Crystal Structure Theory and Applications
Vol.2 No.4(2013), Article ID:41115,9 pages DOI:10.4236/csta.2013.24023
Synthesis, Spectroscopic Investigations, Quantum Chemical Studies (Ab-initio & DFT) and Antimicrobial Activities of 3-(3-Chloro-4,5-dimethoxy-phenyl)-1-(4, 5-dimethoxy-2-methyl-Phenyl) prop-2-en-1-one
1Department of Physics, Sardar Patel University, Vallabh Vidyanagar, India
2P. G. Center in Chemistry, Smt. S. M. Panchal Science College, Talod, India
Email: u_h_patel@yahoo.com, *sahajg7@gmail.com
Copyright © 2013 Urmila H. Patel et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received October 25, 2013; revised November 21, 2013; accepted December 9, 2013
Keywords: Chalcones; FTIR; NMR; Single Crystal X-Ray Diffraction; Hydrogen Bond Interactions; Quantum Chemical Calculations; Antimicrobial Activities
ABSTRACT
The chalcones (1,3-diaryl-2-propenones) and their derivatives are important intermediates in organic synthesis and have widespread applications in medicinal industry. The title choloro chalcone derivative, 3-(3-chloro-4,5-dimethoxyphenyl)-1-(4,5-dimethoxy-2-methyl phenyl) prop-2-en-1-one, has been synthesized. It is characterized by FTIR, 1H NMR, 13C NMR and single crystal X-ray diffraction. Title compound crystallizes in monoclinic space group C2/c with a = 23.540(11) Å, b = 9.738(4) Å, c = 17.305(7) Å, β = 106.37 (3)˚, V = 3806(3) Å3 and Z = 8. The mean plane of the two substituted benzene rings is twisted by 66.29 (12)˚ with respect to each other. Ab-initio and density functional Theory (DFT) calculations have been carried out for the title molecule using RHF/6-311G and B3LYP/6-311G basis set respectively. The calculated results show that the predicted geometry can well reproduce structural parameters. In addition, frontier molecular orbitals and Mullikan charge distributions are carried out by using RHF and B3LYP methods. The calculated HOMO and LUMO energies show that charge transfer occurs in the molecule. Numbers of weak but significant interactions like C-H···O, C-H···π and π-π are involved in the stability of the structure. The weak π-π stacked interaction involves the centroids of the methyl phenyl rings with Cg-Cg separation distance of 3.857(2) Å. Synthesized compound has been screened for its antimicrobial activity against different panels of organisms.
1. Introduction
In chalcones, two aromatic rings are linked by an aliphatic three-carbon chain. The synthesis of chalcone derivative has generated vast interest to organics as well as for medicinal chemists. Chalcones are also key precursors in the synthesis of many biologically important heterocycles such as benzothiazepine, pyrazolines, 1,4- diketones and flavones. Chalcones, belonging to flavonoid family, synthesized or the natural one, displayed many interesting properties including antimalarial [1,2], anticancer [3,4], antiviral [5], antibacterial [6], antifungal [7], antihyperglycemic [8] and photocytotoxicity [9] activities. In the chemical structure, three-carbon α – β unsaturated carbonyl system, the back bone of the open chain flavonoids, joins two aromatic rings. The ab-initio and density functional theory (DFT) are applied for the investigation of the optimized molecular structure and few significant spectroscopy properties. As part of our ongoing research on X-ray Crystallographic investigations of drug molecules and theoretical quantum computational studies of synthesized drugs [10,11], we have synthesized and investigated molecular structure of a novel methoxy-chloro substituted chalcone derivative by IR, NMR and X-ray diffraction techniques and optimized the structure by quantum chemistry.
2. Experimental
2.1. Materials and Instrumentations
General chemicals are purchased from Merck, SD Fine and commercial source. All non-aqueous reactions are performed in dry glass ware. Thin layer chromatography (TLC) is performed on pre-coated plates, silica gel 60- F254 (Merck 1.16834, layer thickness 0.25 mm) using toluene/methanol mixtures (8:2) as developing system. The detection of the products on TLC is carried out in iodine vapor. Melting points are determined on a Polmon Instrument Model: M.P.: 96, Range: 25˚C to 350˚C, Resolution: 0.1˚C fitted with a microscope.
2.2. Synthesis
A mixture of 1-(2-methyl-4,5-dimethoxyphenyl) ethanone (0.01 mole) and 3-chloro-4,5-dimethoxy benzaldehyde (0.01 mole) in ethanol (30 ml) are added to a solution of potassium hydroxide (40 ml, 40%) with constant shaking of the reaction flask. The reaction mixture is stirred for a 24 hours on a magnetic stirrer and poured in to crushed ice and acidified with diluted HCl (2N). The solid mass which separated out is filtered, washed with water, dried and crystallized from methanol to give light yellow needles. The reaction scheme of the title molecule is shown in Figure 1.
2.3. FTIR and NMR Analysis
The FTIR spectrum of the compound is recorded in the KBr phase in the frequency region of 400 - 4000 cm−1 using Bruker FTIR spectrometer. 1H-NMR and 13C-NMR spectra are recorded with a Fourier transform instrument at 400 MHz (Bruker AVANCE 400). The details of FTIR and NMR analysis are given below.
IR (KBr): ν 1447 (C-H def (asym) alkyl), 1375 (C-H def (sym) alkyl), (C-H str. arom.), 1565 (C=C str. arom.), 1113 (C-H i.p.def arom.), 820 (C-H o.o.p.def. arom.), 1217 (C-O-C (sym) ether), 1077 (C-O-C (asym) ether), 1641 (C=O str., chalcone), 1565 (C=C, chalcone), 3273 (OH, phenol), 978 (CH=CH def. chalcone), 3087 (CH=CH str. chalcone), 1641 (C=C str. chalcone), 694 (C-Clstr.) cm−1.
1H NMR (CDCl3) d ppm: 2.44 (s, 3H), 3.89 (s, 3H, OCH3), 3.91 (s, 6H, OCH3), 3.93 (s, 3H, OCH3), 6.754 (s, 1H), 6.99 (d, 1H, J = 1.8 Hz) ,7.05 (m, 1H + 1H chalcone), 7.22 (d, 1H, J = 1.6 Hz), 7.38 (d, 1H, J = 16 Hz, chalcone).
13C NMR (CDCl3) d ppm: 56.01 (C-1), 56.04 (C-2), 55.09 (C-3), 55.04 (C-4), 15.38 (C-5), 194.56 (C-6), 122.75 (C-7), 141.03 (C-8),150.75 (C-9), 146.25 (C-10), 155.27 (C-11),144.89 (C-12), 114.05 (C-13), 131.39 (C-14), 112.21 (C-15), 110.51 (C-16), 130.68 (C-17), 130.20 (C-18), 127.08 (C-19), 126.31 (C-20). (mp: 142˚C - 148˚C; Yield: 80%. Analysis: C20H21ClO5, Found: C: 63.74%, H: 5.61%, O: 21.24%, Cl: 9.41%, Calculated: C: 63.75%, H: 5.62%, O: 21.23%, Cl: 9.42%).
2.4. X-Ray Diffraction Study
A suitable sample of single crystal of size (0.6 × 0.4 × 0.1) mm3 is selected for the crystallographic study. All diffraction measurements are performed at room temperature (296 K) using graphite monochromated MoKα radiation of wavelength 0.71073 Å. The crystal structure is solved by direct methods and refined by full-matrix least square technique on F2, using SHELX-97 set of program [12]. All non-hydrogen atoms are refined anisotropically. The structure is refined to R = 0.0612 for the observed reflections 4383 and Goodness of fit S = 1.048 by using the intensity (I) values of 3132 reflections satisfying the I > 2σ (I) criterion and 4383 reflections in refinement for 235 crystallographic parameters. Highest and lowest electron density peaks “∆ρ” are 0.758 and −0.546 eÅ−3 respectively. The crystallographic data and details of the data collection and structure refinements are listed in Table 1.
2.5. Computational Details
The quantum chemical study of the title compound has been performed within the framework of Hartree Fock [13] and the density functional theory with Becke’s three-parameter hybrid exchange functional with LeeYang-Parr correlation functional (B3LYP) employing 6-311G basis set [14,15]. All quantum chemical calculations are performed using computer software Gaussian- 09 [16] and Gauss-View molecular visualization program [17].
3. Results and Discussion
3.1. X-Ray Crystallography
The ORTEP diagram of the title compound, 3-(3-chloro-4,

Figure 1. Chemical reaction scheme of the title compound.

Table 1. Crystal data and structure refinement parameters of the title compound.
