Pharmacology & Pharmacy
Vol.06 No.07(2015), Article ID:58414,11 pages
10.4236/pp.2015.67035
Epidermal Growth Factor Enhances Orthovanadate-Induced Contraction via Src and Myosin Phosphatase Target Subunit 1 in Rat Vascular Smooth Muscle
Tomoya Sasahara, Natsumi Ohkura, Mariko Shin, Akira Onodera, Katsutoshi Yayama*
Laboratory of Cardiovascular Pharmacology, Department of Biopharmaceutical Sciences, Kobe Gakuin University, Kobe, Japan
Email: *yayama@pharm.kobegakuin.ac.jp
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 25 June 2015; accepted 25 July 2015; published 29 July 2015
ABSTRACT
Inhibition of protein tyrosine phosphatase by orthovanadate induces vasoconstriction, which is mediated by the Rho kinase-dependent inactivation of myosin light chain phosphatase (MLCP) via signaling downstream of Src-induced activation of the epidermal growth factor (EGF) receptor. The present study investigated the potential role of EGF in orthovanadate (OVA)-dependent vasoconstriction. OVA-induced aortic contraction significantly increased in the presence of EGF, and was abolished by inhibitors of Rho kinase (Y27632), extracellular signal-regulated kinase 1 and 2 (Erk1/2) (FR180204), Erk1/2 kinase (PD98059), EGF receptor (AG1478), and Src (PP2). Treatment of the rat endothelium-denuded thoracic aorta with either EGF or OVA augmented the phos- phorylation of myosin phosphatase target subunit 1 (MYPT1) at Thr-853 and of the EGF receptor at Tyr-1173. The phosphorylation of MYPT1 was further increased by co-stimulation with EGF and OVA. EGF receptor phosphorylation at Tyr-845 was also increased by EGF or OVA; this effect was augmented by co-stimulation with EGF and OVA, and was abolished by Src inhibition. In addition, Erk1/2 was phosphorylated by EGF or by co-treatment with EGF and OVA; this was abolished by an EGF receptor inhibitor, but not by Src inhibition. These results suggested that OVA-induced EGF- related contraction was mediated by the Rho kinase-dependent inactivation of MLCP via two different signaling cascades: Src-dependent phosphorylation of the EGF receptor at Tyr-845 and EGF- dependent phosphorylation of Erk1/2.
Keywords:
Epidermal Growth Factor, Myosin Light Chain Phosphatase, Mitogen-Activated Kinase, Orthovanadate

1. Introduction
Smooth muscle contraction is regulated by Ca2+-dependent and Ca2+-independent pathways. The increase in cytosolic Ca2+ in smooth muscle cells activates myosin light chain kinase (MLCK), which phosphorylates myosin light chain (MLC), leading to smooth muscle contraction [1] . Phosphorylated MLC is dephosphorylated by MLC phosphatase (MLCP), allowing the contracted smooth muscle to return to the relaxed state [2] . MLCP consists of three subunits: a 37-kDa catalytic subunit, a 20-kDa variable subunit, and a 110 - 130-kDa myosin phosphatase target subunit 1 (MYPT1) [3] . Phosphorylation of MYPT1 by activated Rho kinase reduces MLCP activity, leading to prolonged smooth muscle contraction [4] [5] .
The function of vascular smooth muscle is also regulated by the balance of protein tyrosine kinases and protein tyrosine phosphatases. Disruption of the cellular tyrosine phosphorylation equilibrium causes several diseases, and the activities of both protein tyrosine kinases and phosphatases are therefore tightly controlled [6] . Vanadium compounds such as sodium orthovanadate (OVA) are well-characterized inhibitors of protein tyrosine phosphatase [7] . In addition, vanadates have several other biochemical and pharmacological properties, such as the inhibition of ATPases [8] , epidermal growth factor (EGF)-like mitogenic activity [9] , insulin-mi- metic properties [10] , anti-apoptotic activities [11] , and antitumor or carcinogenic properties [12] [13] . Vanadium compounds can affect signaling pathways in cells, including phospholipase D, focal adhesion kinase, phospholipase C-γ, Src, phosphatidyl inositol 3-kinase, and extracellular signal-regulated kinase 1 and 2 (Erk1/2) [14] -[19] . In the vasculature, vanadium compounds also enhance the production of vasoactive metabolites such as prostaglandin I, nitric oxide (NO), and endothelium-derived hyperpolarizing factor, leading to relaxation of vascular smooth muscle [20] -[23] . In contrast, several studies reported that vanadates induced vascular and non- vascular smooth muscle contraction; this was observed in the guinea pig taenia coli, trachea, and gallbladder smooth muscle [24] -[26] and in the rat gastric longitudinal muscle and myometrium [27] [28] . Vanadate-in- duced contraction of guinea pig ileal longitudinal smooth muscle was regulated by the activation of a Rho kinase-dependent pathway, resulting in an increase in MLC phosphorylation [29] . Recently, we reported that OVA caused Rho kinase-dependent contraction of the rat thoracic aorta and phosphorylation of MYPT1 in vascular smooth muscle cells [30] . These reports showed that inhibition of protein tyrosine phosphatases by vanadates induced smooth muscle contraction through Rho kinase-dependent inactivation of MLCP. We have shown that OVA-induced vasoconstriction was mediated by the Rho kinase-dependent inactivation of MLCP via signaling downstream of Src-induced transactivation of the EGF receptor (EGFR) [30] . We also showed that EGF caused Ca2+ sensitization in the rat thoracic aorta by Rho kinase-dependent inactivation of MLCP through the Erk1/2 kinase (MEK) pathway [31] . However, EGF did not induce contraction of the normotensive rat thoracic aorta. In addition, the relationships amomg Src, EGF, and tyrosine phosphatase involved in smooth muscle contraction were unclear. The present study therefore assessed whether activation of Src by OVA-mediated inhibition of tyrosine phosphatase was involved in EGF-related contraction of rat vascular smooth muscle.
2. Materials and Methods
2.1. Inhibitors
The inhibitors used in this study are described in Table 1 and were purchased from Merck-Millipore (Tokyo, Japan). OVA was purchased from Nacalai Tesque (Kyoto, Japan). The concentration of inhibitors were selected based our previous studies [30] [31] .
