Pharmacology & Pharmacy
Vol.06 No.11(2015), Article ID:61448,14 pages
10.4236/pp.2015.611055
Influence of CYP2B6 516G > T and Long Term HAART on Population Pharmacokinetics of Efavirenz in Rwandan Adults on HIV and Tuberculosis Cotreatment
Emile Bienvenu1*, Michael Ashton2, Angela Äbelö2
1School of Medicine, College of Medicine and Health Sciences, University of Rwanda, Kigali, Rwanda
2Unit for Pharmacokinetics and Drug Metabolism, Department of Pharmacology, Sahlgrenska Academy at University of Gothenburg, Göteborg, Sweden

Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).


Received 18 June 2015; accepted 22 November 2015; published 25 November 2015

ABSTRACT
Aim: To describe the pharmacokinetic parameters of efavirenz and estimate its clearance (CL/F) accounting simultaneously for drug-drug interactions and CYP2B6 genetic polymorphism. Methods: Genotyping of 516G > T single nucleotide polymorphism of CYP2B6 was performed using a PCR-based technology and plasma efavirenz concentrations were measured by high performance liquid chromatography on blood samples from 76 HIV adults co-infected with tuberculosis who had received an efavirenz-based regimen. Data were analyzed using population modeling with NONMEM. Results: The absorption rate constant and the apparent volume of distribution in the final model were 1.9 h−1 and 580 L/70kg, respectively. The CL/F at baseline was 11.8 L/h/70kg, 8.8 L/h/70kg and 3.9 L/h/70kg for patients carrying the G/G, G/T and T/T genotypes of CYP2B6 516G > T, respectively, in patients who were administered tuberculosis (TB) treatment prior to HIV treatment (Group A); and 16.7 L/h/70kg, 10.6 L/h/70kg and 1.8 L/h/70kg for G/G, G/T and T/T genotype patients respectively, in patients with previous exposure to HIV treatment (Group B). The CL/F at baseline and steady state was always higher in Group B compared to Group A patients. Expectedly, carriers of CYP2B6 516G/G and T/T genotypes exhibited higher and lower CL/F, respectively. Conclusion: Our results indicated that the CL/F of efavirenz in the population studied was predictably different due to whether the patients were mono-treated for TB with HAART deferred or for HIV before initiation of TB therapy, and to CYP2B6 516G > T variant, implying that both CYP2B6 genetic polymorphisms and previous efavirenz-based HAART should be taken into account when adjusting efavirenz dose.
Keywords:
Clearance, CYP2B6, Efavirenz, HIV, RBT, Tuberculosis

1. Introduction
Human immunodeficiency virus (HIV) has been identified as the primary cause of acquired immunodeficiency syndrome (AIDS). The primary and essential receptor for HIV is the CD4 lymphocyte cell which is involved in the induction of most immunologic functions. As a result, the immune system may get slowly damaged and individuals may develop symptoms of the late phase of HIV disease (AIDS) where individuals are susceptible to other opportunistic infections, such as infections with Mycobacterium tuberculosis, Pneumocystis carinii, toxoplasmosis and candidiasis, etc. Infected individuals are diagnosed as having AIDS status when their plasma HIV load is high and the CD4 cell counts falls below 200 cells/mm3 (normal CD4 counts are between 500 and 1.600 cells/mm3) [1] -[3] .
Tuberculosis (TB) is the most common opportunistic infection among people infected with HIV [4] . TB is an infection caused by a bacterium called Mycobacterium tuberculosis that affects the lungs, following transmission after expectoration from an infected individual. Mycobacterium tuberculosis sometimes causes illness soon after the initial exposure, but an immune-competent system can prevent Mycobacterium tuberculosis from causing illness with the involvement of CD4 lymphocyte cells [5] -[7] . As a result, people infected with HIV are much more likely to develop TB than those who are HIV-negative [4] [8] . World Health Organization (WHO) reported that 13% of people who developed TB worldwide in 2012 were HIV-positive, with 75% of these being of African region origin [4] . The standard regimens for the management of each of these diseases combine two to four drugs [9] ; as a result, the medical management of HIV/TB co-infection is challenged by interactions between these drugs once co-administered [10] .