5-dimethoxy-phenyl)-1-(4,5-dimethoxy-2-methyl phenyl) prop-2-en-1-one with thermal ellipsoids drawn at a 50% probability is shown in Figure 2.
Single crystal X-ray diffractions confirms the molecular structure of the title molecule, C20H21ClO5, 2-methoxy, 1-methyl substituted phenyl ring is joined by a prop-2- en-1-one group to 3-chloro-4,5-dimethoxy substituted phenyl ring. Both the phenyl rings (C1-C6) and (C16- C21) are planar. The mean plane of two phenyl rings are twisted by 66.30(12)˚ with respect to each other. The

Figure 2. The ORTEP view of the title molecule shows atomic labelling scheme and 50% probability level displacement ellipsoids.
dihedral angle between the mean plane of prop-2-en1- one group (C12-C14\O15) with the mean plane of the chloro phenyl ring (C1-C6) and methyl phenyl ring (C16-C21) are 29.85 (11)˚ and 38.46 (9)˚ respectively. Molecular conformation about C6-C12, C12-C13, C13- C14 and C14-C16 bonds described by the torsional angles C5-C6-C12-C13, C6-C12-C13-C14, C12-C13-C14- C16 and C13-C14-C16-C21 are 18.1 (4)˚, −176.6 (2)˚, −166.0 (2)˚ and −142.3 (2)˚ respectively and the geometry of the molecule about these bonds are very well comparable with other reported structure [10]. Intra molecular interaction involving C12-H12 with carbonyl oxygen O15 in C12-H12···O15 generates a pseudo ring of S(5) graph set motif [18] (Figure 2). Molecular packing is due to C-H···O, C-H···π and π-π intramolecular hydrogen bond interactions in the structure (Table 2).
In the crystal structure, the Chloro phenyl ring of methoxy group carbon C11 via H113 acts as potential donor to the prop-2-en-1-one group of oxygen O15 of x−1−y, 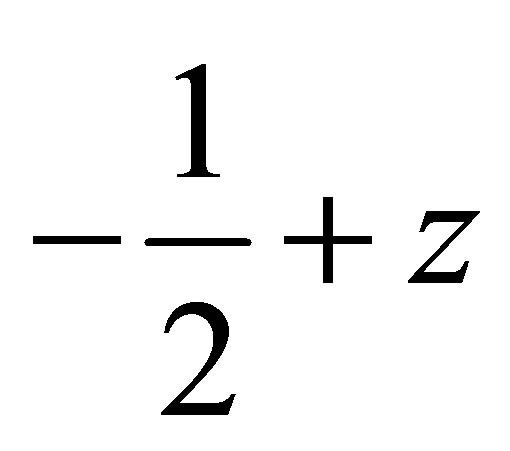 molecule and other phenyl ring of methoxy group carbon C26 via H261 link to the oxygen O15
molecule and other phenyl ring of methoxy group carbon C26 via H261 link to the oxygen O15
(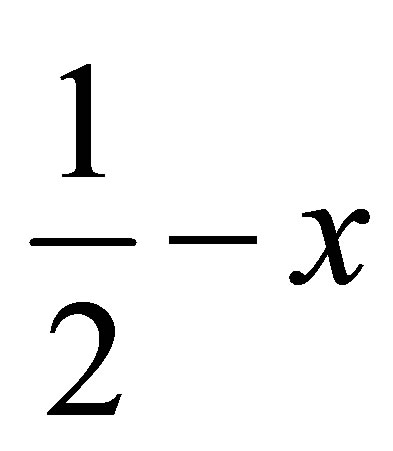 ,
,  ,
, ) forming a chain parallel to the ac plane (Figure 3). The C-H···p interaction of type-I [19] involves chloro phenyl ring carbon C1 via H1 to the centroid (Cg(2)) of the symmetry (
) forming a chain parallel to the ac plane (Figure 3). The C-H···p interaction of type-I [19] involves chloro phenyl ring carbon C1 via H1 to the centroid (Cg(2)) of the symmetry (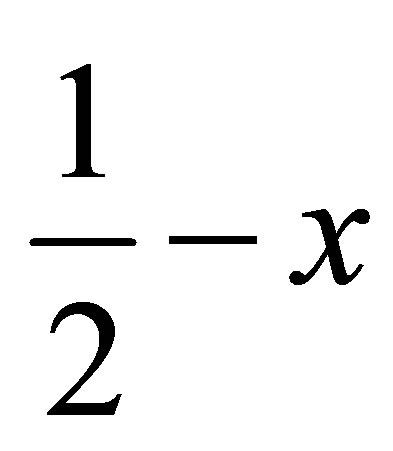 ,
, 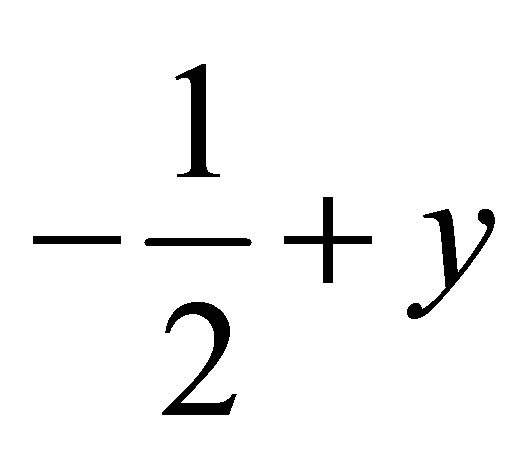 , 2-z)
, 2-z)
related methoxy phenyl ring (C16-C21) where C1- H1···Cg(2) = 3.6613(3) Å.
Face to face π-π stacked interaction observed between the centroids of symmetry related [ ,
, 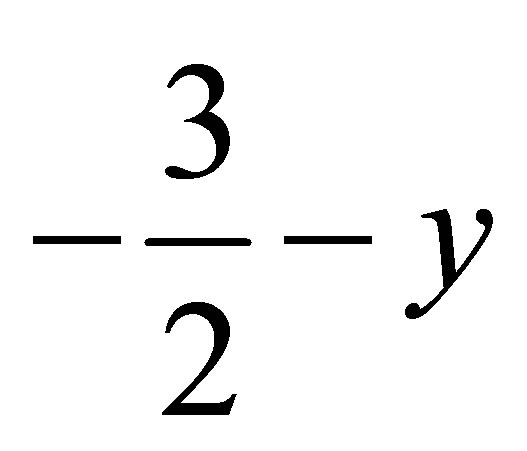 , 2-z]
, 2-z]
methyl phenyl ring (C16-C21), with Cg–Cg separation distance of 3.857(2) Å (α = 0˚), further contributes to the molecular packing (Figure 4).
3.2. Ab-Initio and DFT Studies
The ab-initio and Density Functional Theory (DFT) with Gaussian-09 program package employing B3LYP (Becke

Table 2. Intra and intermolecular interactions (distances in Å, angles in ˚).

Figure 3. Molecular packing showing C-H···O interactions along ac plane, for a shake of celerity some hydrogen atoms removed.
