadehyde. The osmotic pump was placed in a subcutaneous pocket on the animal’s back, with a subcutaneous catheter connecting the pump to the cannula. After placement of the pump, catheter, and cannula, the scalp incision was closed with interrupted sutures. This surgery was performed on the animals at the age of 10 weeks (70 days old mice). After four weeks, the pump was replaced with a new pump containing fresh liposomes. After this, the pumps were left in place to minimize stress to the animals. Cannula placement was confirmed histologically in all of the animals at the end of the experiment.
2.4.3. Motor Performance and Endpoint
Motor performance was evaluated three times a week using a rotarod apparatus rotating at 15 rpm. Mice were habituated to the rotarod over the course of a five day learning period beginning at 8 weeks of age. A mouse was considered to fail the test when it could not stay on the rotating rod for >1 s.e.m. of the number of times it fell during the pre-symptomatic phase [20] . The endpoint of the study was defined as the age at which a mouse could right itself in 30 seconds when placed on its side or the inability to maintain basic grooming and feeding behavior [25,34] .
2.4.4. Data Analysis and Statistics
Statistical significance was determined by one-way ANOVA with Tukey’s post hoc test. For Kaplan-Meier survival plots, statistical significance was determined using a logrank test (GraphPad Prism 4.03). Significance for all analyses was defined as p < 0.05.
3. Results
3.1. Particle Size, Charge and Composition
The average liposome size was found to be 129 nm (PDI = 0.101) and 118 nm (PDI = 0.110) for targeted and nontargeted liposomes, respectively (Figure 2). The low PDI clearly indicates a narrow size distribution. Mean zeta potentials were –29 mV for both targeted and nontargeted liposomes. LPS minocycline liposomes had a LPS concentration of 403 ng/mL which is 5.8 fold higher than the basal LPS level in the liposomes of similar size and composition without LPS (69 ng/mL).
3.2. In Vitro Cellular Uptake of Liposomes
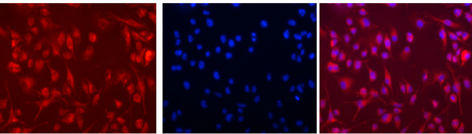
Internalization of the rhodamine labeled targeted liposomes
(LPS modified liposomes) in the BV-2 cells could be demonstrated with fluorescent microscopy (Figure 3).
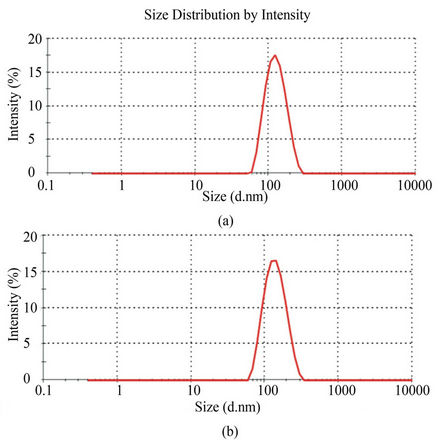
Figure 2. Polydispersity index (PDI) and size distribution of liposomes as determined by dynamic light scattering analysis. (a) The size distribution for non-targeted liposomes was found to have a mean size of 118 nm and PDI of 0.110. (b) The size distribution for targeted liposomes had mean size of 129 nm with PDI of 0.101. Average zeta potentials for both were –29 mV.
To compare the amount of uptake between targeted and untargeted liposomes, a flow cytometry analysis was performed revealing more intracellular cell fluorescence in targeted liposome-treated cells as compared to cells treated with non-targeted liposomes (Figure 4(a)). Normalization of the signals to endogenous fluorescence revealed there was a difference of 29% more fluorescence (p < 0.01) when the cells were exposed to targeted liposomes compared to non-targeted liposomes (Figure 4 (b)).
To determine whether the uptake of minocycline via the liposomes was functional, we measured concentrations of TNF-α secreted from activated and non-activated BV-2 cells. To establish an activation baseline for secretion, cells were exposed to IFN-γ for 48 hours and a significant increase (p < 0.001) over baseline of TNF-α secretion was observed (Figure 5(a)).
