Open Journal of Synthesis Theory and Applications
Vol.2 No.1(2013), Article ID:27202,5 pages DOI:10.4236/ojsta.2013.21005
Competition of Aromatic Bromination and Nitration in Concentrated Sulfuric Acid
Research Institute of Organic Intermediates and Dyes (NIOPIK), Moscow, Russia
Email: gorelik@niopik.ru
Received November 30, 2012; revised December 28, 2012; accepted January 8, 2013
Keywords: Bromination; Nitration; Aromatic Compounds; 5-Bromo-1,3-dinitrobenzene; 5-Bromo-2,4,7-trinitrofluorenone; 4-Bromo-2,7-dinitrophenanthrenquinone
ABSTRACT
Composition of bromine and nitric acid in concentrated sulfuric acid is effective for bromination of strongly deactivated aromatic compounds and for nitration and bromination of moderately deactivated compounds in one stage. The question why brominating agent is more reactive than nitrating agent in relation to strongly deactivated aromatic compounds and is less reactive in relation to moderate deactivated ones is discussed.
1. Introduction
Aromatic compounds with bromine in the ring are of considerable importance. Bromine is introduced into the aromatic ring to give desired properties to the final product or to attain the required reactivity for further syntheses. Substances of the former group are produced in industrial scale as fire retardants, fuel additives, dyes, pharmaceutical and agricultural chemicals [1]. Compounds of the latter group are much more diverse as bromoarenes are capable to wide range of the reactions connected with an exchange of bromine. Their significance has especially increased owing to progress last decades cross-coupling reactions catalyzed with palladium complexes [2,3]. The principal method of bromine introduction into aromatic molecules is substitution of hydrogen atom under the action of an electrophilic brominating agent. Electrophilic bromination easily proceeds when the activating electron-donating groups are present in the aromatic ring. In case of presence of deactivating electron-withdrawing substituents special methods are required. This problem is considered in the review [4].
Severe difficulties arise at brominating of strongly deactivated aromatic substrate containing two or more electron-withdrawing groups. The most popular model compound of this type is 1,3-dinitrobenzene. Poorly available reagents have been used for transformation of 1,3-dinitrobenzene into 5-bromo-l,3-dinitrobenzene. They are bromine and silver sulfate [5] or trifluoromethanesulfonate [6] in sulfuric acid, N,N’-dibromocyanuric acid in sulfuric acid or oleum [7], bromine and BrF3 in chloroform [8]. We have found a simple method for brominetion of strongly deactivated aromatic compounds by the action of bromine in concentrated sulfuric acid in the presence of nitric acid or some other oxidizing agent [9]. More recently bromination of deactivated aromatic compounds with sodium bromine and sodium periodate in sulfuric acid is reported [10].
Reaction medium containing bromine, nitric acid and concentrated sulfuric acid has both brominating and nitrating capacity which relative contribution depends on a structure of substrate. The competition of these reactions is studied and discussed on an example of benzene and some polycyclic nitro compounds in the present work.
2. Experimental Section
2.1. Reagents and Analytics
Bromine, fuming nitric acid and aromatic substrates, including nitrobenzene derivatives, phenanthrene and fluorenone, were obtained from commercial suppliers and were used without further purification. 1Н and 13C NMR spectra were determined in CDCl3 and acetone-d6 on a Jeol ECX-400 NMR spectrometer, the chemical shifts were reported in δ ppm relative to TMS as internal standard. IR spectra were recorded on a Perkin-Elmer-598 spectrometer in KCl pellets. Preparative chromatography was carry out on a column with Silicagel L 40/100μ (Chemapol). The products were analyzed by GLC on a Cbrom-5 instrument with flame detector (metallic colomn 1000 × 3 mm, 5% Silicon XE-60 on Chromaton N-AW-DCS 0.25 - 0.31 mm, nitrogen 90 ml/min). The melting points were measured using Buchi appartatus and were uncorrected. Reactions and purity of the formed products were monitored by TLC on Silufol UV-254 plates with development by solutions of SnCl2 and 4-dimethylaminobenzaldehyde in aqueous ethanol containing HCl.