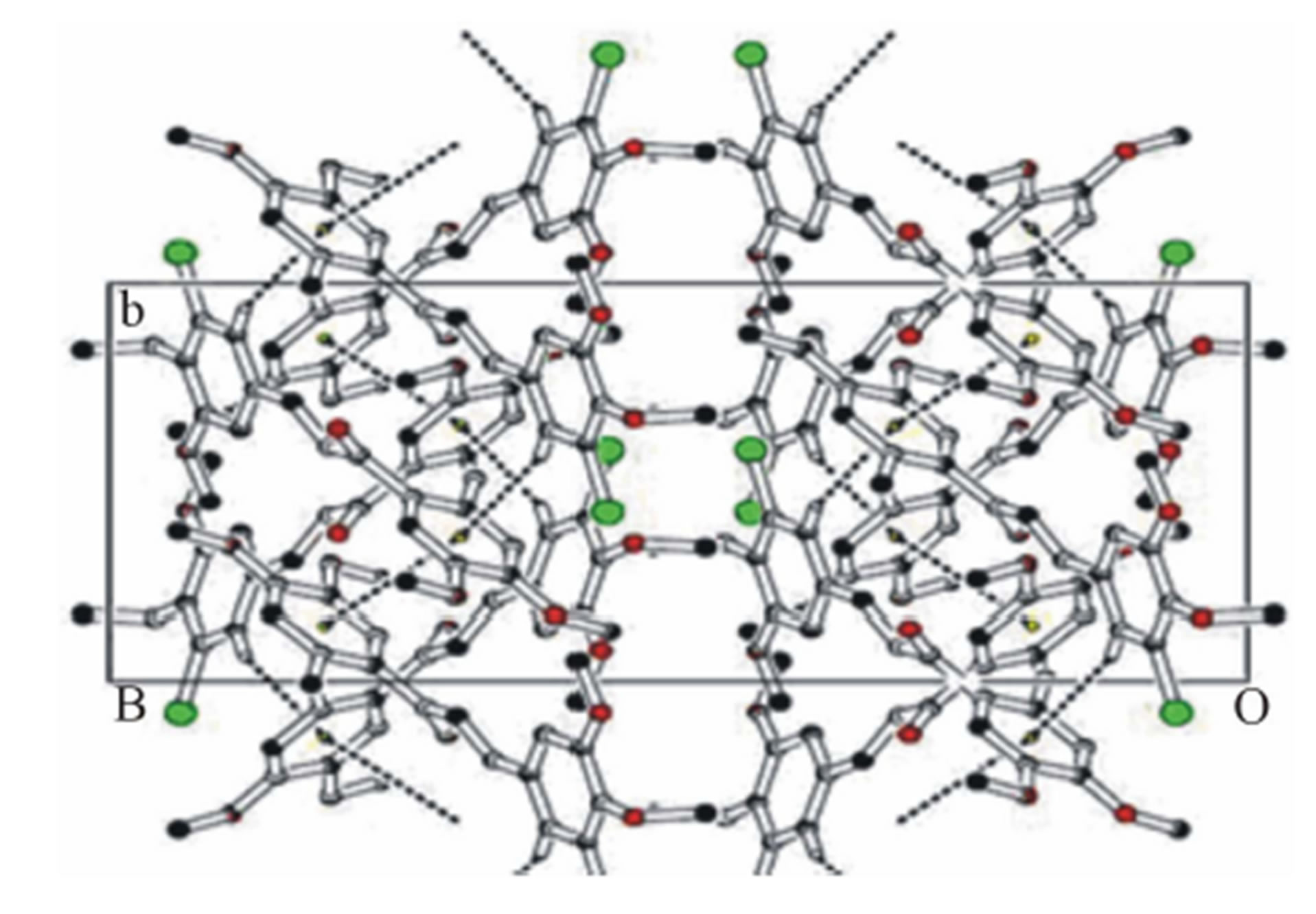
Figure 4. Molecular packing depicting π-π and C-H···π interactions along ab plane, for shake of clarity some hydrogen atoms are removed.
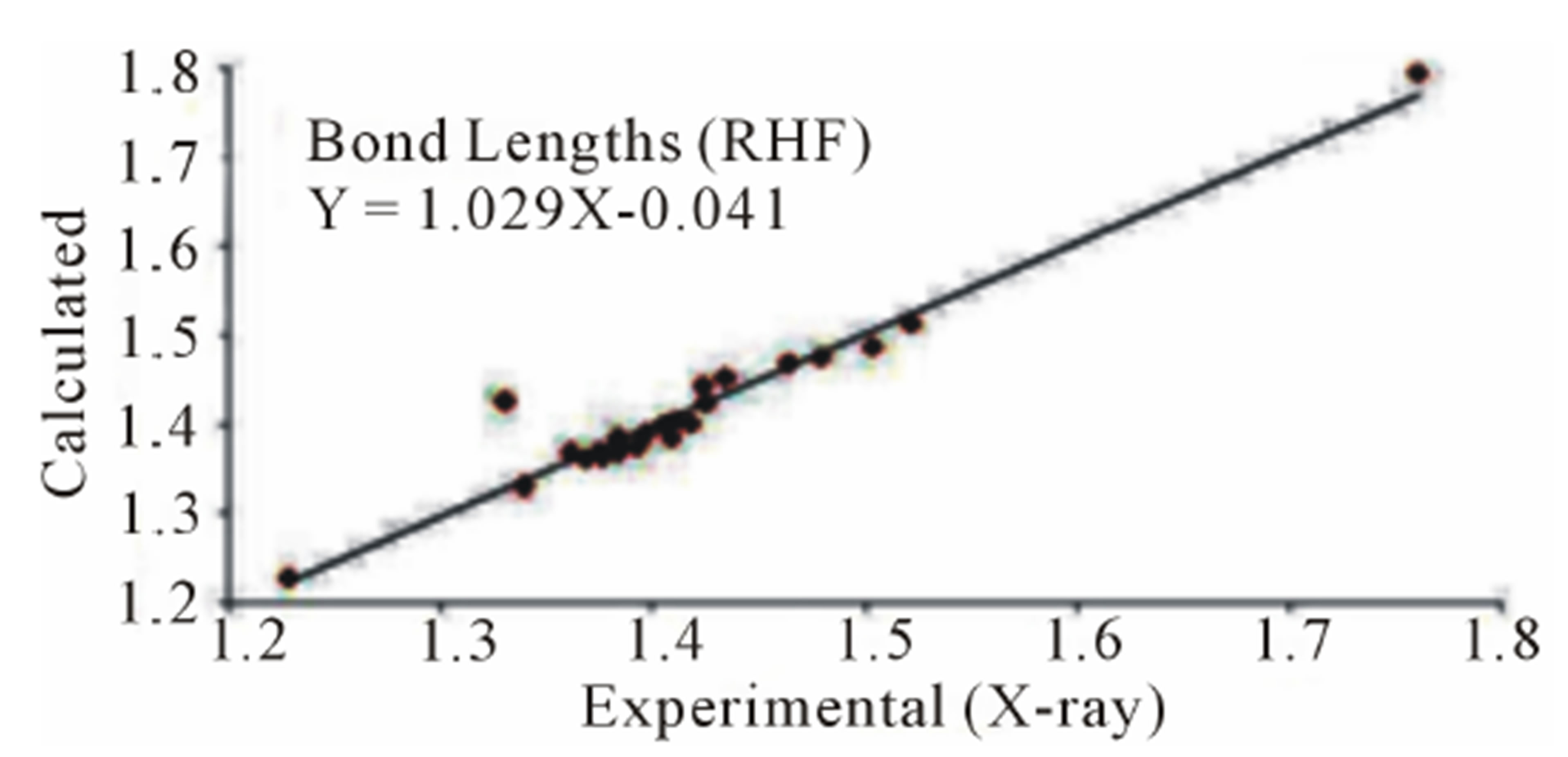
three parameter Lee-Yang-Parr) method with 6-311G* basis set is used to determine optimized bond lengths and angles. Table 3 compares the experimental data with those obtained by theoretical study which reveals that all optimized bond lengths and bond angles are slightly larger than the experimental values. The highest bond length difference is 0.0979 Å and 0.1238 Å for the C11-O10 bond at RHF and B3LYP respectively, where the biggest bond angle deviation occurred in the C3-O8- C9 angle 4.0313˚ in RHF method and 4.1322˚ in B3LYP method. The root mean square error (RMSE) and the correlation coefficient are calculated and the results are presented graphically as shown in Figure 5. The RMSE is found to be about 0.022 Å for RHF and 0.038 Ǻ for B3LYP, indicating that the bond lengths obtained by the HF method shows the strongest correlations with the experimental values. The correlation coefficient in bond lengths by RHF and B3LYP are 0.9699 and 0.9277 respectively. For bond angles, the root mean square error is found 1.361˚ and 1.232˚ for RHF and B3LYP respectively. The correlation coefficient in bond angles by RHF and B3LYP are 0.8966 and 0.9157 respectively, which reveals that the correlation coefficient for bond angles obtained by RHF method are smaller than those determined by B3LYP method as observed in reported research paper [20].