2.2. Organ Chamber Experiments
All animal experiments were performed in accordance with the Guidelines of the Kobe Gakuin University Experimental Animal Care. Male Wistar rats, 7 - 8 weeks old and weighing 170 - 200 g, were anesthetized with diethyl ether. The thoracic aorta was excised, placed in Krebs-Henseleit solution (118.4 mM NaC1, 4.7 mM KC1, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 25.0 mM NaHCO3, and 11.1 mM glucose; pH 7.4), and then

Table 1. Inhibitors used in this study.
cleaned of adherent tissue. The aorta was cut into 5-mm rings, and the endothelium was removed by carefully rotating a manipulator inside the lumen of the rings. Six rings, obtained from one animal, were fixed vertically under a resting tension of 1.0 g in 5-ml organ chambers (UC-5A; Medical Kishimoto, Kyoto, Japan) filled with Krebs-Henseleit solution (37˚C, pH 7.4) and aerated continuously with a gaseous mixture of 95% O2 and 5% CO2 for 60 min. Isometric tension changes were measured by a force displacement transducer (AP-5; Medical Kishimoto) coupled to a dual-channel chart recorder (SS-250F; SEKONIC, Tokyo, Japan). All tissue rings were exposed to 60 mM KCl for 30 min for measurement of the maximal contractile force.
Prior to isotonic measurements of vascular contractility, arteries were allowed to equilibrate for another 60 min. After equilibration, OVA was added to the bath solution to achieve a final concentration of 0.01 - 1 mM. EGF (1 or 10 nM) was dissolved in Krebs-Henseleit solution supplemented with 5% trehalose; this was then added to the organ bath 5 min prior to the addition of OVA.
The inhibitors used in the present study were dissolved in dimethyl sulfoxide and added to the organ chambers in 10-μL volumes 20 min before the addition of OVA. The contractile effect of OVA was expressed as a percentage of the maximal force evoked by 60 mM KCl.
2.3. Western Blotting
Aortic rings were equilibrated as described above and then treated with EGF (10 nM) and/or OVA (0.5 mM) for 5 min. In some experiments, inhibitors were added to the organ bath 20 min before EGF and/or OVA treatment. The rings were homogenized in 50 μL lysis buffer, comprised of 50 mM Tris-HCl (pH7.4), 25 mM CHAPS, protease inhibitor cocktail (Nacalai Tesque), and phosphatase inhibitor cocktail (Nacalai Tesque). Samples were centrifuged at 15,000 × g for 10 min at 4˚C, and the concentration of soluble protein in the supernatant was determined using a BCA Protein Assay Kit (Thermo Scientific, Waltham, MA, USA). Equal amounts of protein (1.5 μg/lane) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinyldifluoride (PVDF) membranes (Immobilon-P; Millipore, Billerica, MA, USA). The blots were then blocked in 5% skimmed milk in Tris-buffered saline (0.1% Tween 20 in 10 mM Tris-HCl, pH 7.5, containing 100 mM NaCl) and incubated overnight at 4˚C in Tris-buffered saline with rabbit antibodies against MYPT1 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA), Thr-853-phosphorylated MYPT1 (1:200; Santa Cruz Biotechnology), Erk1/2 (1:1000; Cell Signaling, Danvers, MA, USA), Thr-202/Tyr-204-phosphorylated Erk1/2 (1:1000; Cell Signaling), EGFR (1:1000; Cell Signaling), and EGFR that was phosphorylated at Tyr-1173 (1:1000; Cell Signaling) and at Tyr-845 (1:1000; Cell Signaling). The PVDF membranes were then washed with Tris-buffered saline, and incubated with horseradish peroxidase-conjugated goat anti-rabbit antibodies (1:2000; Bio-Rad, Hercules, CA, USA) in Tris-buffered saline for 1 h at room temperature. After washing twice in Tris-buffered saline, the blots were visualized using an enhanced chemiluminescence detection system (GE Healthcare Japan, Tokyo, Japan). Immunoblots were quantified using densitometry with Versa Doc 5000 MP (Bio-Rad) and Quantity One software (Bio-Rad).
2.4. Statistical Analysis
All data are expressed as the mean ± the standard error of the mean (SEM). Statistical comparisons were performed using one-way analysis of variance with pair-wise comparisons made using the Bonferroni-Dunn method. Comparisons of concentration-response curves were made using repeated-measures analysis of variance followed by the Bonferroni-Dunn test using the Graph Pad Prism 6 software. Differences were considered statistically significant at P < 0.05.
3. Results
3.1. EGF Enhanced OVA-Induced Contraction of Rat Endothelium-Denuded Thoracic Aorta
OVA activates endothelial NO synthase [23] and it is likely that endothelium-derived NO attenuates the contractile effects of OVA in aortic rings. Since the aim of this study was to evaluate the roles of OVA and EGF in rat aortic smooth muscle contraction, we measured the contractile force in endothelium-denuded aortic rings in order to remove any NO-induced effects. The concentration-response curves for OVA significantly increased in endothelium-denuded rings exposed to stepwise increases in the concentration of OVA (Figure 1). Pretreatment of endothelium-denuded aortic rings with EGF (1 or 10 nM) further increased vasoconstriction, as compared with OVA alone. However, EGF alone (1 or 10 nM) did not induce contraction of endothelium-denuded rat thoracic aorta (data not shown).
3.2. Effects of Src, EGFR, and Rho Kinase Inhibitors on OVA- and EGF-Induced Aortic Contraction
To explore the signaling pathways involved in vasoconstriction induced by co-stimulation with EGF and OVA, the effects of various inhibitors on the contractile force generated in the presence of EGF (10 nM) and OVA (0.5 mM) were investigated in endothelium-denuded aortic rings. OVA was previously shown to activate Src, EGFR, and Rho kinase in rat thoracic smooth muscle, resulting in increased contraction [30] . Therefore, we studied the effects of Rho kinase inhibitors on aortic contraction induced by EGF and OVA. As shown in Figure 2, the contractile effects of EGF and OVA were abolished by the Rho kinase inhibitor, Y27632. The specific Src inhibitor, PP2, or the EGFR inhibitor, AG1478, also significantly attenuated the contractile effects of EGF and OVA (Figure 2). However, the MLCK inhibitor, ML-7, did not affect the contraction of endothelium-denuded thoracic aortic rings induced by EGF and OVA.
3.3. Effects of MEK, Erk1/2, p38 Mitogen Activated Protein Kinase (MAPK p38), and c-Jun N-Terminal Kinase (JNK) Inhibitors on OVA- and EGF-Induced Aortic Contraction
EGF was previously reported to induce MLCP inactivation through Erk1/2 dependent Rho kinase activation [31] .

Figure 1. Effect of epidermal growth factor (EGF) on sodium orthovanadate (OVA)- induced contraction of endothelium-denuded rat thoracic aortic ring preparations. OVA concentration-response curves were constructed by measuring the force after each stepwise increase in the OVA concentration and are shown in the absence or presence of EGF (as indicated), added to the organ bath 5 min before OVA. Contractile force was expressed as a percentage of the maximal force evoked by 60 mM KCl. Data are presented as mean ± SEM for aortic rings from 5 rats; *P < 0.05 vs. control (without EGF).