When activation with INF-γ was coupled with exposure to either free minocycline or targeted or nontargeted liposomes containing minocycline, the TNF-α response was attenuated. The ability of minocycline to inhibit TNF-α secretion was 22.3% for targeted liposomes, 39.1% for non-targeted liposomes, and 34.9% for free minocycline. There was no statistically significant difference between targeted liposome and free minocycline conditions on TNF-α release; however, the difference in the amount of TNF-α released when the cells were exposed to targeted versus non-targeted liposomes was significant (Figure 5(b)). Again, the concentration of
 (a) (b) (c)
(a) (b) (c)
Figure 3. Uptake of LPS-modified rhodamine-labeled liposomes into mouse microglia-derived BV-2 cells. The cells were exposed to liposomes for 2 hours at 37˚C. A, Uptake of rhodamine labeled LPS-modified liposomes by BV-2 cells is clearly visible by the fluorescent microscopic image. B, Cells were also treated with 4,6-diamidino-2- phenylindole (DAPI) nuclear staining. C, Overlay of rhodamine (red) and DAPI (blue) staining indicates the internalization and localization of the targeted liposomes in the cytoplasm in major proportion.
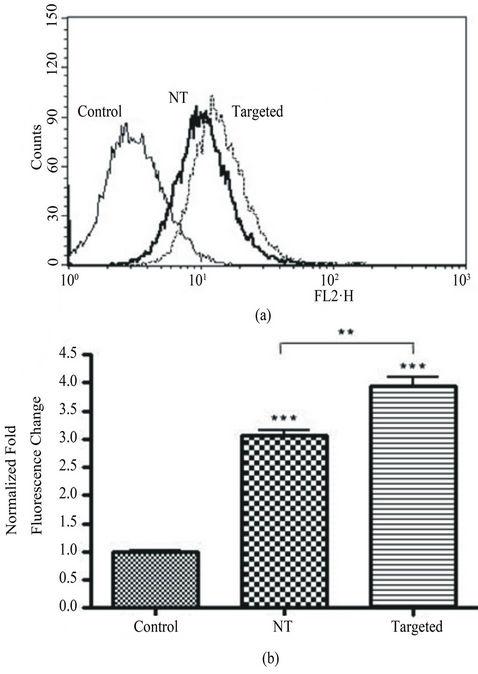
Figure 4. Flow cytometric analysis of liposome accumulation in BV-2 cells. (a), representative overlay of the fluorescence intensity of BV-2 cells treated with targeted versus non-targeted (NT) liposomes. Targeted liposomes with the LPS moiety resulted in a right shift of the curve, hence an increased accumulation of liposomes over a 2 hour exposure. (b), after normalization of the endogenous fluorescent signal (control), analysis reveals a significant increase (p < 0.01) of 29% in the fluorescent change from non-targeted to targeted treatments. Each condition has n = 3, and was run in duplicate. The control conditions are BV-2 cells devoid of treatment.
minocycline for all three treatment groups remains constant (40 μM).
3.3. In Vivo Therapeutic Efficacy
SOD1G93A mice treated with non-targeted minocycline liposomes had significantly (p < 0.01) extended time to rotarod failure and disease endpoints as compared to mice receiving no treatment. There was a significantly prolonged latency to disease endpoint, but not to rotarod failure, in the targeted liposome-treated group when compared to untreated mice. As seen in Table 1, the mean age for rotarod failure in the untreated control mice was 103.0 days, whereas the mean age for the targeted minocycline liposome group was 111.1 days and the non-targeted group was 116.0 days (p < 0.01).
Survival was significantly increased (p < 0.05) to 133.0 and 135.0 days with both targeted and non-targeted therapies, respectively, compared to 124.3 days in the control group. These delays represent an increase of 8% (targeted group) and 13% (non-targeted group) for age at rotarod failure, and 7% and 9% latency to disease endpoint, respectively. Likewise, the Kaplan-Meier plot of the cumulative probability of rotarod failure displays a clear and statistically significant difference (logrank test, p < 0.05) between control and non-targeted liposome groups (Figure 6(a)). The survival plot of disease endpoints for both targeted and non-targeted treatment groups shows a significant difference (logrank test, p < 0.05) as compared to controls (Figure 6(b)).
4. Discussion
The present study was designed to establish the proof of concept that delivery of nanovesicles containing a therapeutic agent via intracerebroventricular infusion could delay symptom onset and prolong survival in the ALS animal model.