The molecular conformation described by the torsional angles obtained from X-ray data are C6-C12-C13-C14, C12-C13-C14-C16 and C13-C14-C16-C21 as −176.6(2)˚ [−178.2701˚ & −177.5469˚], −166.0 (2)˚ [−172.7265˚ & −174.4626˚] and −142.3 (2)˚ [−140.9733˚ & −147.4643˚] respectively. The values in the square bracket are those obtained by RHF and B3LYP method respectively. The angle between the mean plane of prop-2-en 1-one group
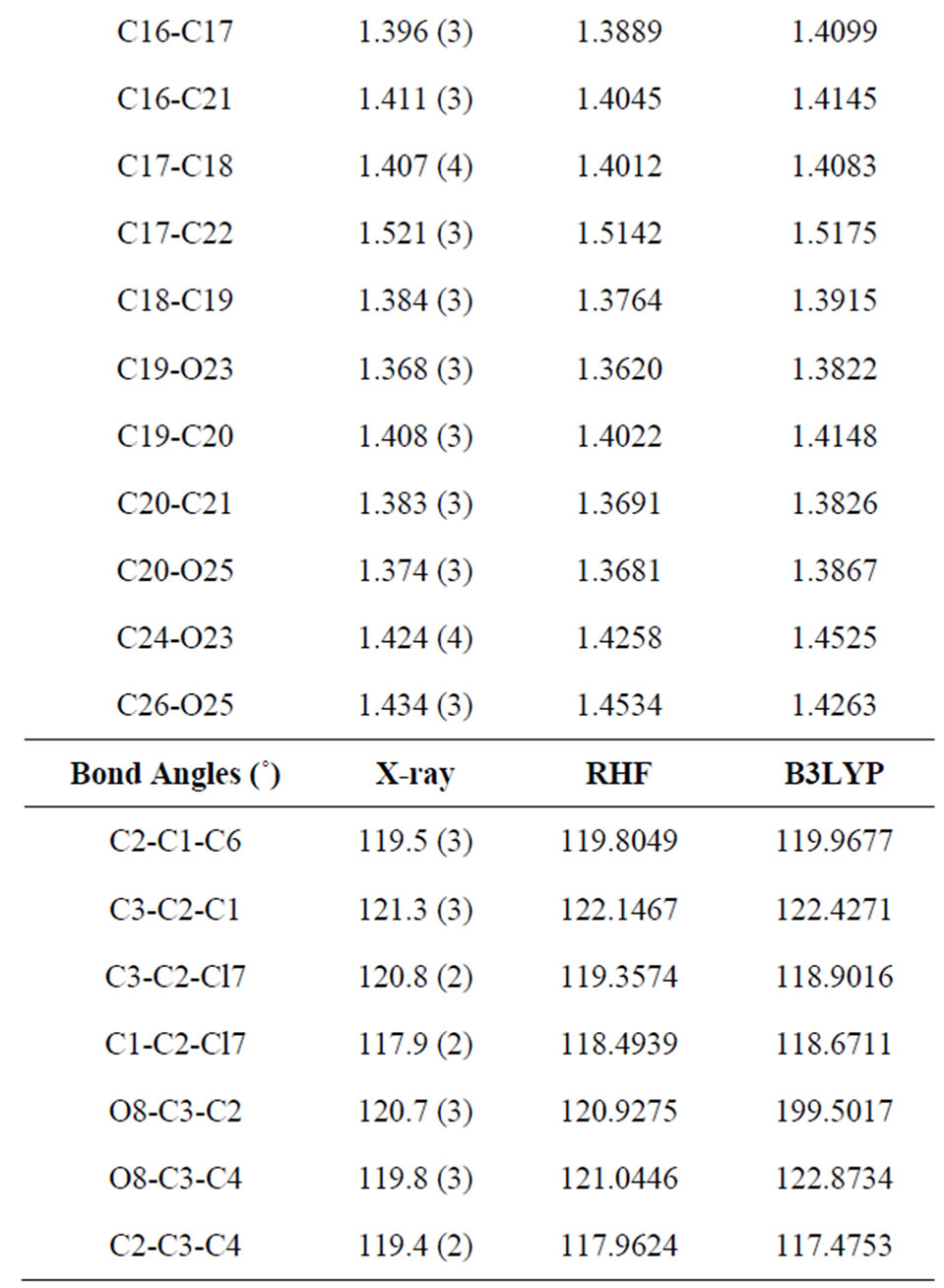

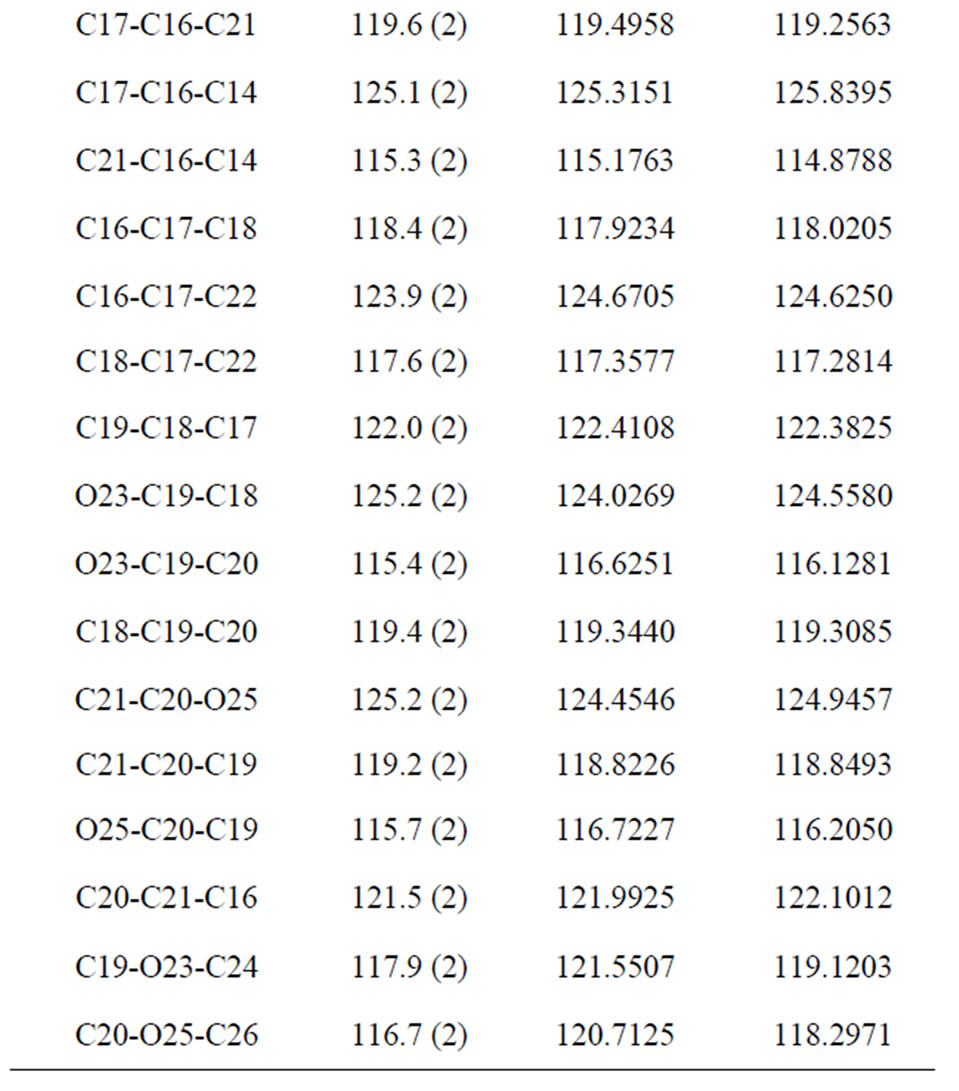
Table 3. Bond lengths (Å) and angles (˚) by X-ray and theoretical calculations for title compound at the RHF/6-311G and B3LYP/6-311G levels of theory.