2.2. Syntheses
General procedure for bromination in the presence of nitric acid. The substrate (0.05 mol) and bromine (4.40 g, 0.027 mol, 0.055 eqiv.) was dissolved in 60 ml of 96% sulfuric acid, and 13 ml (0.3 mol) of nitric acid (1.51 d) was added dropwise over 1 h at 60˚C. The mixture was stirred at temperature 60˚C - 80˚C for 3 - 6 h until the initial compound was absent. After cooling the reaction mixture was poured onto ice/water (300 g). Precipitate was filtered off, washed with cold water and dried. The procedure may be applied in reduced scale.
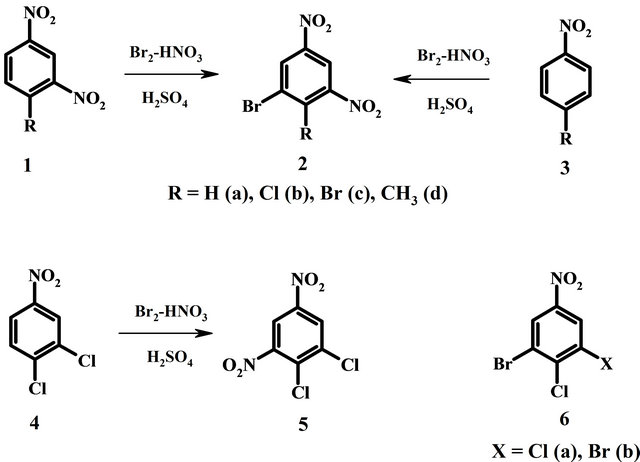
According this procedure 1,3-dinitrobenzenes 1a-d were transformed to 5-bromo-1,3-dinitrobenzenes 2a-d in yield no less than 90%. In the absence of nitric acid the substrates do not change. Treatment of 1,3-dinitrobenzenes 1b-d and nitrobenzenes 3b-d gave identical dinitro compounds 2b-d. Product of reaction of 4,5-dichloronitrobenzene 4 did not contain bromine and was identified as 4,5-dichloro-1,3-dinitrobenzene 5. Compounds 6a and b were synthesized from 4 and 2b correspondingly using hydrogen peroxide instead of nitric acid [9]. Structures of the products were confirmed by comparison of their spectral data and physical constants with those of authentic samples.
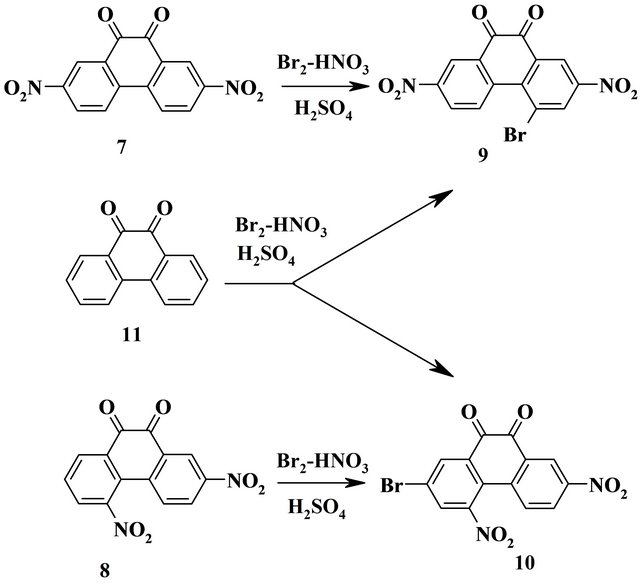
Nitroderivatives of phenanthrenequinone 7 and 8 and of fluorenone 12 and 13 were prepared by nitration of appropriating unsubstituted compounds 11 and 16. Characteristics of brominated products are shown below.
4-Bromo-2,7-dinitro-9,10-phenanthrenequinone (9). Yellow crystals (AcOH), yield 63%, mp > 300˚C (dec.). IR, ν, cm−1: 1700 (С=О), 1515, 1340 (NO2). Found, %: С 44.29, Н 1.58, Br 21.32, N 7.62, С14Н5, BrN2О6. Calculated, %: С 44.59, Н 1.34, Br 21.19, N 7.43.