The purpose of this study was two-fold: 1) to determine if the intracerebroventricular approach had therapeutic efficacy and 2) to test the hypothesis that an LPS-targeting moiety would be more effective than non-targeted liposomes. To our knowledge the use of a targeted liposome with specificity for microglia, as presented in this paper, has not been previously shown. Previous studies have shown that TLR4 is not expressed on neurons, astrocytes, or oligodendrocytes [29,30] , thus TLR4 meets the criteria for selective targeting to microglia. Our data showed a relative increase in uptake and accumulation of targeted nanovesicles compared to nontargeted into BV-2 cells in vitro providing evidence to support the concept put forth in this study. Moreover, encapsulating the drug in a liposome did not limit its efficacy. Both targeted and non-targeted liposomes
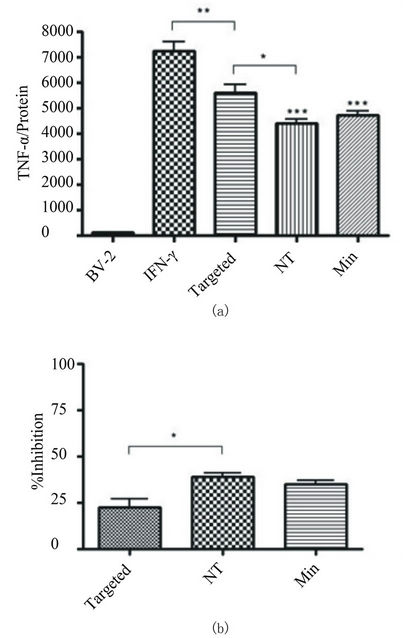
Figure 5. Quantification of TNF-α by standard mouse ELISA kit. (a) BV-2 cells were exposed to 10 ng/ml IFN-γ in media and simultaneously treated with 40 μM minocycline encapsulated in targeted or non-targeted (NT) liposomes, or as free drug (Minocycline). Exposure duration was 48 hours. The control group consisted of BV-2 cells without activation or treatment condition. There was a significant decrease (p < 0.001 NT and Min, p < 0.01 for targeted) in TNF-α levels with all three treatment groups as compared to activated cells alone. All conditions were performed in triplicate and repeated twice. (b) The relative TNF-α inhibition as compared to activated cells receiving no treatment was 22.3%, 39.1%, and 34.9% for targeted, NT, and Minocyline treatment respectively.
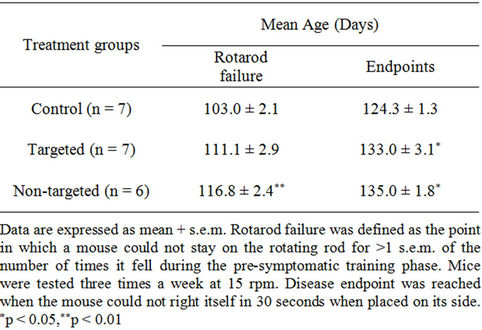
Table 1. Mean age at time of rotarod failure and endpoint stages.
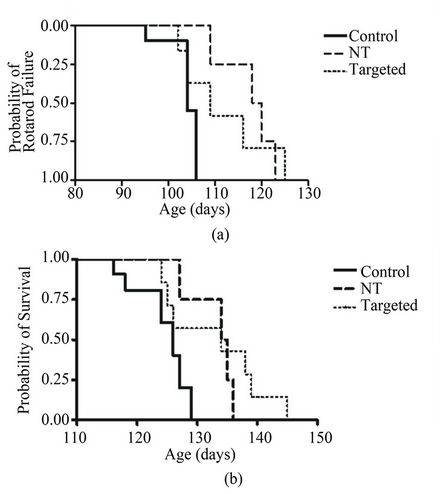
Figure 6. Kaplan-Meier plots of the cumulative probability of rotarod failure and death. A, The therapeutic effect of non-targeted liposomes on motor performance of SOD1G93A mice as compared to controls can be seen by the right shift in the survival curve. Analysis with a logrank test reveals a significant shift (p < 0.05) in the curve of the non-targeted liposome treated group. The right shift for the targeted liposome group did not reach statistical significance (p > 0.05). B, Analysis of the age at disease endpoint for SOD1G93A mice receiving both targeted and NT liposome treatments revealed a significant difference (p < 0.05) in age at death compared to that of controls for both treatment groups.
resulted in decreased TNF-α secretion in activated BV-2 microglial cells as compared to activated cells receiving no treatment. The non-targeted liposomes had a greater effect than the targeted liposomes in reducing the levels of TNF-α released by the BV-2 cells. Although the most parsimonious explanation is that there does not appear to be any correlation between the rate of uptake and the effect on cytokine secretion, an alternative explanation is that targeting the TLR4/LPS complex may have attenuated the efficacy of the minocycline by activating the TLR4 signaling pathway. Increased activation of the microglia may provide an explanation for the increased uptake of targeted liposomes. TNF-α elevation in the microglial cells will subsequently lead to NFκB nuclear translocation which leads to microglial activation by functioning as transcription factor (Figure 7). Minocycline has been previously reported to inhibit the nuclear translocation of NFκB [35,36] in microglia which is one of the rationale for choosing this drug for our study.