The TB therapy recommended in the WHO guidelines involves the combination of ethambutol, isoniazid, rifampicin and pyrazinamide, while streptomycin may be used when one of these drugs is contraindicated [11] . On the other hand, the management of HIV/AIDS usually involves the use of multiple antiretroviral drugs, known as highly active antiretroviral therapy (HAART). HAART combines two nucleoside reverse transcriptase inhibitors and a non-nucleoside reverse transcriptase inhibitor (NNRTI). The recommended HAART consists of an NNRTI (either nevirapine or efavirenz) plus two NRTIs, one of which being lamivudine or emtricitabine and the other zidovudine or tenofovir. HAART involving efavirenz (EFV) as NNRTI is the preferred treatment for HIV-associated TB, because of good tolerance with superior clinical outcomes. EFV acts by binding to the viral-specific enzyme reverse transcriptase and blocks its action in a noncompetitive manner [9] [10] . EFV is metabolised into hydroxylated metabolites primarily by the cytochrome P450 (CYP) 2B6, and secondarily by CYP1A2, CYP2A6, CYP3A4 and CYP3A5 with a minor contribution [12] -[14] . The suggested metabolic pathways of EFV are shown in Figure 1. CYP2B6 makes up to 2% - 10% of the total CYP content and is coded by a polymorphic gene at chromosome 19, which spans a region of approximately 28 kb and contains nine exons. A large number of CYP2B6 variants have been reported, among which 516G > T associated with reduced expression and activity. CYP2B6 metabolizes a number of xenobiotics and is highly inducible by several drugs and other xenobiotics [15] . Drug-drug interactions (DDIs) affecting EFV and involving TB drugs have been reported, enzyme induction caused by rifampicin TB drug being the most documented [15] [16] .
Enzyme induction can be due to co-administration of drugs with induction capability, or a drug inducing its own metabolism (auto-induction), or a combination of the former and the latter [17] . This last combination of events is normally common in the co-management of HIV/TB co-infection, where EFV-based HAART is concomitantly administered with rifampicin-based TB treatment (RBT). The induction effect of rifampicin on EFV metabolizing enzymes (CYP2B6 and CYP3A4) has been demonstrated in different populations [15] [16] [18] . Moreover, EFV has been shown to induce CYP2B6 and CYP3A4, accelerating its own metabolism [17] [19] [20] .
Furthermore, the main EFV metabolizing enzymes, CYP2B6 is characterized by interindividual differences in expression and activity, leading to altered EFV disposition [21] -[23] . In the context of co-management of HIV/TB co-infection, DDIs come as an additional factor that may affect EFV pharmacokinetics (PK). We

Figure 1. Suggested metabolic pathways for efavirenz and its catalytic hepatic enzymes.
recently reported differences in single nucleotide polymorphisms (SNPs) between a Rwandan population and other African populations [24] ; today, no study has characterized the PK of EFV in this population when undergoing both HIV and TB treatment. Besides, PK interactions between antiretroviral and TB drugs, in particular between EFV and rifampicin, as well as their clinical implications are still not well understood given that available data are conflicting, varying from one cohort to another [25] [26] . In addition, contrary to several previous reports, there is a need to characterize the PK of EFV by taking into account a real clinical situation. Therefore, the aim of this study was to describe the PK parameters of EFV in patients under both HIV and TB treatments, and to estimate the clearance by including CYP2B6 genetic polymorphism as a covariate and by taking into account the nature of the treatment previously administered to the patients. We hypothesized that patients may present with different baseline EFV clearances depending on the potential for the previous treatment to induce EFV metabolizing enzymes.
2. Methods
Study design and population: An open-label and observational study was conducted in Rwandan adult HIV/AIDS patients co-infected with TB. Medications were administered in accordance with the Rwandan guidelines for the management of HIV/AIDS [27] . HAART combination was composed of either abacavir or tenofovir or stavudine or zidovudine added to EFV/lamivudine. TB treatment involved rifampicin (R), isoniazid (H), pyrazinamide (Z), and ethambutol (E). The fixed dose-combination regimen was 2(RHZE)7/4(RH)3 (numbers before the letters indicate the months of duration of the treatment for the intensive treatment phase; subscripts indicate how often the treatment was taken each week). Both treatments were administered in a pre- existing directly observed therapy (DOT) program. The study protocol was approved by the National Ethics Committee of the Ministry of Health in Rwanda (No. 148/RNEC/2009).
Patients were all adults (21 - 65 years of age), HIV antibody positive, diagnosed for TB, anti-tubercular naive, on or initiating HAART (naive). Previous exposure to HAART was allowed as in the previous design by ter Heine et al. [28] . Of 147 screened patients, 105 patients meeting inclusion criteria including provision of written informed consent were recruited. Twenty five patients were withdrawn from the study during the follow up period in accordance with the study protocol. Eighty patients completed the study, among which 76 patients had received EFV-based HAART. Naive patients (Group A) were administered TB treatment, with HIV treatment to be initiated after a period ranging between two to eight weeks if CD4 counts are below 500 cells/µL, in accordance with the local treatment guidelines. A second category (Group B) was composed of patients who were diagnosed for TB after varying time of HIV treatment from weeks to years. The day of initiation of TB treatment was the day of enrolment in the study. A schematic representation of the flow of the recruitment process of the patients including the sample size for each group is shown in Figure 2. Demographic variables were recorded at enrolment and relevant clinical chemistry values measured.
Blood sample collection: The patients studied had taken an EFV dose (600 mg) in the evening as per the current treatment guidelines. Samples were collected in the morning at arrival at the clinic, at the convenient time of the patient, approximately 13 hours after the previous evening EFV dose. After initiation of both HIV and TB treatments, patients were monitored for six weeks to collect sparse blood samples for determination of EFV blood concentrations, CD4 cell counts and HIV-RNA copies. Two additional samples were collected at enrolment for genotyping and clinical chemistry tests. At initiation of the treatment after enrolment, rich sampling was performed at pre-dose and after 1, 2, 3, 4, 6 and 8 hours of initiation of HAART. All blood samples were collected into EDTA tubes (4 ml) and stored at −80˚C until analysis. Samples for determination of EFV concentrations were centrifuged (10 minutes at 10,500 g) before storage.