2-Bromo-4,7-dinitro-9,10-phenanthrenequinone (10). Yellow crystals (AcOH), yield 53%, mp 253.5 - 254.5˚C. IR, ν, cm−1: 1685 (С=О), 1525, 1350 (NO2). 1H NMR, δ, J, Hz: 7.85d (1Н, Н5, 3J 8.2), 8.43d (1Н, Н3, 4J 2.0), 8.52d (1Н, Н1, 4J 2.0), 8.60d (1Н, Н6, 3J 8.2, 4J 1.8), 8.84d (1Н, Н8, 4J 1.8). Found, %: С 44.39, Н 1.90, Br 20.92, N 7.40, С14Н5BrN2О6. Calculated, %: С 44.59, Н 1.34, Br 21.19, N 7.43.
Products of bromination of phenanthrenequinone 11 were separated by column chromatography. Compound 10 was isolated as the first fraction in yield 26% and compound 9 as the second fraction in yield 39%.
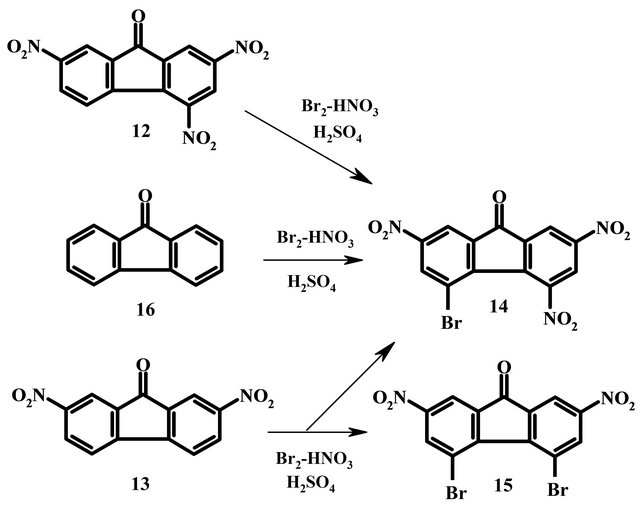
5-Bromo-2,4,7-trinitrofluorenone (14). This compound was obtained according to given above procedure from 2,4,7-trinitrofluorenone 12 in yield 96%, from 4,5-dibromo-2,7-dinitrofluorenone 15 in yield 60%, from fluorenone 16 in yield 70%. Pale yellow crystals (EtOH), m.p. 192˚C - 193˚C (lit. mp 191˚C - 192˚C [11]). IR, ν, cm−1: 1729 (С=О), 1533, 1343 (NO2). 1H NMR: 8.15d (1Н, Н8, 4JН8Н6 1.3), 8.62d (1Н, Н1, 4JН1Н3 1.4), 8.75d (1Н, Н6, 4JН6Н8 1.3), 9.80d (1Н, Н3, 4JН3Н1 1.4). 13C NMR (CDCl3): 118.4 (С8), 121.0 (С5), 122.0 (С1), 126.1 (С3), 135.6 (С6), 149.6 (С7), 150.1 (С2), 138.9, 139.7, 139.8, 144.6, 146.1 (С4,С4a,С4b,С8a,С9а), 185.3 (С9). Found, %: С 39.58, Н 1.05, Br 20.26, N 10.35%, С13Н4BrN3О7. Calculated, %: С 39.62, Н 1.02, Br 20.28, N 10.66.