Therefore, we assessed whether the enhancement of the vasoconstrictive effects of OVA (0.5 mM) by EGF (10 nM) was affected by MEK or Erk1/2 inhibitors. We observed that treatment of these aortic rings with a MEK inhibitor (PD98059) or an Erk1/2 inhibitor (FR180204) significantly reduced the effects of EGF on OVA-in- duced contraction (Figure 3). However, inhibitors of JNK (AS601245) and MAPK p38 (SB203580) did not significantly affect the vasoconstriction observed in the presence of EGF (10 nM) and OVA (0.5 mM) (Figure 3).
3.4. Effects of Various Inhibitors on EGF- and/or OVA-Induced MYPT1 Phosphorylation
To determine whether Rho kinase is a downstream effector of Src and/or EGFR during EGF (10 nM)-mediated enhancement of OVA (0.5 mM)-induced vasoconstriction, we measured the levels of phosphorylated MYPT1

Figure 2. Effect of inhibitors of myosin light chain kinase (ML-7), Src (PP2), epidermal growth factor (EGF) receptor (AG1478), or Rho kinase (Y27632) on sodium orthovanadate (OVA)-induced contraction of EGF-treated rat endothelium-denuded thoracic aortic ring preparations. EGF (10 nM) was added 5 min before the addition of OVA, and ML-7 (10 µM), PP2 (3 µM), AG1478 (10 µM) or Y27632 (10 µM) was added to the organ bath 20 min before the addition of OVA. The concentration-response curves for OVA were constructed by measuring the force after each stepwise increase in the OVA concentration. Contractile force was expressed as a percentage of the maximal force evoked by 60 mM KCl. Data are expressed as mean ± SEM for aortic rings taken from 5 animals; *P < 0.05 vs. EGF only.

Figure 3. Effects of inhibitors of mitogen-activated protein kinase (MAPK p38), extracellular signal-regulated kinases 1 and 2 (Erk1/2), Erk1/2 kinase (MEK), or c-jun N- terminal kinase (JNK) on sodium orthovanadate (OVA)-induced contraction of epidermal growth factor (EGF)-treated rat endothelium-denuded thoracic aortic ring preparations. EGF (10 nM) was added 5 min before the addition of OVA, and inhibitors of MEK (PD98059; 10 µM), Erk1/2 (FR180204; 10 µM), MAPK p38 (SB203580; 10 µM), or JNK (AS601245; 10 µM) were added to the organ bath 20 min before the addition of OVA. The concentration-response curves for OVA were constructed by measuring the force after each stepwise increase in the OVA concentration. Contractile force was expressed as a percentage of the maximal force evoked by 60 mM KCl. Data are expressed as mean ± SEM for aortic rings taken from 5 animals; *P < 0.05 vs. EGF only.
(Thr-853) in rat endothelium-denuded thoracic aorta after stimulation in the presence or absence of various inhibitors. Since co-stimulation with EGF and OVA produced rapid contraction of endothelium-denuded aortic rings, presenting as a hyperbolic rise in tension development for 5 min followed by linear rise (data not shown) and treating aortic rings with OVA or EGF increased the ratio of phosphorylated to total EGFR, Erk1/2, and MYPT1 rapidly within 2 min, and the increased levels were maintained for 10 min [30] [31] , we measured the levels of phosphorylated MYPT1 5 min after exposure to OVA. When rings were treated with only EGF, we measured phosphorylation levels of proteins 5 min after stimulation of EGF. As shown in Figure 4, the ratio of phosphorylated to total MYPT1 was increased after treatment with 10 nM EGF or 0.5 mM OVA (P < 0.05 for both treatments, as compared with control), and further increased by co-treatment with 10 nM EGF and 0.5 mM OVA (P < 0.05 versus EGF only and versus OVA only). The increased phosphorylation of MYPT1 induced by co-stimulation with EGF and OVA was significantly reduced by inhibitors specific for Src (PP2), EGFR (AG1478), Rho kinase (Y27632), MEK (PD98059), and Erk1/2 (FR180204). However, inhibitors of JNK (AS601245) and MAPK p38 (SB203580) did not affect the phosphorylation of MYPT1 induced by EGF and OVA. These results suggested that this EGF- and OVA-induced phosphorylation of MYPT1 in rat endothelium- denuded thoracic aorta was mediated by Src-, EGFR-, MEK-, and Erk1/2-dependent activation of Rho kinase.
3.5. Effects of Various Inhibitors on EGF- and/or OVA-Induced Erk1/2 Phosphorylation
To determine whether Erk1/2 is the downstream effector of EGFR during EGF-mediated enhancement of OVA- induced Rho kinase activation, we measured the level of Erk1/2 that was phosphorylated at Thr-202/Tyr-204 in rat endothelium-denuded thoracic aortic rings in the presence or absence of various inhibitors. As shown in Figure 5, the ratio of phosphorylated to total Erk1/2 was increased following exposure to 10 nM EGF or 0.5 mM OVA (P < 0.05 for both treatments, as compared with control). Erk1/2 phosphorylation was further increased by co-stimulation with EGF and OVA, as compared with OVA only (P < 0.05). The increase in Erk1/2 phosphorylation induced by co-stimulation with EGF and OVA was significantly reduced in the presence of inhibitors of EGFR (AG1478), MEK (PD98059), and Erk1/2 (FR180204), but not by inhibition of Src, JNK, MAPK p38, and Rho kinase (Figure 5).
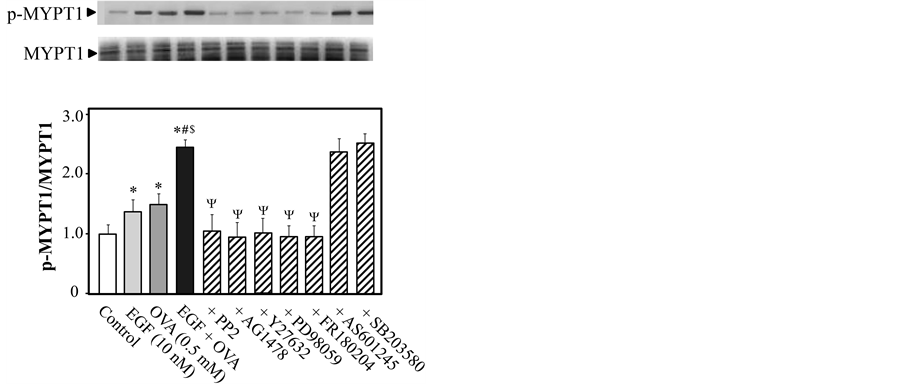
Figure 4. Inhibitor effects on sodium orthovanadate (OVA)- and/or epidermal growth factor (EGF)-induced phosphorylation of myosin phosphatase target subunit 1 (MYPT1) in rat endothelium-denuded thoracic aortic rings. Phosphorylated (Thr-853) and total MYPT1 were measured by western blotting 5 min after treatment with EGF (10 nM) and/or OVA (0.5 mM). The aortic rings were exposed to PP2 (3 μM), AG1478 (10 μM), Y27632 (10 μM), PD98059 (10 μM), FR180204 (10 μM), AS601245 (10 μM), SB203580 (10 μM), or control (10 μL dimethyl sulfoxide) for 15 min prior to EGF and/or OVA treatment. The bar graphs show the densitometric data as the ratio of Thr-853-phosphorylated MYPT1 to total MYPT1. Data are presented as mean ± SEM from 4 independent experiments; *P < 0.05 vs. control; #P < 0.05 vs. EGF, $P < 0.05 vs. OVA; ΨP < 0.05 vs. EGF plus OVA.