(C12-C14\O15) and the mean plane of the chloro phenyl ring (C1-C6) and methyl phenyl ring (C16-C21) are 29.85 (11)˚ and 38.46 (9)˚ respectively. The calculated data are collected in gas phase and no molecular interactions are considered, whereas the experimental data are acquired in the solid state and crystal field interactions, e.g. Van der Waals forces, crystal packing force and hydrogen bond interactions. The observed discrepancies between the theoretical and experimental results may be attributed to different environments of the molecule, being isolated state in gas phase for theoretical study,
 (a)
(a)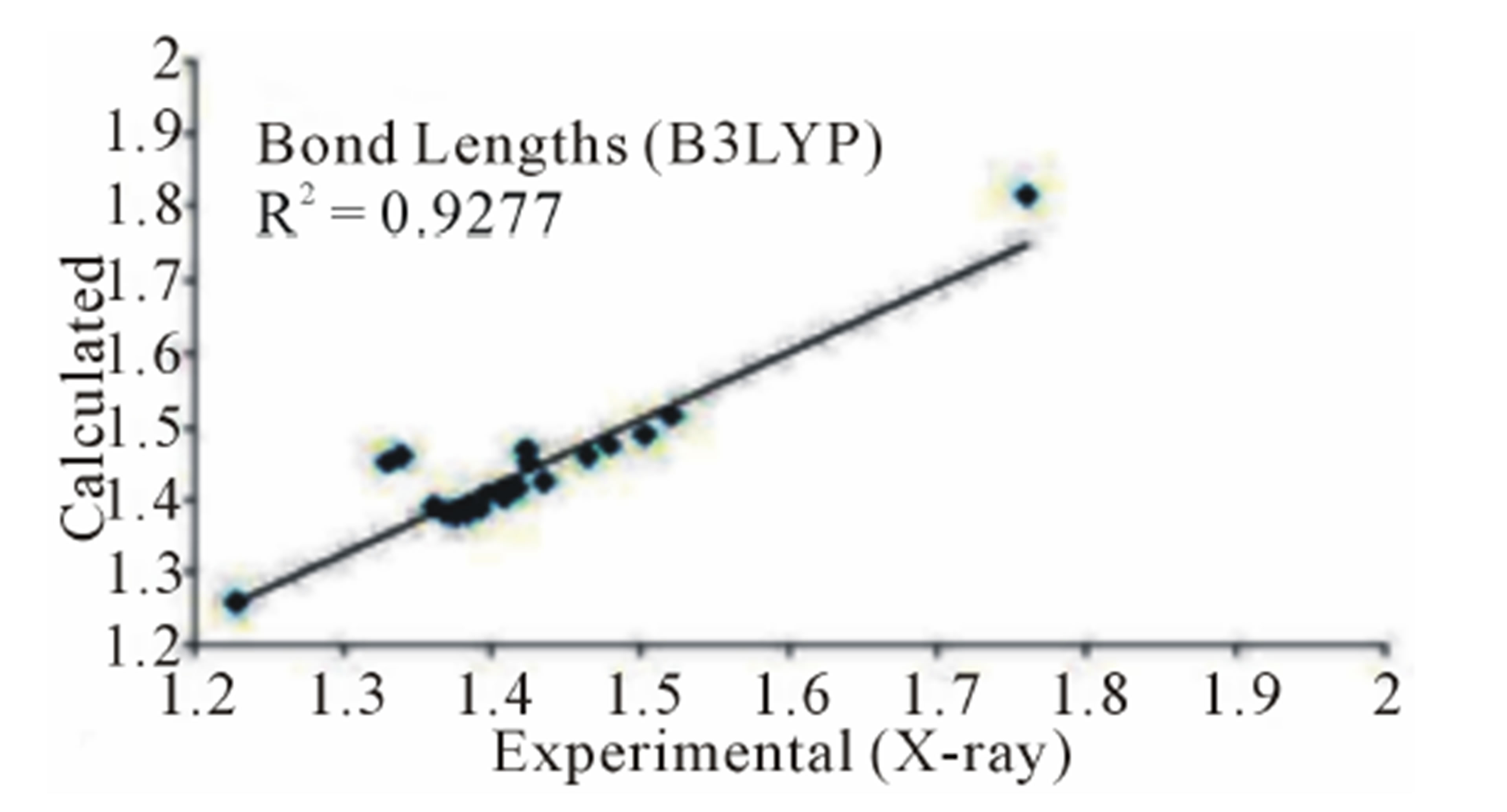 (b)
(b) (c)
(c)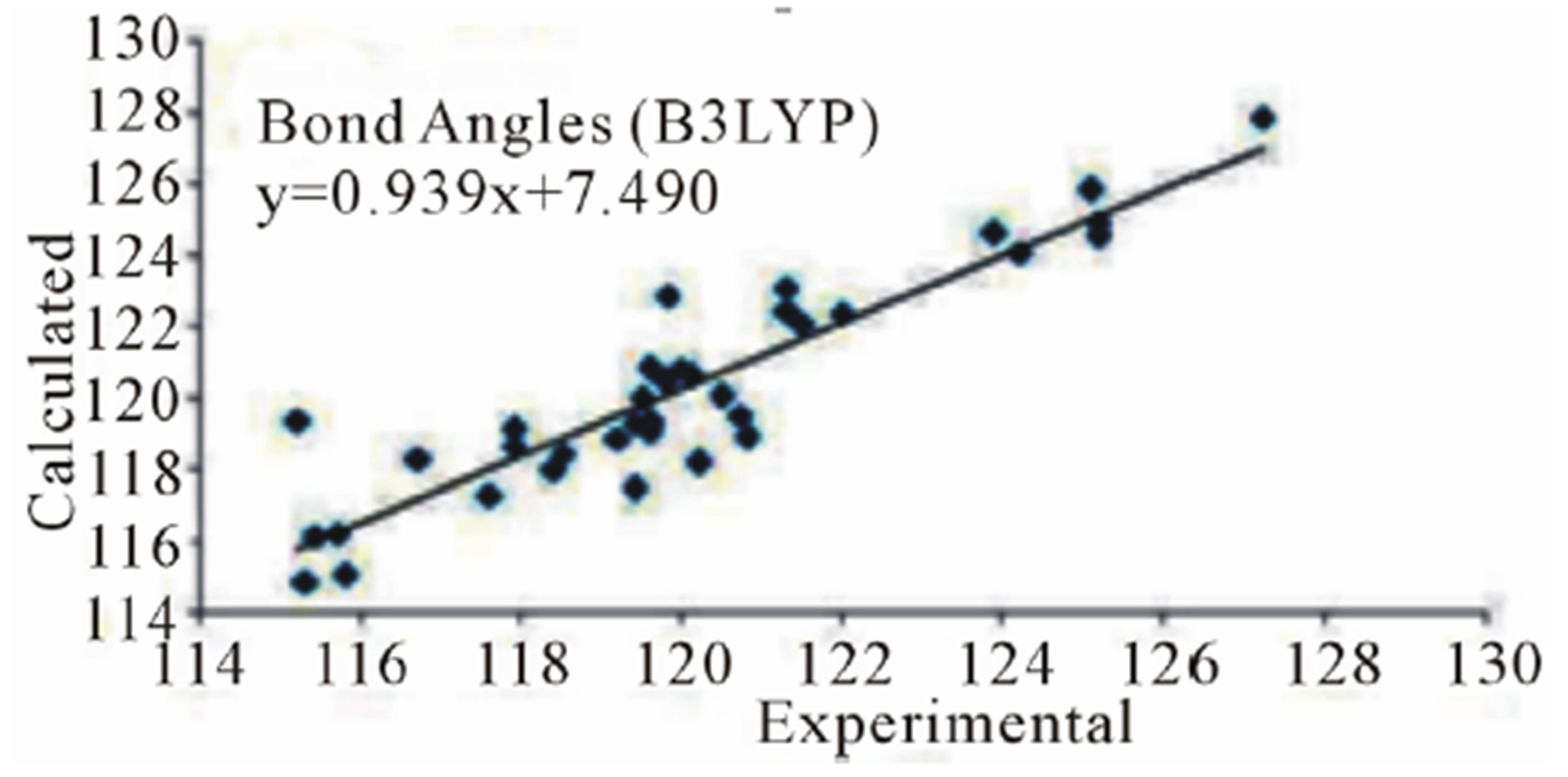 (d)
(d)
Figure 5. Correlation studies of calculated and experimental bond lengths and bond angles.
whereas the experimental values are of the molecule in solid state there by subjected to the intermolecular forces, which are present into π-π, C-H···p and C-H···O interactions.