4,5-Dibromo-2,7-dinitrofluorenone (15). Mixture of 2.70 g (0.001 mol) of 2,7-dinitrofluorenone 13, 60 ml of conc. H2SO4, 10 ml HNO3 (d 1.51) and 1 ml Br2 was heated 4 h at 60˚C. After cooling precipitate of compound 14 was filtered, yield 60%. Filtrate was poured onto ice/water (200 g) and pecipitate of compound 15 was separated and recrystallized from acetic acid, yield 20%. Yellow crystals, mp 226˚C - 227˚C (lit. mp 227˚C - 228˚C [12]). IR, ν, cm−1: 1739 (С=О), 1533, 1344 (NO2). 1H NMR (DMSO-d6), δ, J, Hz: 8.35d (2Н, Н1, Н8, 4JН1(8)Н3(6) 1.8), 8.72d (2Н, Н3, Н6, 4JН3(6)Н1(8) 1.8). Found, %: С 36.58, Н 1.08, Br 37.42, N 6.35, С13Н4Br2N2О5. Calculated, %: С 36.48, Н 0.94, Br 37.34, N 6.55.
3. Results and Discussion
Mixture of nitric and concentrated sulfuric acids is the most popular and powerful reagent for aromatic nitration. 1,3-Dinitrobenzene 1a is so strongly deactivated that can not be nitrated to 1,3,5-trinitrobenzene with nitrating mixture in usual manner. It requires very hard conditions, for example heating in mixture of oleum and fuming HNO3 at 140˚C - 150˚C during one week [13]. Addition of bromine into nitrating mixture and heating a few hours at 60˚C - 80˚C lead to 5-bromo-1,3-dinitrobenzene 2a in excellent yield [9,14]. Similarly 4-substituted 1,3-dinitrobenzenes 1b-d turn into 5-bromo derivatives 2b-d.
Consequently brominating agent has here essentially greater reactivity in relation to 1,3-dinitrobenzene than nitrating agent. It was reasonable to expect predominance of bromination for less deactivated compounds. However it has appeared that moderately deactivated compounds containing one nitro group are first nitrated. 4-Substituted nitrobenzenes 3b-d change directly into 5-bromo- 1,3-dinitro compounds 2b-c identical obtained from 4- halogeno-1,3-dinitrobenzenes 1b-d owing to initial nitration and subsequent bromination (Scheme 1).
If bromination is impossible only nitration takes place. 4,5-Dichloronitrobenzene 4 converted completely to 4,5-dichloro-1,3-dinitrobenzene 5 but not to 3-bromo- 4,5-dichloronitrobenzene 6a. Compounds 6a manages to be obtained from dichloronitrobenzene 4 with bromine in sulfuric acid using other oxidizer instead of nitric acid, in particular hydrogen peroxide or potassium iodate [9].
Scheme 1. Action of bromine and nitric acid in concentrated sulfuric acid on benzene nitro derivatives.
Similar but with double quantity of bromine 3,5-dibromo-4-chloronitrobenzene 6b can be obtained from 4-chloronitrobenzene 3b.
Combining nitration and bromination in a single preparative stage is of practical significance. So new route to the production of 6-bromo-2,4-dinitroaniline (a diazo component for dyes) from 4-chloronitrobenzene 3b [15] is based on ammonolysis of 5-bromo-l,3-dinitro-4-chlorobenzene 2b and includes two stages in comparison with the traditional three-stage method through 2,4-dinitrochlorobenzene and 2,4-dinitroaniline.
Method of bromination in the presence of nitric acid was extended to deactivated polycycic nitro compounds, such as nitro derivatives of 9,10-phenanthrenequinone and 9-fluorenone. Isomeric 2,7-dinitro-9,10-phenanthrenequinone 7 and 2,5-dinitro-9,10-phenanthrenequinone 8 were prepared by nitration of phenanthrenequinone with following separation [16]. On heating of dinitro compound 7 and 8 with bromine and nitric acid in concentrated sulfuric acid at 55˚C 4-bromo-2,7-dinitro-9,10-phenanthrenequinone 9 and 2-bromo-4,7-dinitro-9,10-phenanthrenequinone 10 wear obtained (Scheme 2). 2,4,7-Trinitro-9,10-phenanthrenequinone does not change in this conditions. The position of bromine atom in compound 9 was determined by X-ray study of its solvate [17], structure of compound 10 is confirmed with 1H NMR spectra. Both these compounds are formed together at similar treatment of unsubstituted 9,10-phenanthrenequinone 11. It means that at simultaneous presence of brominated and nitrated agents in the reaction media phenanthrenequinone 11 and resulting its mononitro derivatives are nitrated while formed more deactivated dinitro derivatives 9 and 10 are brominated.