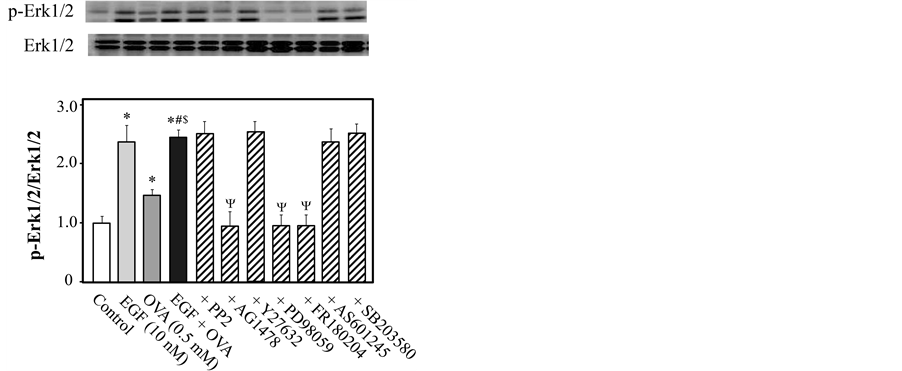
Figure 5. Inhibitory effects on sodium orthovanadate (OVA)- and/or epidermal growth factor (EGF)-induced phosphorylation of extracellular signal-regulated kinases 1 and 2 (Erk1/2) in rat endothelium-denuded thoracic aortic rings. Phosphorylated (p-Erk1/2; Thr-202/Tyr-204) and total Erk1/2 were measured by western blotting 5 min after treatment with EGF (10 nM) and/or OVA (5 mM). The aortic rings were exposed to Y27632 (10 μM), AG1478 (10 μM), PP2 (3 μM), PD98059 (10 μM), FR180204 (10 μM), or control (10 μL dimethyl sulfoxide) for 15 min prior to EGF and/or OVA treatment. The bar graphs show the densitometric data as the ratio of p-Erk1/2 to total Erk1/2. Data are presented as the mean ± SEM of 4 independent experiments; *P < 0.05 vs. control; #P < 0.05 vs. EGF, $P < 0.05 vs. OVA; ΨP < 0.05 vs. EGF plus OVA.
3.6. EGF and OVA Induced EGFR Phosphorylation
We investigated whether the combination of EGF (10 nM) and OVA (0.5 mM) induced phosphorylation of the EGFR at Tyr-845 and Tyr-1173. Western blotting using a phospho-specific antibody for EGFR Tyr-1173 revealed a significantly increased signal in endothelium-denuded rat thoracic aortic rings treated with EGF or OVA, and in tissue co-treated with EGF and OVA (Figure 6(a)). The phosphorylation of EGFR at Tyr-845 was also significantly increased following exposure of the thoracic aortic rings to EGF or OVA (P < 0.05 for both treatments, as compared with control). This Tyr-845 phosphorylation was further increased by co-treatment with EGF and OVA (P < 0.05 versus EGF only and versus OVA only; Figure 6(b)). The increase in EGFR Tyr-845 phosphorylation induced by co-stimulation with EGF and OVA was significantly reduced in the presence of the Src inhibitor, PP2 (Figure 6(b)).
4. Discussion
In the present study, we demonstrated that EGF enhanced OVA-induced contraction of the rat endothelium-de- nuded thoracic aorta and that this effect was mediated by Src, EGFR, MEK, Erk1/2, and Rho kinase. Treatment of the rat endothelium-denuded thoracic aorta with EGF or OVA augmented the phosphorylation of MYPT1. This was further increased by co-stimulation with EGF and OVA, and was blocked by pretreatment with inhibitors of Src, EGFR, MEK, and Erk1/2. The phosphorylation of EGFR at Tyr-1173 in rat endothelium-denuded thoracic aorta was increased by treatment with EGF or OVA, and by co-treatment with EGF and OVA. Phosphorylation of EGFR at Tyr-845 was also augmented by exposure to EGF or OVA. This was further increased by co-stimulation with EGF and OVA, and was reduced by inhibition of Src.
Vanadium compounds such as OVA are tyrosine phosphatase inhibitors, which induce contraction of several tissues including guinea pig ileal longitudinal smooth muscle [29] and rat thoracic arteries [30] . The present study indicated that OVA-induced aortic smooth muscle contraction was mediated by the EGFR, because an EGFR inhibitor (AG1478) abolished this effect. Previous research has demonstrated that vanadium compound-induced smooth muscle contraction was mediated by Rho kinase activation [24] -[27] [32] -[36] . Mori and Tsushima identified a role for Rho kinase in OVA-induced smooth muscle contraction in guinea pig ileal longitudinal smooth muscle, where a Rho kinase inhibitor (Y27632) blocked OVA-induced contraction and MLC
 (a) (b)
(a) (b)
Figure 6. Effect of sodium orthovanadate (OVA)- and/or epidermal growth factor (EGF)-induced phosphorylation of EGF receptor (EGFR) at Tyr-845 and at Tyr-1173 in the rat endothelium-denuded thoracic aorta. (a) Rat endothelium-denuded thoracic aortic rings were treated with EGF (10 nM) and/or OVA (0.5 mM) for 5 min prior to analysis of phosphorylated EGFR (p-EGFR) at Tyr-845 and at Tyr-1173 by western blotting. (b) PP2 (3 μM) was added 15 min before treatment with EGF (10 nM) and/or OVA (0.5 mM) for 5 min and subsequent analysis of p-EGFR by western blotting. The bar graphs show the densitometric quantification of the ratio of p-EGFR to total EGFR. Data are presented as the mean ± SEM of four independent experiments; *P < 0.05 vs. control; #P < 0.05 vs. EGF, $P < 0.05 vs. OVA; ΨP < 0.05 vs. EGF plus OVA.
phosphorylation [29] . This was consistent with our earlier study, where we showed that OVA-induced smooth muscle contraction was Rho kinase-dependent, since Rho kinase inhibition abolished OVA-induced aortic contraction, whereas MLCK inhibition by ML-7 did not affect this process [30] . In addition, several studies have reported that OVA- or pervanadate-induced contraction of smooth muscle cells was not accompanied by a rise in cytosolic Ca2+, but instead occurred as a result of an increased Ca2+ sensitivity of the contractile apparatus [36] -[38] . Activation of Rho kinase inhibits MLCP by phosphorylating MYPT1; this causes Ca2+ sensitization and thereby enhances contraction in the absence of a change in the cytosolic Ca2+ levels [29] .