The potential therapeutic efficacy of the targeted and non-targeted minocycline liposomes was tested in SOD1G93A mice. The non-targeted nanovesicles signifi-
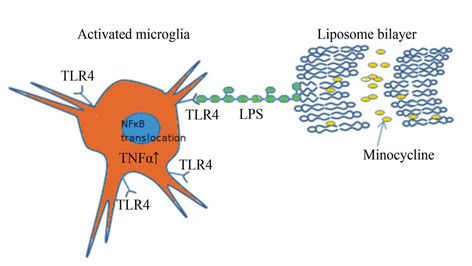
Figure 7. Schematic representation of LPS modified liposomes carrying minocycline targeting the activated microglia. Lipopolysaccharide moiety interacts with the TLR4 receptors in the activated microglia and internalize and deliver the minocycline, which might have an impact on the TNFα levels and NFκB translocation.
cantly increased the latency to rotarod failure and both targeted and non-targeted nanovesicles significantly delayed disease endpoints. While there was an increase in the mean age to time of rotarod failure for the targeted liposome-treated versus the control groups, the difference (8%) was not statistically significant. The difference in the outcomes for the targeted and non-targeted liposomes represent an exciting area to explore because it is not clear from our present study about the cell types which accumulate the minocycline from the non-targeted liposomes.
The non-targeted liposomes did accumulate in the BV- 2 cells in culture so it is likely that some of the non-targeted liposomes were taken up by microglia in vivo. Indeed, given the phagocytotic nature of these cells it can be expected. Thus, at least some part of the positive therapeutic effect in vivo of non-targeted liposomes is likely attributable to microglial uptake, but it is also possible that astrocytes accumulated some of the liposomes. The onset and progression of ALS symptoms are represented by distinct disease phases that can be characterized by mutant SOD1 action within different cell types [5,6] and raise the possibility that targeted and non-targeted liposomes may be used in combination or in sequence to combat different stages of the disease.
Comparative analysis of the present study to previous studies involving oral or injected minocycline in SOD1G93A mice reveals a similar effect in the latency to disease endpoints (Table 2). For example when free minocycline was delivered to the SOD1G93A mice at a dose of 15 mg/kg body weight once a day for every weekday, there was an increase in the life-span of about 16% when compared to untreated mice [20] . However, important to this analysis is the improved efficacy of the nanovesicle delivery approach when compared to
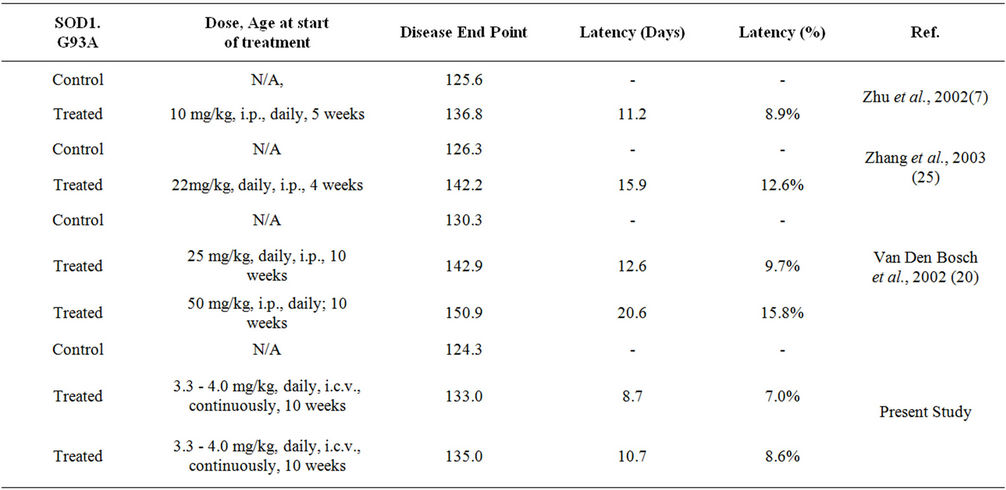
Table 2. Comparison of present study with previous SOD1G93A mouse models and minocycline therapy.