Bioanalysis: EFV was quantitated using a validated high-performance liquid chromatography method with ultraviolet detection, as described previously [29] . Briefly, chromatographic separation was carried out using a C18 analytical column equipped with a security guard column. The mobile phase consisted of the mixture acetonitrile-water (75:25% v/v; pH of water adjusted at 3.2 using 0.1% formic acid), and was pumped at a flow rate of 0.3 mL/min. EFV and ritonavir (internal standard) were monitored at 247 nm. Plasma proteins were precipitated by centrifugation. The lower limit of quantitation was set to 0.06 lg/mL with deviation from the nominal concentrations being <20%, in accordance with the bioanalytical method validation guidelines of The United States Food and Drug Administration. The response was linear with a correlation coefficient of 0.9997, a slope of 0.189 and y intercept of 0.003. The relative standard deviation for the slope was 5.474%. The accuracy ranged between 98% and 115% (intraday) and between 99% and 117% (interday). The precision ranged from 1.670% to 4.087% (intraday) and from 3.447% to 13.347% (interday). Recovery ranged from 98% to 132%.

Figure 2. A schematic representation of the flow of the recruitment process of the patients.
Stability ranged between 99% and 123%. The selectivity was proven by analysis of drugs used for the management of HIV/AIDS and TB.
CYP2B6 genotyping: Patients participating in this study were genotyped using a PCR-based technology with respect to different SNPs including CYP2B6 516G > T. The background, genotyping methodology and results including the frequency of alleles and genotypes were described elsewhere [24] . In summary, whole blood sample was collected for genotyping. DNA was extracted at LGC Genomics GmbH (Berlin, Germany) using the patented PLUS XL manual kit (catalog: catalogue number 40,801 and 40,810). Selected SNPs including CYP2B6 516G > T were genotyped at LGC Genomics Ltd., (Hoddesdon, UK) using the patented KASP assay (Kompetitive Allele Specific PCR), which is a polymerase chain reaction based, homogeneous, fluorescent, endpoint-genotyping technology. The KASP genotyping system was comprised of the SNP-specific assay (a combination of three unlabelled primers) and the universal reaction mix, which contains all other required components including the universal fluorescent reporting system and a specially developed Taq polymerase. The Hardy-Weinberg equilibrium (HWE) testing was performed for each of the SNP to validate the quality of genotyping, and all the SNPs conformed to HWE.
Population PK analysis: The population PK analysis was performed in NONMEM computer program, Version 7.1.2 and goodness-of-fit plots were constructed with Xpose4 library (version 4.3.2, 2010) run in R program (version 2.13.0, 2011). Preliminarily, rich and sparse data were graphically examined. All data was then combined and used in the modeling. The data used in the modeling was log-transformed. A one-compartment model with first-order absorption and elimination was used to describe the data, using the NONMEM subroutine ADVAN5/TRANS1 and first order conditional estimation (FOCE) method. The PK parameters were allometrically scaled by a standard body weight (70 kg) as previously applied by Gengiah et al. [30] . The scaling factor was a priori set to 0.75 for clearance (CL/F) and to 1 for volume of distribution (V/F). Inter-individual variability (ω2) was estimated for all the structural parameters of the model using an exponential error model, assuming that the individual value for a structural parameter was log-normally distributed. Residual variability (σ2) was estimated as an additive residual error on log-transformed data corresponding to an approximately exponential residual model.
Population PK parameters including CL/F, V/F and absorption constant rate (Ka) with a lag time (alag) were first estimated in the basic PK model. Given that CL/F of EFV has been shown to increase over time because of auto-induction [10] or the presence of an inducing agent, like the TB drug rifampicin [9] , CL/F values were, in the final model, estimated taking into account the potential for the previous treatment to induce EFV. Hence CL/F values were estimated at baseline and at steady state to account for drug-drug interactions caused by enzyme induction. CL/F at baseline corresponded to the CL/F of the first day of initiation of HAART for Group A patients (naive) after some weeks of RBT, and the CL/F at the day of enrolment in the study after several months of EFV-based HAART in Group B patients. CL/F at steady state corresponded to the CL/F estimated for the two patient groups over six weeks with patients being under concomitant HIV and TB treatments. Different CL/F for each of the 3 CYP2B6 516G > T genotypes were estimated in the two groups in order to quantify the impact of genetic polymorphism in that gene on the metabolism of EFV. The CL/F at baseline and at steady state was modelled according to the following equations:
At baseline (time = 0):
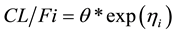
At steady-state (time > 0):
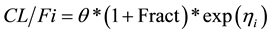
where
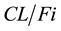 is the apparent clearance of the ith individual; q is the typical value of clearance in the population;
is the apparent clearance of the ith individual; q is the typical value of clearance in the population;
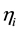 is the inter-individual variability in clearance; and Fract represents the fractional increase in baseline clearance.
is the inter-individual variability in clearance; and Fract represents the fractional increase in baseline clearance.