OVA-induced contraction of the rat thoracic aorta is mediated by Rho kinase-dependent inactivation of MLCP via signaling downstream of Src-induced transactivation of EGFR [30] . The binding of EGF to the EGFR induces receptor dimerization, which triggers autophosphorylation of five specific tyrosine residues (Tyr- 1173, -1148, -1086, -1068, and -992) in the intracellular carboxy-terminal region of the EGFR. EGFR at Tyr- 1173 represents the major autophosphorylation site [39] . Src functions as a co-transducer of transmembrane signals emanating from a variety of polypeptide growth factor receptors, including the EGFR [40] . Src can activate the EGFR either directly, by phosphorylation of EGFR at Tyr-845 in the cytoplasm [40] [41] , or indirectly via the metalloproteinase-catalyzed release of heparin-binding (HB)-EGF from pro-HB-EGF [42] . In our previous study, OVA increased the phosphorylation of Src at Tyr-416 in rat vascular smooth muscle cells and this effect was blocked in the presence of a Src inhibitor. Furthermore, two inhibitors of pro-EGF shedding (TAPI-0 and CRM 197) blocked not only OVA-induced contraction of the rat endothelium-denuded thoracic aorta, but also OVA-induced phosphorylation of MYPT1 and of EGFR at Tyr-1173 and at Tyr-845 in rat vascular smooth muscle cells [30] . The observation that OVA-induced EGF phosphorylation at Tyr-1173 and Tyr-845 was blocked by Src inhibition suggests that the transactivation of EGF via pro-HB-EGF processing depends on Src kinase activity in vascular smooth muscle cells [30] . In the present study, we showed that EGF and/or OVA induced EGFR phosphorylation at Tyr-845 and Tyr-1173 in rat endothelium-denuded thoracic aorta. The contraction and the phosphorylation of EGFR at Tyr-845 were further increased by co-treatment with EGF and OVA, and were abolished by treatment with a Src inhibitor. The aortic contraction and the phosphorylation of MYPT1 induced by OVA or EGF were further augmented by co-stimulation with EGF and OVA, and inhibited by Src inhibition. The phosphorylation of EGFR at Tyr-845 is a major Src-dependent phosphorylation site that is associated with increased receptor function. Src-catalyzed phosphorylation of EGFR at this residue is thought to be essential for mitogenic signaling via the EGFR, since cells expressing Y845F mutant EGFR showed markedly decreased DNA synthesis in response to EGF [40] [43] [44] . Therefore, it is likely that the activation of Src by inhibitors of protein tyrosine phosphatase (such as OVA) induces phosphorylation of EGFR at Tyr-845, which in turn results in increased signaling via the EGFR and activation of Rho kinase dependent MYPT1 phosphorylation in rat endothelium-denuded thoracic aorta.
The MAPK family members, such as Erk1/2, MAPK p38, and JNK, are the major transducers of signals generated by EGF [45] . We used inhibitors to explore the role of MAPKs in EGF- and OVA-induced aortic contraction and Rho kinase-dependent MYPT1 phosphorylation. The increase in MYPT1 phosphorylation was abolished by inhibitors of Src, EGFR, MEK, Erk1/2, and Rho kinase, but not of JNK or MAPK p38. Subsequently, we showed that OVA and/or EGF induced Erk1/2 phosphorylation in endothelium-denuded rat thoracic aorta, an effect that was abolished by inhibitors targeting the EGFR, MEK, and Erk1/2, but not by Src and Rho kinase inhibition. Binding of EGF to EGFR leads to the phosphorylation of Erk1/2 [46] . Src can phosphorylate the EGFR either directly or indirectly [40] -[42] , resulting in vascular smooth muscle contraction [30] . The vasoconstrictive effects of co-stimulation of rat endothelium-denuded thoracic aorta with EGF and OVA were blocked by Src and Erk1/2 inhibitors. Src inhibition also blocked the EGF and OVA-induced EGFR phosphorylation at Tyr-845 in this preparation, but failed to inhibit Erk1/2 phosphorylation. Taken together, OVA-induced Src activated the phosphorylation of MYPT1 by phosphorylating the EGFR at Tyr-845, leading to smooth muscle contraction, whereas EGF activated MYPT1 phosphorylation through Erk1/2 phosphorylation. Thus, it seems that two different pathways regulate MYPT1 phosphorylation in smooth muscle exposed to EGF and OVA.
Vascular smooth muscle contraction in response to EGF was observed in thoracic aorta preparations from deoxycorticosterone acetate (DOCA)-salt hypertensive rats [47] and from a one kidney, one-clip hypertensive rat model [48] . Kim et al. reported that EGF-induced aortic contraction in DOCA-salt hypertensive rats was regulated by Erk1/2 and Rho kinase. However, EGF did not induce contraction of vascular smooth muscle from normotensive rats [49] . These reports suggested that EGF-induced vascular smooth muscle contraction was regulated by MEK, Erk1/2, and Rho kinase in hypertensive rats, but not in normotensive rats [49] . The phosphorylation of Src, EGFR, and Erk1/2 was significantly higher in vascular smooth muscle cells from spontaneously hypertensive rats, as compared to those in Wistar-Kyoto rats [50] [51] . Touyz et al. reported that angiotensin II increased the synthesis of DNA in vascular smooth muscle cells derived from small peripheral resistance arteries from hypertensive patients, but not in those from normotensive patients. Inhibition of Src reduced Erk1/2 activity and normalized Erk1/2 responses in normotensive patients [52] . These studies and our recent findings suggest that Src plays an important role in the regulation of vascular smooth muscle functions.