In the fluorenone series bromination of 2,4,7-trinitrofluorenone 12 and 2, 7-dinitrofluorenone 13 were studied. Treatment of trinitrofluorenone 12 in concentated sulfuric acid with bromine and nitric acid leads to 5-bromo- 2,4,7-trinitrofluorenone 14 in yield 96%. Nitration and
Scheme 2. Action of bromine and nitric acid in concentrated sulfuric acid on phenanthrenequinone derivatives.
bromination of dinitrofluorenone 13, less deactivated than trininrofluorenone 12, proceeds in parallel (Scheme 3). After heating 4 hours at 60˚C 5-bromo-2,4,7-trinitrofluorenone 14 and 4,5-dibromo-2,7-dinitrofluorenone 15 were obtained in yield 60% and 20% correspondingly. At prolongation of heating compound 14 becomes a single product. 4,5-Dibromo-2,7-dinitro-fluorenone 15, isolated in the pure state, turns to 5-bromo-2,4,7-trinitrofluorenone 14 at repeated processing. It allows to synthesize 5-bromo-2,4,7-trinitrofluorenone 14 directly from unsubstituted fluorenone 16 by heating till disappearance of 4,5-dibromo-2,7-dinitrofluorenone 15 instead of nitration and after that bromination as in the work [18]. Transformation proceeds evidently through a stage of bromine elimination (protodebromination) and subsequent nitration.
Thus both in benzene and in polycyclic series strongly deactivated aromatic compounds are brominated while moderately deactivated compounds are nitrated at combined action of bromine and nitric acid in concenrated sulfuric acid. It attracts an attention to the question on nature of brominating species.
Participation of nitric acid is necessary for success of bromination since in absence of nitric acid bromination does not proceed. Concentration of sulfuric acid acid is also important. Decrease in concentration to 90% reduces brominating capacity of the system and decrease in concentration below 85% cancels bromination. Nitric acid exists in sulfuric acid with concenration of 90% and more completely as nitronium cation [19]. Therefore interaction of nitronium cation with bromine is a key stage in the formation of brominating agent. Stoichiometry of bromination includes expense of 0.5 mol of bromine to 1 mol of aromatic substrate and reduction of nitronium cation to nitrosonium cation.
The predominance of nitration over bromination for moderately deactivated compounds is not connected with slow formation of brominating agent. The preliminary stirring of a mixture of bromine, nitric acid and sulfuric acid for a long time does not lead to bromination instead of nitration. Replacement of bromine in aromatic ring by nitro group noted above for fluorenone compounds 15 ® 14 (Scheme 3) does not play determinant role since 3,5- dibromo-4-chloronitrobenzene 6b does not turn to 5-bromo-l,3-dinitro-4-chlorobenzene 2b in conditions when 4- chloronitrobenzene 3b turns to 2b.
Greater activity of brominating agent to strongly deactivated compounds and on the contrary smaller activity to moderately deactivated compounds at competition with nitrating agent is caused by smaller sensitivity of bromination to electronic structure of substrate. Reaction of bromination has smaller substrate selectivity than nitration in considered conditions. Increase of number of electron‑acceptor substituents in the aromatic ring complicates nitration much more than bromination. At the certain step the substrate becomes more inert to attack of nitronium cation, than to attack of brominating species. There is an inversion of relative rates and bromination becomes prevailing reaction.
The absolute value of a constant ρ in Hammett equation serves a quantitative measure of sensitivity to structural factors [20,21]. High sensitivity to electronic effects of substituents is typical for nitration in sulfuric acid (ρ = −9.7 for benzene compounds [22]). Qualitatively at absence of kinetic data it is possible to estimate decrease in nitration reactivity from comparison of preparative conditions. So, mononitro phenanthrenequinones turn into dinitro derivatives at action of 65% HNO3 in conc Н2SO4 at 50˚C in 30 min whereas transformation
Scheme 3. Action of bromine and nitric acid in concentrated sulfuric acid on fluorenone derivatives.
of 2,7-dinitrophenanthrenequinone into 2,4,7-trinitrophenanthrenequinone requires many hours heating (14.5 h) in fuming HNO3 and conc. Н2SO4 at 100˚C and 16% remains unchaned [16].