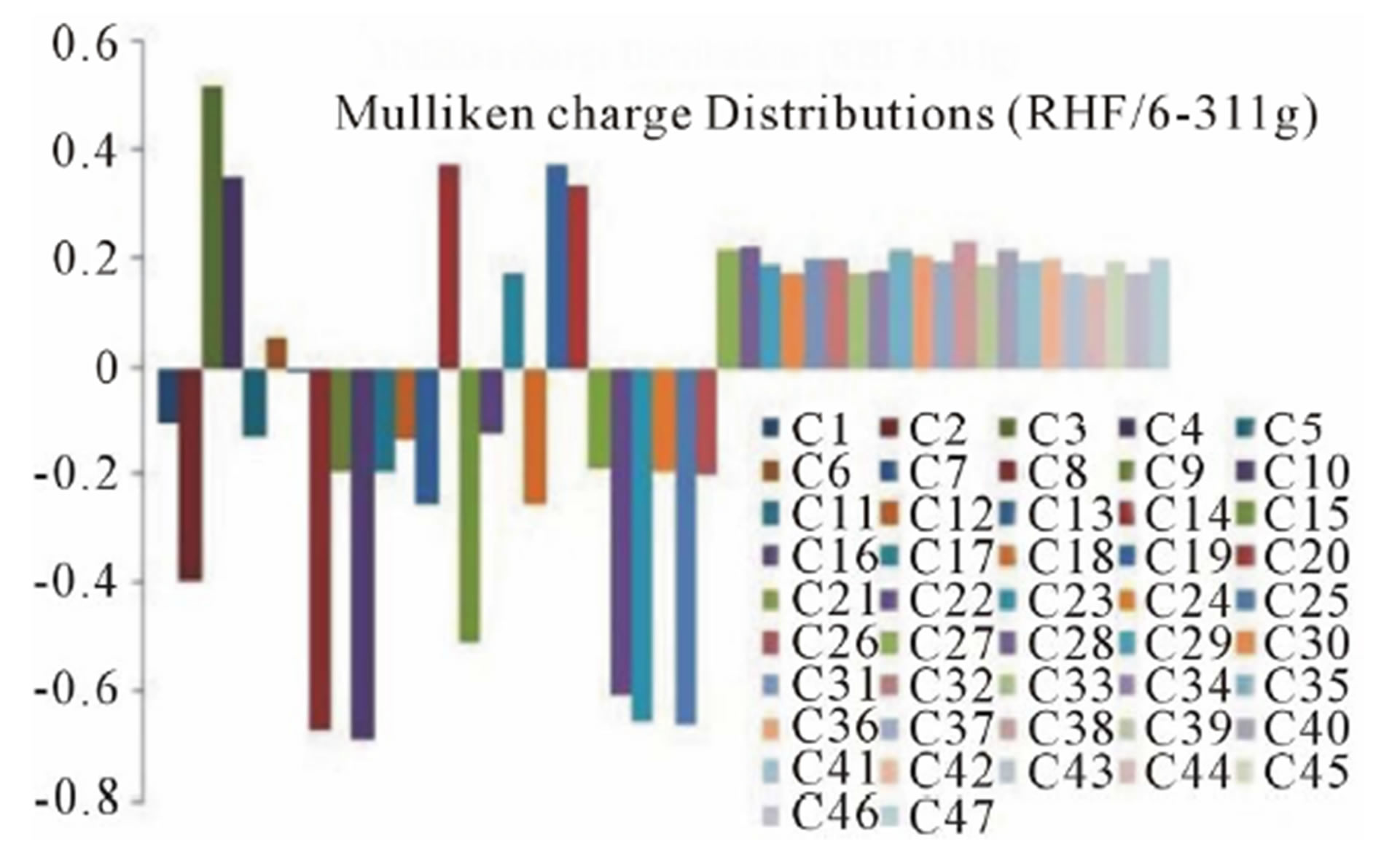
3.3. Mulliken Charge Distributions
The Mulliken charge distributions of the title compound have been calculated using RHF and B3LYP methods with 6-311G level (Table 4) and graphically shown in Figure 6. It may be noted that the all oxygen atoms have negative charge and all hydrogen atoms have positive charge. The oxygen O10 (−0.691300 and −0.531309 from RHF and B3LYP respectively) atom has more
 (a)
(a)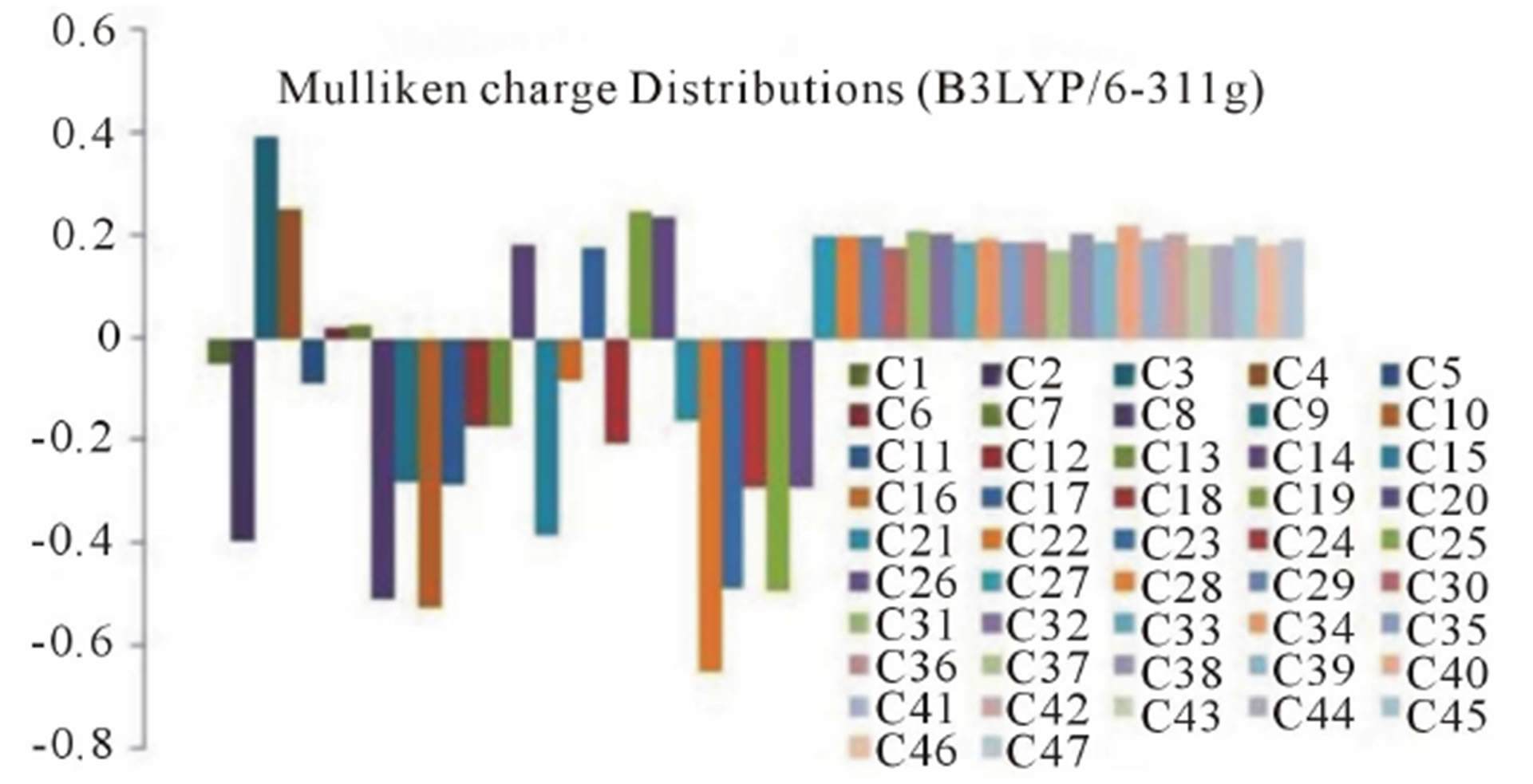 (b)
(b)
Figure 6. Graphical reorientations of Milliken charge distributions of title compound.
negative charge than other oxygen atoms, whereas the hydrogen atom H21 (0.230926 and 0.204517 from RHF and B3LYP respectively) has more positive charge than the other hydrogen atoms. The results suggest that the oxygen atoms are electron acceptor and charge transfer takes place from H to O. The carbon atom C3 (0.519465 and 0.393703 from RHF and B3LYP respectively) is more positive than the other positive carbon atoms and C22 (−0.611291 and −0.655711 from RHF and B3LYP respectively) is the more negative than the other negative carbons in the title molecule, due to electron-donating substituent at that position. The presence of donor and acceptor atoms suggests the existence of both intra and inter-molecular hydrogen bonding in the crystalline phase.
3.4. Total Energies, Dipole Moments and Molecular Orbitals Analysis
The RHF/6-311G and B3LYP/6-311G methods are used to predict the dipole moment of the title molecule 6.1795 and 6.8948 (Table 5) at a total energy of −1602.7509 (RHF) and −1610.6173 (B3LYP) respectively. The high value of the dipole moments suggests that the title compound is reactive and attractive for further interactions with other system. The HOMO and LUMO plots by B3LYP method of the title compound are shown in Figure 7. HOMO of the title compound presents a charge density localized mainly on both the phenyl rings and all oxygen atoms where as LUMO is mostly on plane of prop-2-en-1-one group and few carbon atoms of both the phenyl rings. The energy value of HOMO and LUMO is computed at −0.30717 and 0.04286 by RHF method and −0.21689 eV and −0.08978 eV by B3LYP method respectively. The HOMO and its orbital play role of electron donors, and the LUMO and its orbital play role of electron acceptors. The energy gap of HOMO-LUMO explains the charge transfer interaction within the molecule.
4. Study of Antimicrobial Activities
The Minimum Inhibition Concentrations (MICs) of the title compound are determined by the microdilution method as described by the National Committee for Clinical Laboratoty Standard (NCCLS-1992) [21]. The antimicrobial activities tested against the different panel of organisms like S. aureus MTCC 96, S. pyogenus MTCC 443, E. coli MTCC 442 and P. aeruginosa MTCC 441 and antifungal strains C. albicans MTCC 227 using Gentamycin and K. nystatin as reference standards respectively. Each test organism with title compound is run in duplicate. The test plates are incubated at 35˚C - 37˚C for 24 h. The MIC is taken as the minimum concentration of the dilutions that inhibited the growth of the test microorganism. The concentration of the solvents used in the following assays is maintained at less than 2% so that no inhibition of organisms or interference occurred. The Serial dilution technique is followed by micro method as per NCCLS-1992 manual [21]. The observed MIC values, for bacterial and fungal strains of the title compound are presented in Table 6. The data reveals that the response of the drug is significant in S. aureus compare to other panel of organisms.