In summary, the present study provides the novel finding that Src phosphorylates the EGFR at Thr-845, which in turn activates MYPT1 phosphorylation via an unknown signaling cascade, leading to smooth muscle contraction and vessel constriction. On the other hand, EGF activates Erk1/2 phosphorylation, which phosphorylates MYPT1, resulting in smooth muscle contraction. Although EGF alone does not induce vascular smooth muscle contraction, it enhances OVA-induced contraction. Interestingly, both the activation of Src by OVA and of Erk1/2 by EGF result in MLCP inactivation via MYPT1 phosphorylation, leading to smooth muscle contraction via different signaling pathways. However, the OVA target molecule involved in Src-dependent vessel contraction is unclear, as it is the signaling cascade involved in Rho kinase dependent MYPT1 phosphorylation. Further studies are required to comprehensively elucidate the role of protein tyrosine phosphatase regulation in the mechanisms underlying vasoconstriction.
Acknowledgments
This study was partially supported by a Grant-in-Aid for Scientific Research (C) (No. 15K07984) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Conflict of Interest
There are no conflicts of interest to declare.
Cite this paper
TomoyaSasahara,NatsumiOhkura,MarikoShin,AkiraOnodera,KatsutoshiYayama, (2015) Epidermal Growth Factor Enhances Orthovanadate-Induced Contraction via Src and Myosin Phosphatase Target Subunit 1 in Rat Vascular Smooth Muscle. Pharmacology & Pharmacy,06,329-340. doi: 10.4236/pp.2015.67035
References
- 1. Kamm, K.E. and Stull, J.T. (1985) The Function of Myosin and Myosin Light Chain Kinase Phosphorylation in Smooth Muscle. Annual Review of Pharmacology and Toxicology, 25, 593-620.
http://dx.doi.org/10.1146/annurev.pa.25.040185.003113 - 2. Lin, G., Fandel, T.M., Shindel, A.W., Wang, G., Banie, L., Ning, H., Lue, T.F. and Lin, C.S. (2011) Modulation of Smooth Muscle Tonus in the Lower Urinary Tract: Interplay of Myosin Light-Chain Kinase (MLCK) and MLC Phosphatase (MLCP). BJU International, 108, E66-E70.
http://dx.doi.org/10.1111/j.1464-410x.2010.09819.x - 3. Arimura, T., Suematsu, N., Zhou, Y.B., Nishimura, J., Satoh, S., Takeshita, A., Kanaide, H. and Kimura, A. (2001) Identification, Characterization, and Functional Analysis of Heart-Specific Myosin Light Chain Phosphatase Small Subunit. The Journal of Biological Chemistry, 276, 6073-6082.
http://dx.doi.org/10.1074/jbc.M008566200 - 4. Somlyo, A.P. and Somlyo, A.V. (2003) Ca2+ Sensitivity of Smooth Muscle and Nonmuscle Myosin II: Modulated by G Proteins, Kinases, and Myosin Phosphatase. Physiological Reviews, 83, 1325-1358.
http://dx.doi.org/10.1152/physrev.00023.2003 - 5. Sward, K., Mita, M., Wilson, D.P., Deng, J.T., Susnjar, M. and Walsh, M.P. (2003) The Role of RhoA and Rho-Associated Kinase in Vascular Smooth Muscle Contraction. Current Hypertension Reports, 5, 66-72.
http://dx.doi.org/10.1007/s11906-003-0013-1 - 6. Tiganis, T. and Bennett, A.M. (2007) Protein Tyrosine Phosphatase Function: The Substrate Perspective. Biochemical Journal, 402, 1-15.
http://dx.doi.org/10.1042/BJ20061548 - 7. Swarup, G., Cohen, S. and Garbers, D.L. (1982) Inhibition of Membrane Phosphotyrosyl-Protein Phosphatase Activity by Vanadate. Biochemical and Biophysical Research Communications, 107, 1104-1109.
http://dx.doi.org/10.1016/0006-291X(82)90635-0 - 8. Cantley Jr., L.C. Josephson, L., Warner, R., Yanagisawa, M., Lechene, C. and Guidotti, G. (1977) Vanadate Is a Potent (Na,K)-ATPase Inhibitor Found in ATP Derived from Muscle. The Journal of Biological Chemistry, 252, 7421-7423.
- 9. Chen, Y. and Chan, T.M. (1993) Orthovanadate and 2,3-Dimethoxy-1,4-naphthoquinone Augment Growth Factor-Induced Cell Proliferation and c-fos Gene Expression in 3T3-L1 Cells. Archives of Biochemistry and Biophysics, 305, 9-16.
http://dx.doi.org/10.1006/abbi.1993.1387 - 10. Mehdi, M.Z., Pandey, S.K., Theberge, J.F. and Srivastava, A.K. (2006) Insulin Signal Mimicry as a Mechanism for the Insulin-Like Effects of Vanadium. Cell Biochemistry and Biophysics, 44, 73-81.
http://dx.doi.org/10.1385/CBB:44:1:073 - 11. Morita, A., Yamamoto, S., Wang, B., Tanaka, K., Suzuki, N., Aoki, S., Ito, A., Nanao, T., Ohya, S., Yoshino, M., Zhu, J., Enomoto, A., Matsumoto, Y., Funatsu, O., Hosoi, Y. and Ikekita, M. (2010) Sodium Orthovanadate Inhibits p53-Mediated Apoptosis. Cancer Research, 70, 257-265.
http://dx.doi.org/10.1158/0008-5472.CAN-08-3771 - 12. Wozniak, K. and Blasiak, J. (2004) Vanadyl Sulfate Can Differentially Damage DNA in Human Lymphocytes and HeLa Cells. Archives of Toxicology, 78, 7-15.
http://dx.doi.org/10.1007/s00204-003-0506-3 - 13. Sabbioni, E., Pozzi, G., Pintar, A., Casella, L. and Garattini, S. (1991) Cellular Retention, Cytotoxicity and Morphological Transformation by Vanadium(IV) and Vanadium(V) in BALB/3T3 Cell Lines. Carcinogenesis, 12, 47-52.
Http://Dx.Doi.Org/10.1093/Carcin/12.1.47 - 14. Soeda, S., Shimada, T., Koyanagi, S., Yokomatsu, T., Murano, T., Shibuya, S. and Shimeno, H. (2002) An Attempt to Promote Neo-Vascularization by Employing a Newly Synthesized Inhibitor of Protein Tyrosine Phosphatase. FEBS Letters, 524, 54-58.
http://dx.doi.org/10.1016/S0014-5793(02)03002-8 - 15. Carr, A.N., Davis, M.G., Eby-Wilkens, E., Howard, B.W., Towne, B.A., Dufresne, T.E. and Peters, K.G. (2004) Tyrosine Phosphatase Inhibition Augments Collateral Blood Flow in a Rat Model of Peripheral Vascular Disease. AJP: Heart and Circulatory Physiology, 287, H268-H276.
http://dx.doi.org/10.1152/ajpheart.00007.2004 - 16. Natarajan, V., Scribner, W.M. and Vepa, S. (1997) Phosphatase Inhibitors Potentiate 4-Hydroxynonenal-Induced Phospholipase D Activation in Vascular Endothelial Cells. American Journal of Respiratory Cell and Molecular Biology, 17, 251-259.
http://dx.doi.org/10.1165/ajrcmb.17.2.2623 - 17. Yuan, Y., Meng, F.Y., Huang, Q., Hawker, J. and Wu, H.M. (1998) Tyrosine Phosphorylation of Paxillin/pp125FAK and Microvascular Endothelial Barrier Function. American Journal of Physiology, 275, H84-H93.