The final model selection was based on standard goodness-of-fit plots, agreement in the observed and individual and population predicted data, lack of trend or pattern in scatter plots of weighted residuals versus predicted concentrations or conditional weighted residuals versus time, the precision of the parameter estimates expressed as 95% confidence interval (CI) , agreement between the observed data and the 95% CI of the data and comparison of NONMEM objective function values (OFV; it is expressed for convenience as minus twice the log of the likelihood, and is a single number that provides an overall summary of how closely the model predictions match the data; the lower the OFV, the best the fit). A reduction in OFV of at least 3.84 was considered significant (p = 0.05). Model evaluation was performed using bootstrap with 500 re-sampled datasets, and a simulation-based visual predictive check (VPC) to evaluate whether model predictions adequately described the observed data [31] .
3. Results
Characteristics of the population studied: The baseline characteristics of participating patients are presented in Table 1. The mean ± SD age for the patients participating in this study was 38.0 ± 8.1 years, varying between 21 and 52 years for group A patients and between 26 and 57 years for group B patients. The gender ratio (male: female) was 1.06 (37:35) with 22 and 15 males in groups A and B, respectively, and 17 and 18 females in Groups A and B, respectively. Among the 72 patients participating in the present analysis, thirty nine patients were in Group A, with RBT for an average period of six weeks prior to EFV-based HAART. Thirty three patients with previous exposure to EFV-based HAART belonged to Group B. Among the 64 individuals with available genetic data in CYP2B6 516G > T, there was equal number (45.3%) of extensive (CYP2B6 516G/G genotype carriers) and intermediate (CYP2B6 516G/T genotype carriers) metabolizers of EFV, while poor metabolizers (CYP2B6 516T/T genotype carriers) represented 9.4%.
Efavirenz plasma concentrations and the dataset: Sparse EFV plasma concentrations were measured for 76 patients who were treated with EFV-based HAART. Only 72 patients (Figure 2) were considered in the present analysis, removed ones had either undetectable or below the lower limit of quantitation plasma concentrations. Of the remaining patients, 42 had undergone rich sampling. Finally, a total of 740 plasma concentrations from 72 individuals, of which 64 individuals with genetic data in CYP2B6 available (Table 1) were used to build a dataset merging sparse and rich data for this population PK analysis. EFV plasma concentrations were highly variable, as shown by plots of the sparse and rich concentrations used in the present analyses (Figure 3); they ranged between 0.08 and 15.0 mg/L. Consequently, one patient had concentrations that were constantly very high at all follow up occasions (8.0 - 10.2 mg/L for rich occasions, and 10.2 - 15.0 mg/L for sparse occasions), and 14 concentrations from different patients were very low (0.08 - 0.19 mg/L), and appeared as outliers in diagnostic and VPC plots. Twelve of them were part of rich data.
Pharmacokinetic parameters: The final PK model was a one compartment model with first order absorption and elimination, and with inter-individual variability on CL/F, V/F, Ka and alag. CL/F values were estimated according to the three CYP2B6 516G > T genotypes in each of the patient groups, at baseline and at steady state. The final population PK parameter estimates are detailed in Table 2 and Table 3. The absorption rate constant and the apparent volume of distribution in the final model were 1.9 h−1 and 580 L/70kg, respectively (Table 2). The CL/F at baseline was 11.8 L/h/70kg, 8.8 L/h/70kg and 3.9 L/h/70kg for patients carrying the G/G, G/T and T/T genotypes of CYP2B6 516G > T, respectively, in patients who initiated TB treatment for some weeks prior to HAART; and 16.7 L/h/70kg, 10.6 L/h/70kg and 1.8 L/h/70kg for G/G, G/T and T/T genotype patients respectively, in patients with previous exposure to HAART (Table 3). The fractional increases of CL/F were 0.51 and 0.55 in patients who had initiated TB treatment prior to HAART and in group of those who had previous months

Table 1. Baseline characteristics of the patients participating in this study.
*Mean ± SD (range); ¨Median (range).
 (a)
(a) (b)
(b)
Figure 3. Observed EFV plasma concentrations over a 6-week follow up (sparse sampling) in 72 patients on concomitant HIV and TB treatment (a) and over 8 hours (rich sampling) in 42 patients after initiation of HAART (b). Naive patients who initiated HAART after some weeks of TB treatment (Group (A)) are represented by solid lines. Patients who have been on HAART since several months before initiation of TB treatment (Group (B)) are represented by dashed lines.
Table 2. Population pharmacokinetic parameter estimates for efavirenz.