The greatest electronic influence of substituents on the reaction center exists in the case of ionic or coming close to ionic reactions. High sensitivity of nitration, proceeding at attack of the nitronium cation, to electronic effects of substituents is caused by a polar transition state, similar to the ionic σ-complex having the full positive charge. Transition state of bromination reaction, less sensitive to electronic effects of substituents, is probably less polar and is formed at attack of less polar agent. The interaction of bromine with nitronium cation does not lead, apparently, to electron transfer with generating of bromonium cation, and is limited to polarization of bromine molecule due to association with nitronium cation. It is possible to present brominating agent as a complex of type A, stabilized by solvation with sulfuric acid. Bromine atom of the polarized bond Br-Br in the complex, possessing deficiency of electronic density, is the attacking electrophilic site.
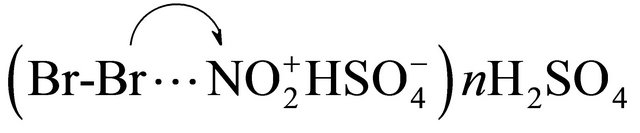 (A)
(A)
The assumption of a complex A as brominating agent explains absence of correlation between the strength of an oxidizer, characterized in value of standard redox potential Ео, and efficiency in bromination reaction [9]. Potential Ео, measured electrochemically in relation to a standard hydrogen electrode, does not reflect capacity for donor-acceptor interaction with bromine responsible for formation of the brominating agent. Nitronium cation activates molecule of bromine for electrophilic attack as Lewis acid, and acts as an oxidizer on one of the subsequent stages.
4. Conclusion
Action of bromine and nitric acid in concentrated sulfuric acid is an effective method for bromination of strongly deactivated aromatic compounds. Moderately deactivated aromatic compounds are nitrated at first and then formed strongly deactivated compounds are brominated. That allows to combine nitration and bromination into one preparative stage. Brominating species has higher relative reactivity than nitrating species in relation to strongly deactivated compounds but weaker reactivity in relation to moderately deactivated compounds. Inversion of relative reactivities is explained by smaller substrate selectivity of bromination in comparison with nitration.
REFERENCES
- D. Ioffe and A. Kampf, “Bromine, Organic Compounds,” In: Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley and Sons, Inc., New York, 2007, pp. 340-364.
- A. de Meijere and F. Diederich, “Metal-Catalyzed CrossCoupling Reactions,” 2nd Edition, Wiley-VCH, Weinheim, 2004.
- S. P. Stanforth, “Vinyl and Aryl Halides,” In: A. R. Katritzky and R. J. K. Taylor, Eds., Comprehensive Organic Functional Group Transformations, Elsevier, Amsterdam, 2005, pp. 561-593.
- A. M. Andrievsky and M. V. Gorelik, “Bromination of Deactivated Aromatic Compounds,” Russian Chemical Reviews, Vol. 80, No. 5, 2011, pp. 421-428. doi:10.1070/RC2011v080n05ABEH004178
- D. H. Derbyshire and W. A. Waters, “The Significance of the Bromine Cation in Aromatic Substitution. Part II. Preparative Applicability,” Journal of the Chemical Society, Vol. 1950, No. 2, 1950, pp. 573-577.