- 18. Garcia, J.G., Schaphorst, K.L., Verin, A.D., Vepa, S., Patterson, C.E. and Natarajan, V. (1985) Diperoxovanadate Alters Endothelial Cell Focal Contacts and Barrier Function: Role of Tyrosine Phosphorylation. Journal of Applied Physiology, 89, 2333-2343.
- 19. Suzuki, E., Nagata, D., Yoshizumi, M., Kakoki, M., Goto, A., Omata, M. and Hirata, Y. (2000) Reentry into the Cell Cycle of Contact-Inhibited Vascular Endothelial Cells by a Phosphatase Inhibitor. Possible Involvement of Extracellular Signal-Regulated Kinase and Phosphatidylinositol 3-Kinase. The Journal of Biological Chemistry, 275, 3637-3644.
http://dx.doi.org/10.1074/jbc.275.5.3637 - 20. Shimizu, H., Takayama, H., Lee, J.D., Satake, K., Taniguchi, T., Yamamura, H. and Nakamura, T. (1994) Effects of Vanadate on Prostacyclin and Endothelin-1 Production and Protein-Tyrosine Phosphorylation in Human Endothelial Cells. Thrombosis and Haemostasis, 72, 973-978.
- 21. Helgadottir, A., Halldorsson, H., Magnusdottir, K., Kjeld, M. and Thorgeirsson, G. (1997) A Role for Tyrosine Phosphorylation in Generation of Inositol Phosphates and Prostacyclin Production in Endothelial Cells. Arteriosclerosis, Thrombosis, and Vascular Biology, 17, 287-294.
http://dx.doi.org/10.1161/01.ATV.17.2.287 - 22. Hellermann, G.R., Flam, B.R., Eichler, D.C. and Solomonson, L.P. (2000) Stimulation of Receptor-Mediated Nitric Oxide Production by Vanadate. Arteriosclerosis, Thrombosis, and Vascular Biology, 20, 2045-2050.
http://dx.doi.org/10.1161/01.ATV.20.9.2045 - 23. Nakaike, R., Shimokawa, H., Owada, M.K., Tokunaga, O., Yasutake, H., Kishimoto, T., Imada, C., Shiraishi, T., Egashira, K. and Takeshita, A. (1996) Vanadate Causes Synthesis of Endothelium-Derived NO via Pertussis Toxin-Sensitive G Protein in Pigs. American Journal of Physiology, 271, H296-H302.
- 24. Di Salvo, J., Semenchuk, L.A. and Lauer, J. (1993) Vanadate-Induced Contraction of Smooth Muscle and Enhanced Protein Tyrosine Phosphorylation. Archives of Biochemistry and Biophysics, 304, 386-391.
http://dx.doi.org/10.1006/abbi.1993.1366 - 25. Nayler, R.A. and Sparrow, M.P. (1983) Mechanism of Vanadate-Induced Contraction of Airways Smooth Muscle of the Guinea-Pig. British Journal of Pharmacology, 80, 163-172.
http://dx.doi.org/10.1111/j.1476-5381.1983.tb11062.x - 26. Alcon, S., Camello, P.J., Garcia, L.J. and Pozo, M.J. (2000) Activation of Tyrosine Kinase Pathway by Vanadate in Gallbladder Smooth Muscle. Biochemical Pharmacology, 59, 1077-1089.
http://dx.doi.org/10.1016/S0006-2952(00)00237-9 - 27. Laniyonu, A., Saifeddine, M., Ahmad, S. and Hollenberg, M.D. (1994) Regulation of Vascular and Gastric Smooth Muscle Contractility by Pervanadate. British Journal of Pharmacology, 113, 403-410.
http://dx.doi.org/10.1111/j.1476-5381.1994.tb17003.x - 28. Boulven, I., Robin, P., Desmyter, C., Harbon, S. and Leiber, D. (2002) Differential Involvement of Src Family Kinases in Pervanadate-Mediated Responses in Rat Myometrial Cells. Cellular Signalling, 14, 341-349.
http://dx.doi.org/10.1016/S0898-6568(01)00269-8 - 29. Mori, M. and Tsushima, H. (2004) Vanadate Activates Rho A Translocation in Association with Contracting Effects in Ileal Longitudinal Smooth Muscle of Guinea Pig. Journal of Pharmacological Sciences, 95, 443-451.
http://dx.doi.org/10.1254/jphs.FP0030576 - 30. Yayama, K., Sasahara, T., Ohba, H., Funasaka, A. and Okamoto, H. (2014) Orthovanadate-Induced Vasocontraction Is Mediated by the Activation of Rho-Kinase through Src-Dependent Transactivation of Epidermal Growth Factor Receptor. Pharmacology Research & Perspectives, 2, Article ID: e00039.
http://dx.doi.org/10.1002/prp2.39 - 31. Sasahara, T., Ohkura, N., Kobe, A. and Yayama, K. (2015) Epidermal Growth Factor Induces Ca2+ Sensitization through Rho-Kinase-Dependent Phosphorylation of Myosin Phosphatase Target Subunit 1 in Vascular Smooth Muscle. European Journal of Pharmacology, 762, 89-95.
http://dx.doi.org/10.1016/j.ejphar.2015.05.042 - 32. Rapp, J.P. (1981) Aortic Responses to Vanadate: Independence from (Na,K)-ATPase and Comparison of Dahl Salt-Sensitive and Salt-Resistant Rats. Hypertension, 3, I168-1172.
http://dx.doi.org/10.1161/01.HYP.3.3_Pt_2.I168 - 33. Sanchez-Ferrer, C.F., Marin, J., Lluch, M., Valverde, A. and Salaices, M. (1988) Actions of Vanadate on Vascular Tension and Sodium Pump Activity in Cat Isolated Cerebral and Femoral Arteries. British Journal of Pharmacology, 93, 53-60.
http://dx.doi.org/10.1111/j.1476-5381.1988.tb11404.x - 34. Fox, A.A., Borchard, U. and Neumann, M. (1983) Effects of Vanadate on Isolated Vascular Tissue: Biochemical and Functional Investigations. Journal of Cardiovascular Pharmacology, 5, 309-316.
http://dx.doi.org/10.1097/00005344-198303000-00024 - 35. Shimada, T., Shimamura, K. and Sunano, S. (1986) Effects of Sodium Vanadate on Various Types of Vascular Smooth Muscles. Journal of Vascular Research, 23, 113-124.
http://dx.doi.org/10.1159/000158628 - 36. Spurrell, B.E., Murphy, T.V. and Hill, M.A. (2000) Tyrosine Phosphorylation Modulates Arteriolar Tone but Is Not Fundamental to Myogenic Response. The American Journal of Physiology—Heart and Circulatory Physiology, 278, H373-H382.