CL/F: oral clearance. Ka: absorption constant rate. V/F: apparent volume of distribution. Alag: Absorption lag-time. σadd: residual error. IIV: Inter-individual variability. CI: confidence interval. Bio: oral bioavailability. *95% CIs calculated as the 2.5 and 97.5 percentiles of bootstrap estimates. Parameter estimates are based on population mean values from NONMEM, CI values are based on 460 successful bootstrap runs out of 500 for the final model. §Refer to Table 3 for CL/F (L/h) values of the final model. ªFixed to one.
Table 3. Population clearance of the final model for efavirenz by CYP2B6 516G > T genotype and by nature of previous treatment.
CL/F: oral clearance stratified by CYP2B6 516G > T genotypes, at baseline and at steady state. IIV: Inter-individual variability. CI: confidence interval. HAART: Highly Active Antiretroviral Therapy. TBPREV: Group of patients who had TB treatment prior to HAART. HAARTPREV: Group of patients who had previous exposure to HAART before being diagnosed for TB and initiation TB therapy. *95% CIs calculated as the 2.5 and 97.5 percentiles of bootstrap estimates. Parameter estimates are based on population mean values from NONMEM, CI values are based on 460 successful bootstrap runs out of 500 for the final model.
of exposure to HAART for a long period, respectively.
Table 3 presents the CL/F values of the final model stratified by CYP2B6 516G > T genotypes in each patient group. As can be seen, the CL/F at baseline and steady state was always higher in patients with previous exposure to HAART (HAARTPREV) compared to those who were naive, and who initiated TB treatment prior to HAART (TBPREV). Exception to this was for carriers of CYP2B6 516T/T genotype; probably because of the limited sample size (only one T/T patient in the second group). Carriers of CYP2B6 516G/G genotype (extensive metabolizers) in all patient groups exhibited higher CL/F compared to carriers of other genotypes, whereas lower CL/F were seen with carriers of CYP2B6 516T/T genotype (poor metabolizers). All the above indicates that previous exposure to HAART increased the CL/F, regardless of CYP2B6 516G > T polymorphisms. This is in agreement with the trends of measured EFV plasma concentrations plotted in Figure 3(a), where patients with previous exposure to HAART showed lower concentrations compared to those who had prior TB treatment. Note that one patient who was previously treated with HAART showed the highest sparse and rich concentrations in Figure 3 and was carrier of CYP2B6 516T/T genotype (poor metabolizer).
Ka was estimated with a poor precision probably because of few data for the absorption phase; this difficulty was previously reported in other studies [32] [33] . Other parameters were estimated with a satisfactory precision (Table 2 and Table 3). Allometrically scaling the CL/F and the V/F for weight resulted in improved OFV compared to a model without weight. The final model was evaluated, and the results of the VPC are displayed in Figure 4, as the prediction of the median concentration (solid line), and the 95% CIs (grey area). The VPC results confirmed the adequacy of the model predictions in describing the observed data.
4. Discussion
This study aimed to describe the PK parameters of EFV by taking into account a real clinical situation and to estimate the CL/F of EFV by accounting simultaneously for CYP2B6 genetic polymorphisms and for DDIs caused by induction. Interindividual variability in EFV pharmacokinetics associated with CYP2B6 516G > T genetic polymorphisms has been extensively studied [21] -[23] and DDIs affecting EFV either through auto-induction or involving the TB drug rifampicin documented [15] -[17] [19] [20] . Nevertheless, today no study has characterized the PK of EFV in the studied population, reported to exhibit differences in SNPs to other African population [24] , when undergoing HIV/TB cotreatment. Consequently whether CL/F values of EFV differed according to the current treatment nature is not known. This study reports CL/F values of EFV which differed according to whether the patients were under TB treatment before initiation of EFV-based HAART or whether they had previous exposure to HAART before being diagnosed for TB and initiation of TB therapy, regardless of the CYP2B6 516G > T polymorphisms. CL/F values were also different according to the CYP2B6 516G > T genotype carried by the patients (G/G, G/T and T/T for extensive, intermediate and poor metabolizers, respectively), regardless of the previous treatment received by the patients. Moreover, this study demonstrates that a one-compartment model with first-order absorption and elimination can be used to characterize CL/F of EFV in a patient population with various durations of previous exposure to treatments that have inducing effects.
In patients pre-treated for TB (Group A), it appears that the CL/F of EFV was under the influence of enzyme induction which was elicited by the previous TB treatment before the initiation of EFV-based HAART. While in
 (a)
(a) (b)
(b)
Figure 4. Visual predictive check (95% prediction interval) of the final pharmacokinetic model (a) and for the early rich sampling phase (b). Observed data is depicted as circles. The solid and dashed lines are the median, 2.5th and 97.5th percentile of the predicted and observed data, respectively. The grey shaded areas are the 95 confidence interval of the simulated median, 2.5th and 97.5th percentile, respectively.