- K. Huthmacher and F. Effenberger, “Neue Reaktive Bromierungsagentinen,” Synthesis, Vol. 1978, No. 9, 1978, pp. 693-694. doi:10.1055/s-1978-24861
- W. Gottardi, “Zur Chemie der Bromisocyanursäuren: Eigenschaften und Reaktionen der Dibromisocyanursäure,” Monatshefte für Chemie—Chemical Monthly, Vol. 108, No. 5, 1977, pp. 1067-1084. doi:10.1007/BF00897688
- S. Rosen and O. Lerman, “Bromination of Deactivated Aromatics Using Bromine Trifluoride without a Catalyst,” The Journal of Organic Chemistry, Vol. 58, No. 1, 1993, pp. 239-240. doi:10.1021/jo00053a043
- A. M. Andrievskii, M. V. Gorelik, S. V. Avidon and E. Sh. Al’tman, “Bromination of Deactivated Aromatic Compounds,” Russian Journal of Organic Chemistry, Vol. 29, No. 9, 1993, pp. 1519-1525.
- L. Kumar, T. Mahajan and D. D. Agarwal, “Bromination of Deactivated Aromatic Compounds with Sodium Bromine/Sodium Periodate under Mild Acidic Conditions,” Industrial & Engineering Chemistry Research, Vol. 51, No. 36, 2012, pp. 11593-11597. doi:10.1021/ie202851k
- M. S. Newman and J. Blum, “4-Bromoand 4-Iodo-2,5,7- trinitrofluorenones as Complexing Agents,” Journal of the American Chemical Society, Vol. 86, No. 24, 1964, pp. 5600-5602. doi:10.1021/ja01078a039
- D. Hellwinkel and G. Haas, “4,5-Dihalogen-9-Fluorenone,” Justus Liebigs Annalen der Chemie, Vol. 1978, No. 12, 1978, pp. 1913-1915.
- H. O. Jenkins, “The Dipole Moments of Certain Polynitro-Compounds,” Journal of the Chemical Society, No. 6, 1936, pp. 862-867.
- H. Jian and J. M. Tour, “Preparative Fluorous Mixture Synthesis of Diazonium-Functionalized Oligo(Phenylene vinylene)s,” The Journal of Organic Chemistry, Vol. 70, No. 9, 2005, pp. 3396-3424. doi:10.1021/jo048051s
- A. M. Andrievskii, M. V. Gorelik, S. V. Avidon, E. V. Gordievskaya, E. Sh. Al’tman, G. N. Vorozhtsov and K. M. Dyumaev, “Method of Obtaining 2-Bromo-4,6-dinitroaniline,” Chemical Abstracts, Vol. 113, 1990, Article ID: 190886.
- S. Kato, M. Maezawa, S. Hirano and S. Ishigaku, “Nitration of Phenanthraquinone,” Chemical Abstracts, Vol. 51, 1957, Article ID: 10462.
- G. V. Gridunova, Y. T. Struchkov, R. V. Linko, A. M. Andrievskii and B. E. Zaitsev, “X-Ray Structural and Spectral Study of 1:2 Solvate of 2,7-Dinitro-4-bromo- 10,10-dihydroxy-9-one with Dimethylformamide,” Bulletin of the Russian Academy of Sciences, Vol. 41, No. 4, 1992, pp. 710-715. doi:10.1007/BF01150897
- D. F. Perepichka, M. R. Bryce, I. F. Perepichka, S. B. Lyubchik, C. A. Christensen, N. Godbert, A. S. Batsanov, E. Levillain, E. J. L. McInnes and J. P. Zhao, “A (π-Extended Tetrathiafulvalene)-Fluorene Conjugate. Unusual Electrochemistry and Charge Transfer Properties: The First Observation of a Covalent D2+-σ-A•− Redox State,” Journal of the American Chemical Society, Vol. 124, No. 47, 2002, pp. 14227-14238. doi:10.1021/ja012518o
- K. Schofield, “Aromatic Nitration,” Cambridge University Press, Cambridge, 1980.
- L. P. Hammett, “Physical Organic Chemistry,” McGrawHill, New York, 1970.
- C. D. Johnson, “The Hammett Equation,” Cambridge University Press, Cambridge, 1973.
- A. R. Katritzky, S. Clementi and H. O. Tarhan, “Discussion of Standard Nitration Rates for Benzenoid and Heteroaromatic Compounds,” Journal of the Chemical Society, Perkin Transactions 2, Vol. 1975, No. 14, 1975, pp. 1624-1627.