- 37. Masui, H. and Wakabayashi, I. (2000) Tyrosine Phosphorylation Increases Ca2+ Sensitivity of Vascular Smooth Muscle Contraction. Life Sciences, 68, 363-372.
http://dx.doi.org/10.1016/S0024-3205(00)00942-5 - 38. Murphy, T.V., Spurrell, B.E. and Hill, M.A. (2002) Mechanisms Underlying Pervanadate-Induced Contraction of Rat Cremaster Muscle Arterioles. European Journal of Pharmacology, 442, 107-114.
http://dx.doi.org/10.1016/S0014-2999(02)01498-X - 39. Voldborg, B.R., Damstrup, L., Spang-Thomsen, M. and Poulsen, H.S. (1997) Epidermal Growth Factor Receptor (EGFR) and EGFR Mutations, Function and Possible Role in Clinical Trials. Annals of Oncology, 8, 1197-1206.
http://dx.doi.org/10.1023/A:1008209720526 - 40. Biscardi, J.S., Maa, M.C., Tice, D.A., Cox, M.E., Leu, T.H. and Parsons, S.J. (1999) C-Src-Mediated Phosphorylation of the Epidermal Growth Factor Receptor on Tyr845 and Tyr1101 Is Associated with Modulation of Receptor Function. The Journal of Biological Chemistry, 274, 8335-8343.
http://dx.doi.org/10.1074/jbc.274.12.8335 - 41. Sato, K., Sato, A., Aoto, M. and Fukami, Y. (1995) C-Src Phosphorylates Epidermal Growth Factor Receptor on Tyrosine 845. Biochemical and Biophysical Research Communications, 215, 1078-1087.
http://dx.doi.org/10.1006/bbrc.1995.2574 - 42. Pai, R., Soreghan, B., Szabo, I.L., Pavelka, M., Baatar, D. and Tarnawski, A.S. (2002) Prostaglandin E2 Transactivates EGF Receptor: A Novel Mechanism for Promoting Colon Cancer Growth and Gastrointestinal Hypertrophy. Nature Medicine, 8, 289-293.
http://dx.doi.org/10.1038/nm0302-289 - 43. Tice, D.A., Biscardi, J.S., Nickles, A.L. and Parsons, S.J. (1999) Mechanism of Biological Synergy between Cellular Src and Epidermal Growth Factor Receptor. Proceedings of the National Academy of Sciences of the United States of America, 96, 1415-1420.
http://dx.doi.org/10.1073/pnas.96.4.1415 - 44. Haskell, M.D., Slack, J.K., Parsons, J.T. and Parsons, S.J. (2001) C-Src Tyrosine Phosphorylation of Epidermal Growth Factor Receptor, P190 RhoGAP, and Focal Adhesion Kinase Regulates Diverse Cellular Processes. Chemical Reviews, 101, 2425-2440.
http://dx.doi.org/10.1021/cr0002341 - 45. Cowan, K.J. and Storey, K.B. (2003) Mitogen-Activated Protein Kinases: New Signaling Pathways Functioning in Cellular Responses to Environmental Stress. Journal of Experimental Biology, 206, 1107-1115.
http://dx.doi.org/10.1242/jeb.00220 - 46. Zhang, Y., Wang, L.Y., Zhang, M.M., Jin, M.L., Bai, C.X. and Wang, X.D. (2012) Potential Mechanism of Interleukin-8 Production from Lung Cancer Cells: An Involvement of EGF-EGFR-PI3K-Akt-Erk Pathway. Journal of Cellular Physiology, 227, 35-43.
http://dx.doi.org/10.1002/jcp.22722 - 47. Florian, J.A. and Watts, S.W. (1999) Epidermal Growth Factor: A Potent Vasoconstrictor in Experimental Hypertension. American Journal of Physiology, 276, H976-H983.
- 48. Northcott, C., Florian, J.A., Dorrance, A. and Watts, S.W. (2001) Arterial Epidermal Growth Factor Receptor Expression in Deoxycorticosterone Acetate-Salt Hypertension. Hypertension, 38, 1337-1341.
http://dx.doi.org/10.1161/hy1201.096815 - 49. Kim, J., Lee, C.K., Park, H.J., Kim, H.J., So, H.H., Lee, K.S., Lee, H.M., Roh, H.Y., Choi, W.S., Park, T.K. and Kim, B. (2006) Epidermal Growth Factor Induces Vasoconstriction through the Phosphatidylinositol 3-Kinase-Mediated Mitogen-Activated Protein Kinase Pathway in Hypertensive Rats. Journal of Pharmacological Sciences, 101, 135-143.
http://dx.doi.org/10.1254/jphs.FP0060021 - 50. Li, Y., Levesque, L.O. and Anand-Srivastava, M.B. (2010) Epidermal Growth Factor Receptor Transactivation by Endogenous Vasoactive Peptides Contributes to Hyperproliferation of Vascular Smooth Muscle Cells of SHR. AJP: Heart and Circulatory Physiology, 299, H1959-H1967.
http://dx.doi.org/10.1152/ajpheart.00526.2010 - 51. Sandoval, Y.H., Li, Y. and Anand-Srivastava, M.B. (2011) Transactivation of Epidermal Growth Factor Receptor by Enhanced Levels of Endogenous Angiotensin II Contributes to the Overexpression of Gialpha Proteins in Vascular Smooth Muscle Cells from SHR. Cellular Signalling, 23, 1716-1726.
http://dx.doi.org/10.1016/j.cellsig.2011.06.006 - 52. Touyz, R.M., Wu, X.H., He, G., Salomon, S. and Schiffrin, E.L. (2002) Increased Angiotensin II-Mediated Src Signaling via Epidermal Growth Factor Receptor Transactivation Is Associated with Decreased C-Terminal Src Kinase Activity in Vascular Smooth Muscle Cells from Spontaneously Hypertensive Rats. Hypertension, 39, 479-485.
http://dx.doi.org/10.1161/hy02t2.102909
NOTES

*Corresponding author.