the daily administered dosage of 15 mg/kg body weight [20] . In other words, intracerebroventricular delivery requires less dosage to achieve the same relative effect because of the local drug action, potentially resulting in a lower frequency of adverse treatment-related events. Rotarod failure and disease onset are not listed in the table due to differing definitions in the various studies. Our data showing lower concentrations with similar effects at lower concentrations than those administered via intraperitoneal injecttion as well as the potential for direct uptake by microglia, suggest that perhaps the use of minocycline as a treatment in human ALS should be revisited. The clinical failure of minocycline in human ALS ADDIN EN.CITE ADDIN EN.CITE.DATA [24] may have involved the inadvertent toxicity to motor neurons due to nonselective oral drug administration or the inability to deliver sufficient amounts of drug to the desired site of action. Current treatment for ALS includes only one FDA approved drug (riluzole) that has exhibited modest and variable efficacy [37-40] . In addition to novel pharmaceutical agents, novel delivery platforms of agents that show efficacy should be evaluated and the findings debated in the literature.
5. Conclusion
In this study we have developed TLR4 targeting nanoliposomes by conjugating lipopolysaccharides and loaded them with minocycline a tropic agent and characterized them for their suitability for treating Amyotropic Lateral Sclerosis (ALS).Our in vitro studies in the BV-2 microglial cells demonstrated the specific targeting ability of the LPS modified liposomes through the receptors expressed on the cells. From the in vivo study it is clear that the disease endpoints in the SOD-1 mutant mice are delayed in both targeted and non-targeted liposomes formulations carrying minocycline. Our study also demonstrates that intracerebroventricular administration of minocycline through the liposomes in the osmotic pump has comparable effect in the latency to the disease endpoints when compared to the oral and injected minocycline as reported by others, with a potential to minimize the toxicity associated with frequent administration.
6. Acknowledgements
This work was supported by funds from The Paul and Harriett Campbell Fund for ALS Research and The Zimmerman Family Love Fund. The authors wish to thank the flow cytometry core facility of Penn State College of Medicine as well as Dr. Bill Zinnanti for his assistance in using the HPLC instrument for minocycline analysis.
REFERENCES
- M. E. Gurney, H. Pu, A. Y. Chiu, M. C. Dal Canto, C. Y. Polchow, D. D. Alexander, J. Caliendo, A. Hentati, Y. W. Kwon, H. X. Deng, et al., “Motor Neuron Degeneration in Mice That Express a Human Cu,Zn Superoxide Dismutase Mutation,” Science, Vol. 264, No. 5166, 1994, pp. 1772-1775. doi:10.1126/science.8209258
- T. A. Ferguson and L. B. Elman, “Clinical Presentation and Diagnosis of Amyatrophic Lateral Slerosis,” Neurorehabilitation, Vol. 22, No. 6, 2007, pp. 409-416.
- M. E. Alexianu, M. Kozovska and S. H. Appel, Neurology, “Immune Reactivity in a Mouse Model of Familial ALS Correlates with Disease Progression,” Vol. 57, No. 7, 2001, pp. 1282-1289. doi:10.1212/WNL.57.7.1282
- T. Kawamata, H. Akiyama, T. Yamada and P. L. McGeer, “Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue,” American Journal of Pathology, Vol. 140, No. 3, 1992, pp. 691-707.