patients with previous exposure to EFV-based HAART (Group B), the estimated CL/F of EFV was auto-induced by EFV itself. On one hand, rifampicin, one of the TB drugs has been shown to induce EFV metabolizing enzymes such as CYP2B6 [16] [34] [35] and CYP3A4 [18] [36] . Enzymes metabolizing EFV were reported to be induced to a maximum degree during the early stage (first eight weeks) of RBT, indicating no prolonged enzyme induction to affect EFV kinetics over time [20] . This suggests that rifampicin enzyme induction was still ongoing the day of initiation of EFV-based HAART in Group A patients of our cohort who had less than eight weeks of TB treatment, which was the case for 85% of the patients of this group. This indicates that enzyme induction had not yet reached its maximum, explaining the lower CL/F of EFV at baseline for patients of this group compared to Group B patients. On the other hand when treating HIV alone, EFV was reported to accelerate its own metabolism by inducing CYP2B6 and CYP3A4 up to a prolonged period of 3 months to reach almost its maximum [10] [12] . This indicates that in Group B, EFV auto-induction was still ongoing for patients who have been treated with HAART for a period of less than three months, whereas it has reached its maximum for those who have been treated with HAART for a period of more than three months. Since patients of this group may have been under HIV treatment for several months, EFV metabolizing enzymes were highly induced up to the maximum in patients of this group, explaining the higher CL/F values observed at baseline for Group B compared to Group A patients.
Furthermore, when treating both HIV and TB, Ngaimisi et al. [20] reported an early induction of EFV metabolizing enzymes by RBT and a non-significant additive or synergistic effects for EFV auto-induction over and above ongoing RBT. Our data however, showing an increase of 51% and 55% of the CL/F in Group A and B patients, respectively, argue for a significant contribution of EFV auto-induction to the overall induction. This indicates that the increase of the CL/F observed in this study was dependant on the inducing effects of both RBT and EFV.
The influence of CYP2B6 genetic polymorphism on the CL/F of EFV was observed in this cohort, according to which carriers of G/G genotype had higher CL/F compared to carriers of G/T and T/T genotypes. These results are in agreement with our previous report where carriers of CYP2B6 516T/T genotype were found to present with higher EFV plasma levels [37] . On the other hand, they are comparable to the significantly higher EFV trough concentrations and exposure in T/T than in G/T and G/G genotype patients reported by Ramachandran et al. [35] in Indians, and the longest half-life for T/T genotype individuals reported by To et al. [38] in Chinese; all these implying a lower and higher CL/F of EFV for T/T and G/G genotype patients, respectively. Similar trends were also reported in African populations, such as South Africans [39] , Ghanaians [40] and Zimbabwean patients [41] . Our results imply that genetic polymorphism in CYP2B6 516G > T should be taken into account when adjusting EFV dose and patients under EFV-based treatment monitored closely since carriers of T/T and G/G genotypes may be at risk of supra-therapeutic levels (responsible for neuropsychiatric side effects) and sub-therapeutic levels of EFV (responsible for treatment failure). Overall, estimated PK parameters for EFV were in agreement with those previously reported [32] [33] [41] .
Several population PK models for EFV have been developed; nevertheless, ours is among few attempts to characterize the CL/F of EFV taking into account all categories of the patients that may exist in a real clinical situation, with various durations of previous exposure to treatments that have inducing effects. This made our cohort being composed of patients with previous exposure to treatments that had the potential to induce EFV metabolizing enzymes, implying that the estimated baseline CL/F was already induced and to different extents in patients in both groups of this cohort, while no data for a non-induced phase were available. In addition, we enrolled patients with various durations of exposure to HAART (Group B) and they may also have had differences in compliance to the treatment. Also, sparse data used in our dataset were from mid-dose sampling (sampling performed between doses, usually between 8 - 20 hours). Because EFV is generally taken at bedtime to improve its tolerability, we used mid-dose sampling because it is the most practical in an outpatient setting for patient convenience [42] . Also, mid-dose sampling is usually used in clinical studies of EFV disposition since mid-dose levels of EFV have been reported to be highly correlated with EFV area under the curve values when measured at steady state [43] .
5. Conclusion
The results of this study indicated higher CL/F values in patients with previous exposure to HAART, regardless of the CYP2B6 516G > T polymorphisms, and in carriers of CYP2B6 516G/G genotype, regardless of the previous treatment received by the patients. This suggested that the CL/F of EFV in the population studied was predictably different due to whether the patients were mono-treated for TB with HAART deferred or for HIV before initiation of TB therapy, and to CYP2B6 516G > T variant. Thus, not only should the patient genotype status with respect to CYP2B6 516G > T be taken into account when adjusting EFV dose, but also the current treatment nature with caution to previous exposure to HAART.
Acknowledgements
This work was financially supported by the Swedish International Development Cooperation Agency (Sida) [Agreement 2008-03-04, Decision no 2006-006238, Contribution no 75007334]. The advice and assistance of Dr Kurt-Jürgen Hoffmann during bioanalysis and in reviewing the manuscript are acknowledged.
Conflict of Interest
The authors declare that they have no conflict of interest.
Cite this paper
EmileBienvenu,MichaelAshton,AngelaÄbelö, (2015) Influence of CYP2B6 516G > T and Long Term HAART on Population Pharmacokinetics of Efavirenz in Rwandan Adults on HIV and Tuberculosis Cotreatment. Pharmacology & Pharmacy,06,533-546. doi: 10.4236/pp.2015.611055
References
- 1. Hassan, M.N. (2013) Pathogenesis of HIV Infection. Infectious Disease Reports, 5, 1-6.