Table 4. Mulliken Charge Distributions of the title molecule.
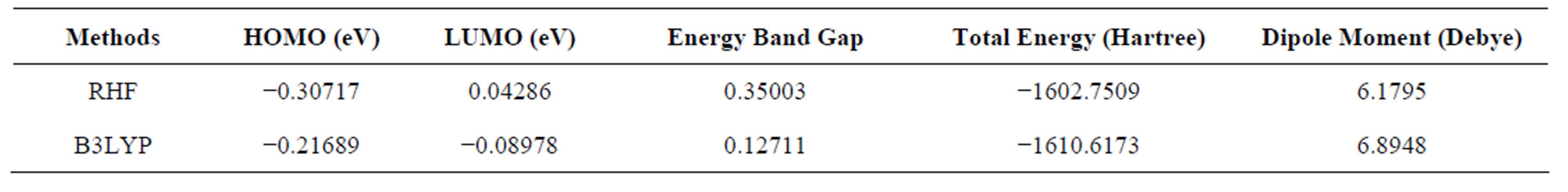
Table 5. HOMO-LUMO, total energies and dipole moments of the title compound.
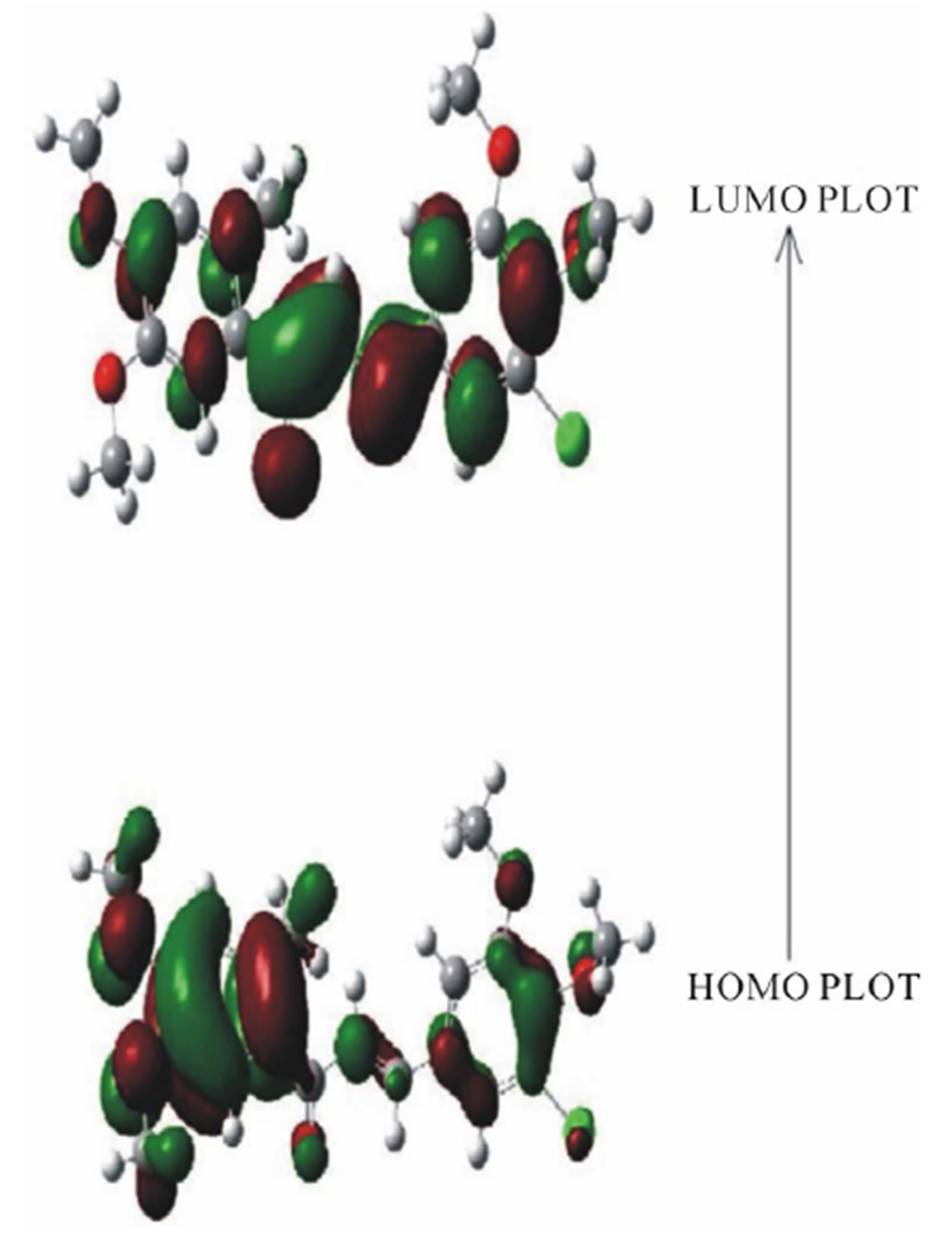
Figure 7. The HOMO and LUMO plots by B3LYP method of the title compound.
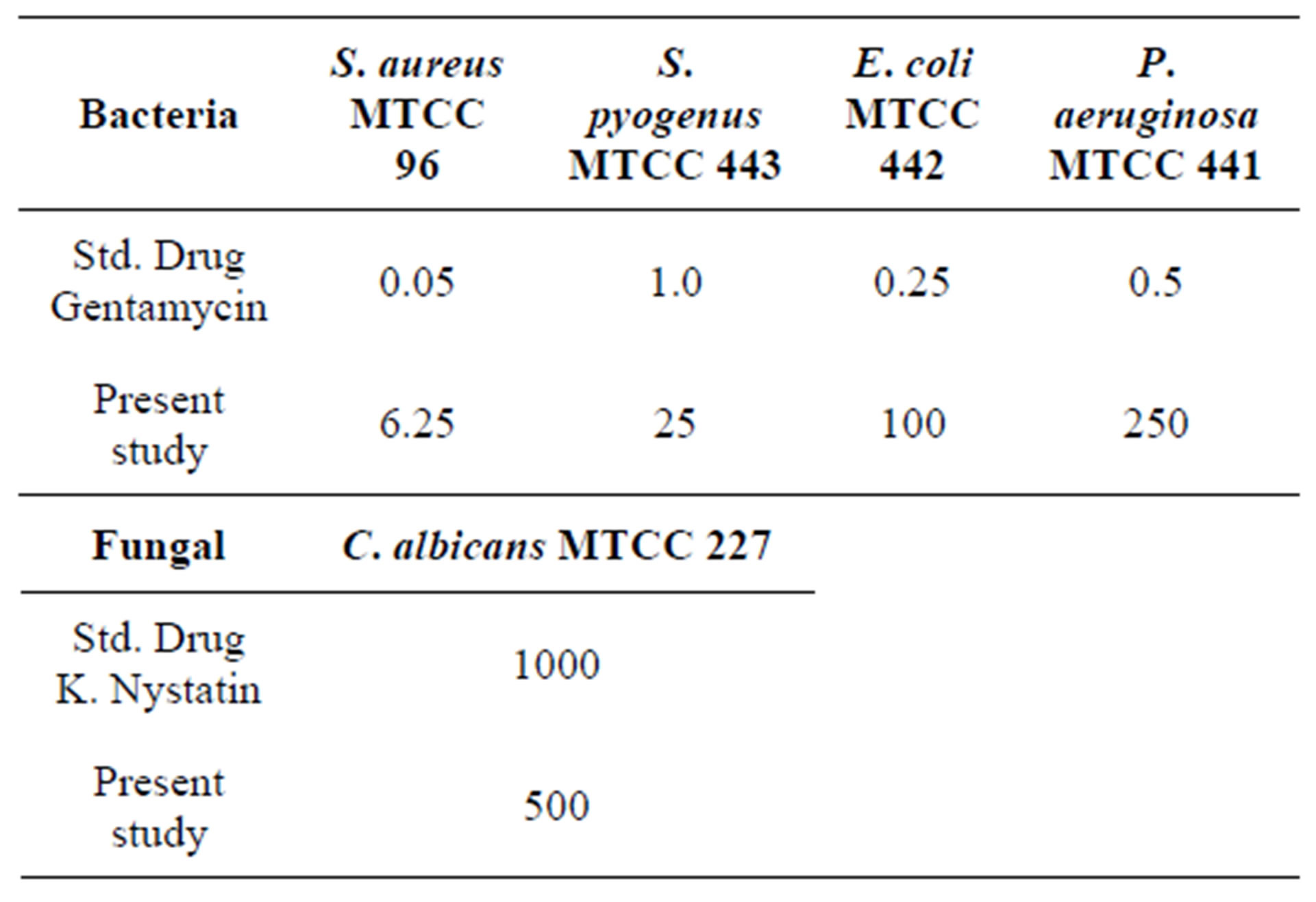
Table 6. Minimal inhibition concentrations of bacterial and fungle strains (MIC) in µg/ml.