- T. M. Miller, S. H. Kim, K. Yamanaka, M. Hester, P. Umapathi, H. Arnson, L. Rizo, J. R. Mendell, F. H. Gage, D. W. Cleveland and B. K. Kaspar, “Gene Transfer Demonstrates That Muscle Is Not a Primary Target for Non-Cell-Autonomous Toxicity in Familial Amyotrophic Lateral Sclerosis,” Proceedings of the National Academy of Science of the United States of America, Vol. 103, No. 51, 2006, pp. 19546-19551. doi:10.1073/pnas.0609411103
- S. Boillee, K. Yamanaka, C. S. Lobsiger, N. G. Copeland, N. A. Jenkins, G. Kassiotis, G. Kollias and D. W. Cleveland, “Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia,” Science, Vol. 312, No. 5778, 2006, pp. 1389-1392. doi:10.1126/science.1123511
- S. Zhu, I. G. Stavrovskaya, M. Drozda, B. Y. Kim, V. Ona, M. Li, S. Sarang, A. S. Liu, D. M. Hartley, D. C. Wu, S. Gullans, R. J. Ferrante, S. Przedborski, B. S. Kristal and R. M. Friedlander, “Minocycline Inhibits Cytochrome C Release and Delays Progression of Amyotrophic Lateral Sclerosis in Mice,” Nature, Vol. 417, No. 6884, 2002, pp. 74-78. doi:10.1038/417074a
- H. Manev and R. Manev, “Interactions with GluR1 AMPA Receptors Could Influence the Therapeutic Usefulness of Minocycline in ALS,” Amyotroph Lateral Scler, Vol. 10, No. 5-6, 2009, pp. 416-417. doi:10.3109/17482960802702288
- J. Yrjanheikki, R. Keinanen, M. Pellikka, T. Hokfelt and J. Koistinaho, “Tetracyclines Inhibit Microglial Activation and Are Neuroprotective in Global Brain Ischemia,” Proceedings of the National Academy of Science of the United States of America, Vol. 95, No. 26, 1998, pp. 15769-15774. doi:10.1073/pnas.95.26.15769
- K. L. Arvin, B. H. Han, Y. Du, S. Z. Lin, S. M. Paul and D. M. Holtzman, “Minocycline Markedly Protects the Neonatal Brain against Hypoxic-Ischemic Injury,” Annals of Neurology, Vol. 52, No. 1, 2002, pp. 54-61. doi:10.1002/ana.10242
- R. O. Sanchez Mejia, V. O. Ona, M. Li and R. M. Friedlander, “Minocycline Reduces Traumatic Brain Injury-mediated Caspase-1 Activation, Tissue Damage, and Neurological Dysfunction,” Neurosurgery, Vol. 48, No. 6, 2001, pp. 1393-1399.
- J. E. Wells, R. J. Hurlbert, M. G. Fehlings and V. W. Yong, “Neuroprotection by Minocycline Facilitates Significant Recovery from Spinal Cord Injury in Mice,” Brain, Vol. 126, No. 7, 2003, pp. 1628-1637. doi:10.1093/brain/awg178
- V. Brundula, N. B. Rewcastle, L. M. Metz, C. C. Bernard and V. W. Yong, “Targeting Leukocyte MMPs and Transmigration Minocycline as a Potential Therapy for Multiple Sclerosis,” Brain, Vol. 125, No. 6, 2002, pp. 1297-1308. doi:10.1093/brain/awf133
- Y. Du, Z. Ma, S. Lin, R. C. Dodel, F. Gao, K. R. Bales, L. C. Triarhou, E. Chernet, K. W. Perry, D. L. Nelson, S. Luecke, L. A. Phebus, F. P. Bymaster and S. M. Paul, “Minocycline Prevents Nigrostriatal Dopaminergic Neurodegeneration in the MPTP Model of Parkinson’s Disease,” Proceedings of the National Academy of Science of the United States of America, Vol. 98, No. 25, 2001, pp. 14669-14674. doi:10.1073/pnas.251341998
- D. C. Wu, V. Jackson-Lewis, M. Vila, K. Tieu, P. Teismann, C. Vadseth, D. K. Choi, H. Ischiropoulos and S. Przedborski, “Blockade of Microglial Activation Is Neuroprotective in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model of Parkinson Disease,” The Journal of Neuroscience, Vol. 22, No. 5, 2002, pp. 1763- 1771.