- 2. Hoffmann, C. and Rockstroh, J.K. (2013) HIV Book 2012/2013. http://hivbook.com/
- 3. Nieves, D.M., Plaud, M., Wojna, V., Skolasky, R. and Melendez, L. (2007) Characterization of Peripheral Blood Human Immunodeficiency Virus Isolates from Hispanic Women with Cognitive Impairment. Journal of NeuroVirology, 13, 315-327.
http://dx.doi.org/10.1080/13550280701361508 - 4. World Health Organization (WHO) (2013) Global Tuberculosis Report 2013. WHO, Geneva.
- 5. Lawn, S.D. and Zumla, A. (2011) Tuberculosis. The Lancet, 378, 57-72.
http://dx.doi.org/10.1016/S0140-6736(10)62173-3 - 6. Frieden, T.R., Sterling, T.R., Munsiff, S.S., Watt, C.J. and Dye, C. (2003) Tuberculosis. The Lancet, 362, 887-899.
http://dx.doi.org/10.1016/S0140-6736(03)14333-4 - 7. Kunnath-Velayudhan, S. and Gennaro, M.L. (2011) Immunodiagnosis of Tuberculosis: A Dynamic View of Biomarker Discovery. Clinical Microbiology Reviews, 24, 792-805.
http://dx.doi.org/10.1128/CMR.00014-11 - 8. Ottenhoff, T.H. and Kaufmann, S.H. (2012) Vaccines against Tuberculosis: Where Are We and Where Do We Need to Go? PLoS Pathog, 8, e1002607.
http://dx.doi.org/10.1371/journal.ppat.1002607 - 9. Pozniak, A.L., Coyne, K.M., Miller, R.F., Lipman, M.C., Freedman, A.R., Ormerod L.P., Johnson, M.A., Collins, S. and Lucas, S.B. (2011) British HIV Association Guidelines for the Treatment of TB/HIV Coinfection 2011. HIV Medicine, 12, 517-524.
http://dx.doi.org/10.1111/j.1468-1293.2011.00954.x - 10. Avihingsanon, A., Hemachandra, A. and van de Lugt, J. (2009) Antiretroviral Therapy for HIV-Associated Tuberculosis. Asian Biomedicine, 3, 73-87.
- 11. World Health Organization (WHO) (2010) Treatment of Tuberculosis: Guidelines. Fourth Edition.
http://www.who.int/tb/features_archive/new_treatment_guidelines_may2010/en/index.html - 12. Bélanger, A.S., Caron, P., Harvey, M., Zimmerman, P.A., Mehlotra, R.K. and Guillemette, C. (2009) Glucuronidation of the Antiretroviral Drug Efavirenz by UGT2B7 and an in Vitro Investigation of Drug-Drug Interaction with Zidovudine. Drug Metabolism and Disposition, 37, 1793-1796.
http://dx.doi.org/10.1124/dmd.109.027706 - 13. Ogburn, E.T., Jones, D.R., Masters, A.R., Xu, C., Guo, Y. and Desta, Z. (2010) Efavirenz Primary and Secondary Metabolism in Vitro and in Vivo: Identification of Novel Metabolic Pathways and Cytochrome P450 2A6 as the Principal Catalyst of Efavirenz 7-Hydroxylation. Drug Metabolism and Disposition, 38, 1218-1229.
http://dx.doi.org/10.1124/dmd.109.031393 - 14. Ward, B.A., Gorski, J.C., Jones, D.R., Hall, S.D., Flockhart, D.A. and Desta, Z. (2003) The Cytochrome P450 2B6 (CYP2B6) Is the Main Catalyst of Efavirenz Primary and Secondary Metabolism: Implication for HIV/AIDS Therapy and Utility of Efavirenz as a Substrate Marker of CYP2B6 Catalytic Activity. Journal of Pharmacology and Experimental Therapeutics, 306, 287-300.
http://dx.doi.org/10.1124/jpet.103.049601 - 15. Kwara, A., Tashima, K.T., Dumond, J.B., Poethke, P., Kurpewski, J., Kashuba, A.D., Court, M.H. and Greenblatt, D.J. (2011) Modest but Variable Effect of Rifampin on Steady-State Plasma Pharmacokinetics of Efavirenz in Healthy African-American and Caucasian Volunteers. Antimicrobial Agents and Chemotherapy, 55, 3527-3533.
http://dx.doi.org/10.1128/AAC.00980-10 - 16. Rodríguez-Nóvoa, S., Barreiro, P., Jiménez-Nácher, I. and Soriano, V. (2006) Overview of the Pharmacogenetics of HIV Therapy. Pharmacogenomics, 6, 234-245.