5. Conclusion
A novel chalcone derivative, 3-(3-chloro-4,5-dimethoxyphenyl)-1-(4,5-dimethoxy-2-methyl phenyl) prop-2-en- 1-one, C20H21ClO5, has been synthesized. The synthesized product has been confirmed by chemical analysis, IR, 1H NMR and 13C NMR, also its three-dimensional structure has been investigated by single crystal X-ray diffraction technique. The predicted optimized molecular conformation is in a good agreement with experimental results. The results of RMSE and correlation coefficient predict the best matching of experimental and theoretical data. The analysis of Milliken charge distributions supports the presence of intra and intermolecular interactions involving the specific atoms. HOMO-LUMO energy band gap further supports the presents of molecular interactions, showing the charge transfer within the molecule. MIC reports reveal that the synthesized drugs are good responses against S. aureus bacteria compared to other panel of organisms.
6. Acknowledgements
We are thankful to DST, New Delhi, India for providing the single-crystal X-ray diffractometer (Kappa Apex-II) at Department of Physics, Sardar Patel University, Vallabh Vidyanagar, Gujarat, India, under FIST facility. Sahaj Gandhi is also thankful to UGC, New Delhi, for the financial support (RFSMS) to carry out the research work.
REFERENCES
- S. K. Awasthi, N. Mishra, B. Kumar, M. Sharma, A. Bhattacharya, L. C. Mishra and V. K. Bhasin, “Potent Antimalarial Activity of Newly Synthesized Substituted Chalcone Analogs in Vitro,” Medicinal Chemistry Research, Vol. 18, No. 6, 2009, pp. 407-420. http://dx.doi.org/10.1007/s00044-008-9137-9
- A. Valla, B. Valla, D. Cartier, R. Le Guillou, R. Labia, L. Florent, S. Charneau, J. Schrevel and P. Potier, “New Syntheses and Potential Antimalarial Activities of New ‘Retinoid-Like Chalcones,’ ” European Journal of Medicinal Chemistry, Vol. 41, No. 1, 2006, pp. 142-146. http://dx.doi.org/10.1016/j.ejmech.2005.05.008
- C. Echeverria, J. F. Santibanez, O. Donoso-Tauda, C. A. Escobar and R. R. Tagle, “Structural Antitumoral Activity Relationships of Synthetic Chalcones,” International Journal of Molecular Sciences, Vol. 10, No. 1, 2009, pp. 221- 231. http://dx.doi.org/10.3390/ijms10010221
- E. Szliszka, Z. P. Czuba, B. Mazur, L. Sedek, A. Paradysz and W. Krol, “Chalcones Enhance TRAIL-Induced Apoptosis in Prostate Cancer Cells,” International Journal of Molecular Sciences,” Vol. 11, No. 1, 2010, pp. 1- 13. http://dx.doi.org/10.3390/ijms11010001
- S. Cheenpracha, C. Karalai, C. Ponglimanont, S. Subhadhirasakul and S. Tewtrakal, “Anti-HIV-1 Protease Activity of Compounds from Boesenbergia Pandurata,” Bioorganic and Medicinal Chemistry, Vol. 14, No. 6, 2006, pp. 1710-1714. http://dx.doi.org/10.1016/j.bmc.2005.10.019
- N. M. Bhatia, K. R. Mahadik and M. S. Bhatia, “QSAR Analysis of 1, 3-Diaryl-2-propen-1-ones and Their Indole Analogs for Designing Potent Antibacterial Agents,” Chemical Papers, Vol. 63, No. 4, 2009, pp. 456-463. http://dx.doi.org/10.2478/s11696-009-0026-6
- K. L. Lahtchev, D. I. Batovska, S. P. Parushev, V. M. Ubiyvovk and A. A. Sibirny, “Antifungal Activity of Chalcones: A Mechanistic Study Using Various Yeast Strains,” European Journal of Medicinal Chemistry, Vol. 43, No. 10, 2008, pp. 2220-2228. http://dx.doi.org/10.1016/j.ejmech.2007.12.027
- M. Satyanarayana, P. Tiwari, B. K. Tripathi, A. K. Srivastava and R. Pratap, “Synthesis and Antihyperglycemic Activity of Chalcone Based Aryloxypropanolamines,” Bioorganic and Medicinal Chemistry, Vol. 12, No. 5, 2004, pp. 883-889. http://dx.doi.org/10.1016/j.bmc.2003.12.026
- V. V. Serra, F Camoes, S. I. Vieira, M. A. F. Faustino, J. P. C. Tomé, D. C. G. A. Pinto, M. G. P. M. S. Neves, A. C. Tomé, A. M. S. Silva, E. F. da Cruz e Silva and J. A. S. Cavaleiro, “Synthesis and Biological Evaluation of Novel Chalcone-Porphyrin Conjugates,” Acta Chemica Solvenica, Vol. 56, 2009, pp. 603-611.
- U. H. Patel, S. A. Gandhi, V. M. Barot and M. C. Patel, “3-(2-Chloro-3-hydroxy-4-methoxyphenyl)-1-(4,5-dimethoxy-2-methylphenyl)-prop-2-en-1-one,” Acta Crystallography, Vol. E68, 2009, pp. o2926-o2927. http://dx.doi.org/10.1107/S1600536812038275
- U. H. Patel and S. A. Gandhi, “Quantum Chemical Studies on Crystal Structures of Sulfacetamide and Sulfasalazine,” Indian Journal of Pure and Applied Physics, Vol. 49, 2008, pp. 263-269.
- G. M. Sheldrick, “A Short History of SHELX,” Acta Crystallographica, Vol. A64, No. 1, 2008, pp. 112-122. http://dx.doi.org/10.1107/S0108767307043930
- A. D. Becke, “A New Mixing of Hartree-Fock and Local Density-Functional Theories,” The Journal of Chemical Physics, Vol. 98, No. 2, 1993, pp. 1372-1377. http://dx.doi.org/10.1063/1.464304
- B. G. Jhonson, P. M. Gill and J. A. Pople, “The Performance of a Family of Density Functional Methods,” The Journal of Chemical Physics, Vol. 98, No. 7, 1993, pp. 5612-5626. http://dx.doi.org/10.1063/1.464906
- C. Lee, W. Yang and R. G. Parr, “Development of the Colic-Salvetti Correlation-Energy Formula into a Functional of the Electron Density,” Physical Review B, Vol. B37, No. 2, 1988, pp. 785-789.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, “Gaussian 09, Revision A.1,” Gaussian, Inc., Wallingford, 2009.
- R. Dennington, T. Keith and J. Millam, “Gauss View Version 5,” Semichem Inc., Shawnee Mission, 2009.
- J. Bernstein, R. E. Davis and L. Shimoni, “Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals,” Angewandte Chemie International Edition in English, Vol. 34, No. 15, 1995, pp. 1555-1573. http://dx.doi.org/10.1002/anie.199515551
- J. F. Malone, C. M. Murray, M. H. Charlton, R. Docherty and A. J. Lavrry, “X-H···π (phenyl) Interactions Theoretical and Crystallographic Observations,” Journal of Chemical Society, Vol. 93, No. 19, 1997, pp. 3429-3436. http://dx.doi.org/10.1039/a700669a
- B. Yılmaz, H. Saracoglu, N. Caliskan, I. Yilmaz and A. Cukurovali, “X-Ray Diffraction and Theoretical Approach to the Molecular Structure of (E)-2-(2-(1,3-dioxoisoindolin-2-yl)-1-(3-phenyl-3-methylcyclobutyl)ethylidene) Hydrazine Carboxamide,” Journal of Chemical Crystallography, Vol. 42, No. 8, 2012, pp. 897-904. http://dx.doi.org/10.1007/s10870-012-0333-6
- National Committee for Clinical Laboratory Standards (NCCLS), “Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts,” Approved Standard, 2nd Edition, NCCLS Document M27-A2, 2002.
NOTES
*Corresponding author.