- Y. He, S. Appel and W. Le, “Minocycline Inhibits Microglial Activation and Protects Nigral Cells after 6-Hydroxydopamine Injection into Mouse Striatum,” Brain Research, Vol. 909, No. 1-2, 2001, pp. 187-193. doi:10.1016/S0006-8993(01)02681-6
- M. Chen, V. O. Ona, M. Li, R. J. Ferrante, K. B. Fink, S. Zhu, J. Bian, L. Guo, L. A. Farrell, S. M. Hersch, W. Hobbs, J. P. Vonsattel, J. H. Cha and R. M. Friedlander, “Minocycline Inhibits Caspase-1 and Caspase-3 Expression and Delays Mortality in a Transgenic Mouse Model of Huntington disease,” Nature Medicine, Vol. 6, No. 7, 2000, pp. 797- 801. doi:10.1038/80538
- X. Wang, S. Zhu, M. Drozda, W. Zhang, I. G. Stavrovskaya, E. Cattaneo, R. J. Ferrante, B. S. Kristal and R. M. Friedlander, “Minocycline Inhibits Caspase-independent and -Dependent Mitochondrial Cell Death Pathways in Models of Huntington’s Disease,” Proceedings of the National Academy of Science of the United States of America, Vol. 100, No. 18, 2003, pp. 10483-10487. doi:10.1073/pnas.1832501100
- J. Kriz, M. D. Nguyen, and J. P. Julien, “Minocycline Slows Disease Progression in a Mouse Model of Amyotrophic Lateral Sclerosis,” Neurobiology of Disease, Vol. 10, No. 3, 2002, pp. 268-278. doi:10.1006/nbdi.2002.0487
- L. Van Den Bosch, P. Tilkin, G. Lemmens and W. Robberecht, “Minocycline Delays Disease Onset and Mortality in a Transgenic Model of ALS,” Neuroreport, Vol. 13, No. 8, 2002, pp. 1067-1070. doi:10.1097/00001756-200206120-00018
- P. N. Leigh, V. Meininger, G. Bensimon, M. Cudkowicz and W. Robberecht, “Minocycline for Patients with ALS,” Lancet Neurol, Vol. 7, No. 2, 2008, pp. 119-120.
- A. Milane, L. Tortolano, C. Fernandez, G. Bensimon, V. Meininger and R. Farinotti, “Brain and Plasma Riluzole Pharmacokinetics: Effect of Minocycline Combination,” Journal of Pharmacy and Pharmaceutical Sciences, Vol. 12, No. 2, 2009, pp. 209-217.
- H. Manev and R. Manev, “Interactions with GluR1 AMPA Receptors Could Influence the Therapeutic Usefulness of Minocycline in ALS,” Amyotroph Lateral Scler, Vol. 10 No. 5-6, 2009, pp. 1-2. doi:10.1080/17482960802702288
- P. H. Gordon, D. H. Moore, R. G. Miller, J. M. Florence, J. L. Verheijde, C. Doorish, J. F. Hilton, G. M. Spitalny, R. B. MacArthur, H. Mitsumoto, H. E. Neville, K. Boylan, T. Mozaffar, J. M. Belsh, J. Ravits, R. S. Bedlack, M. C. Graves, L. F. McCluskey, R. J. Barohn and R. Tandan, “Efficacy of Minocycline in Patients with Amyotrophic Lateral Sclerosis: A Phase Iii Randomised Trial,” The Lancet Neurology, Vol. 6, No. 12, 2007, pp. 1045-1053. doi:10.1016/S1474-4422(07)70270-3
- W. Zhang, Narayanan, M., and Friedlander, R. M. “Additive Neuroprotective Effects of Minocycline with Creatine in a Mouse Model of ALS,” Annals of Neurology, Vol. 53, No. 2, 2003, pp. 267-270. doi:10.1002/ana.10476
- C. Gurley, J. Nichols, S. Liu, N. K. Phulwani, N. Esen and T. Kielian, “Microglia and Astrocyte Activation by Toll-Like Receptor Ligands: Modulation by PPAR-Agonists,” PPAR Research, Vol. 2008, 2008, pp. 1-15. doi:10.1155/2008/453120
- J. C. Chow, D. W. Young, D. T. Golenbock, W. J. Christ and F. Gusovsky, “Toll-Like Receptor-4 Mediates Lipopolysaccharide-Induced Signal Transduction,” The Journal of Biological Chemistry, Vol. 274, No. 16, 1999, pp. 10689-10692. doi:10.1074/jbc.274.16.10689
- D. W. Chung, K. Y. Yoo, I. K. Hwang, D. W. Kim, J. Y. Chung, C. H. Lee, J. H. Choi, S. Y. Choi, H. Y. Youn, I. S. Lee and M. H. Won, “Systemic Administration of Lipopolysaccharide Induces Cyclooxygenase-2 Immunoreactivity in Endothelium and Increases Microglia in the Mouse Hippocampus,” Cell Mol Neurobiol, Vol. 30, No. 4, 2009.
- K. Hoshino, O. Takeuchi, T. Kawai, H. Sanjo, T. Ogawa, Y. Takeda, K. Akeda and S. Akira, “Cutting Edge: Toll-Like Receptor 4 (TLR4)-Deficient Mice Are Hyporesponsive to Lipopolysaccharide: Evidence for TLR4 as the Lps Gene Product,” The Journal of Immunology, Vol. 162, No. 7, 1999, pp. 3749-3752.
- S. Lehnardt, C. Lachance, S. Patrizi, S. Lefebvre, P. L. Follett, F. E. Jensen, P. A. Rosenberg, J. J. Volpe and T. Vartanian, “The Toll-Like Receptor TLR4 Is Necessary for Lipopolysaccharide-Induced Oligodendrocyte Injury in the CNS,” The Journal of Neuroscience, Vol. 22, No. 7, 2002, pp. 2478-2486.
- E. Blasi, R. Barluzzi, V. Bocchini, R. Mazzolla and F. Bistoni, “Immortalization of Murine Microglial Cells by a V-Raf/V-Myc Carrying Retrovirus,” Journal of Neuroimmunology, Vol. 27, No. 2-3, 1990, pp. 229-237. doi:10.1016/0165-5728(90)90073-V
- S. G. Kremlev, R. L. Roberts, and C. Palmer, “Differential Expression of Chemokines and Chemokine Receptors during Microglial Activation and Inhibition,” Journal of Neuroimmunology, Vol. 149, No. 1, 2004, pp. 1-9. doi:10.1016/j.jneuroim.2003.11.012
- M. Nikodemova, I. D. Duncan, and J. J. Watters, “Minocycline Exerts Inhibitory Effects on Multiple MitogenActivated Protein Kinases and IκBα Degradation in a Stimulus-Specific Manner in Microglia,” Journal of Neurochemistry, Vol. 96, No. 2, 2006, pp. 314-323. doi:10.1111/j.1471-4159.2005.03520.x
- M. E. Gurney, F. B. Cutting, P. Zhai, A. Doble, C. P. Taylor, P. K. Andrus and E. D. Hall, “Benefit of Vitamin E, Riluzole, And Gababapentin in a Transgenic Model of Familial Amyotrophic Lateral Sclerosis,” Annals of Neurology, Vol. 39, No. 2, 1996, pp. 147-157. doi:10.1002/ana.410390203
- M. Molina-Hernandez, N. P. Tellez-Alcantara, J. PerezGarcia, J. I. Olivera-Lopez and M. T. Jaramillo-Jaimes, “Antidepressant-Like Actions of Minocycline Combined with Several Glutamate Antagonists,” Progress in NeuroPsychopharmacology and Biological Psychiatry, Vol. 32, No. 2, 2008, pp. 380-386. doi:10.1016/j.pnpbp.2007.09.004
- T. Miyaoka, R. Yasukawa, H. Yasuda, M. Hayashida, T. Inagaki and J. Horiguchi, “Possible Antipsychotic Effects of Minocycline in Patients with Schizophrenia,” Progress in Neuro-Psychopharmacology and Biological Psychiatry, Vol. 31, No. 1, 2007, pp. 304-307. doi:10.1016/j.pnpbp.2006.08.013
- G. Bensimon, L. Lacomblez and V. Meininger, “A Controlled Trial of Riluzole in Amyotrophic Lateral Sclerosis,” The New England Journal of Medicine, Vol. 330, No. 9, 1994, pp. 585-591. doi:10.1056/NEJM199403033300901
- L. Lacomblez, G. Bensimon, P. N. Leigh, P. Guillet and V. Meininger, “Dose-Ranging Study of Riluzole in Amyotrophic Lateral Sclerosis,” Lancet, Vol. 347, No. 9013, 1996, pp. 1425- 1431.
- C. Scherfler, T. Sather, E. Diguet, N. Stefanova, Z. Puschban, F. Tison, W. Poewe and G. K. Wenning, “Riluzole Improves Motor Deficits and Attenuates Loss of Striatal Neurons in a Sequential Double Lesion Rat Model of Striatonigral Degeneration (Parkinson Variant of Multiple System Atrophy),” Journal of Neural Transmission, Vol. 112, No. 18, 2005, pp. 1025-1033. doi:10.1007/s00702-004-0245-5
- J. A. Rocha, C. Reis, F. Simoes, J. Fonseca and J. Mendes Ribeiro, “Diagnostic Investigation and Multidisciplinary Management in Motor Neuron Disease,” Journal of Neuroimmunology, Vol. 252, No. 12, 2005, pp. 1435-1447. doi:10.1007/s00415-005-0007-9
Abbreviations
LPS—LipopolysaccharidesTLR4—Toll like receptor-4SOD-1—Superoxide dismutase-1ALS—Amyotrophic lateral sclerosis,
NOTES
*Corresponding author.