http://dx.doi.org/10.1038/sj.tpj.6500374 - 17. Zhu, M., Kaul, S., Nandy, P., Grasela, D.M. and Pfister, M. (2009) Model-Based Approach to Characterize Efavirenz Autoinduction and Concurrent Enzyme Induction with Carbamazepine. Antimicrobial Agents and Chemotherapy, 53, 2346-2353.
http://dx.doi.org/10.1128/AAC.01120-08 - 18. Szalat, A., Gershkovich, P., Ben-Ari, A., Shaish, A., Liberman, Y., Boutboul, E., Gotkine, M., Hoffman, A., Harats, D., Leitersdorf, E. and Meiner, V. (2007) Rifampicin-Induced CYP3A4 Activation in CTX Patients Cannot Replace Chenodeoxycholic Acid Treatment. Biochimica et Biophysica Acta, 1771, 839-844.
http://dx.doi.org/10.1016/j.bbalip.2007.04.012 - 19. Ngaimisi, E., Mugusi, S., Minzi, O.M., Sasi, P., Riedel, K.D., Suda, A., Ueda, N., Janabi, M., Mugusi, F., Haefeli, W.E., Burhenne, J. and Aklillu, E. (2010) Long-Term Efavirenz Autoinduction and Its Effect on Plasma Exposure in HIV Patients. Clinical Pharmacology & Therapeutics, 88, 676-684.
http://dx.doi.org/10.1038/clpt.2010.172 - 20. Ngaimisi, E., Mugusi, S., Minzi, O.M., Sasi, P., Riedel, K.D., Suda, A., Ueda, N., Janabi, M., Mugusi, F., Haefeli, W.E., Burhenne, J. and Aklillu, E. (2011) Effect of Rifampicin and CYP2B6 Genotype on Long-Term Efavirenz Autoinduction and Plasma Exposure in HIV Patients with or without Tuberculosis. Clinical Pharmacology & Therapeutics, 90, 406-413.
http://dx.doi.org/10.1038/clpt.2011.129 - 21. Holzinger, E.R., Grady, B., Ritchie, M.D., Ribaudo, H.J., Acosta, E.P., Morse, G.D., Gulick, R.M., Robbins, G.K., Clifford, D.B., Daar, E.S., McLaren, P. and Haas, D.W. (2012) Genome-Wide Association Study of Plasma Efavirenz Pharmacokinetics in AIDS Clinical Trials Group Protocols Implicates Several CYP2B6 Variants. Pharmacogenetics and Genomics, 22, 858-867.
http://dx.doi.org/10.1097/FPC.0b013e32835a450b - 22. King, J. and Aberg, J.A. (2008) Clinical Impact of Patient Population Differences and Genomic Variation in Efavirenz Therapy. AIDS, 22, 1709-1717.
http://dx.doi.org/10.1097/QAD.0b013e32830163ad - 23. Sánchez, A., Cabrera, S., Santos, D., Valverde, M.P., Fuertes, A., Domínguez-Gil, A. and García, M.J. (2011) Population Pharmacokinetic/Pharmacogenetic Model for Optimization of Efavirenz Therapy in Caucasian HIV-Infected Patients. Antimicrobial Agents and Chemotherapy, 55, 5314-5324.
http://dx.doi.org/10.1128/AAC.00194-11 - 24. Bienvenu, E., Swart, M., Dandara, C., Ekman, A., Äbelö, A., Wonkam, A. and Ashton, M. (2013) Frequencies of Single Nucleotide Polymorphisms in Cytochrome P450 Genes (CYP1A2, 2A6, 2B6, 3A4 and 3A5) in a Rwandan Population: Difference to Other African Populations. Current Pharmacogenomics and Personalized Medicine, 11, 237-246.
http://dx.doi.org/10.2174/18756921113119990006 - 25. Luetkemeyer, A.F., Rosenkranz, S.L., Lu, D., Marzan, F., Ive, P., Hogg, E., Swindells, S., Benson, C.A., Grinsztejn, B., Sanne, I.M., Havlir, D.V. and Aweeka, F. (2013) Relationship between Weight, Efavirenz Exposure, and Virologic Suppression in HIV-Infected Patients on Rifampin-Based Tuberculosis Treatment in the AIDS Clinical Trials Group A5221 STRIDE Study. Clinical Infectious Diseases, 57, 586-593.
http://dx.doi.org/10.1093/cid/cit246 - 26. Kwara, A., Ramachandran, G. and Swaminathan, S. (2010) Dose Adjustment of the Non-Nucleoside Reverse Transcriptase Inhibitors during Concurrent Rifampicin-Containing Tuberculosis Therapy: One Size Does Not Fit All. Expert Opinion on Drug Metabolism & Toxicology, 6, 55-68.
http://dx.doi.org/10.1517/17425250903393752 - 27. Rwandan Ministry of Health (2009) Guidelines for the Provision of Comprehensive Care to Persons Infected by HIV in Rwanda. MoH, Kigali, 38-41.
- 28. ter Heine, R., Scherpbier, H.J., Crommentuyn, K.M., Bekker, V., Beijnen, J.H., Kuijpers, T.W. and Huitema, A.D. (2008) A Pharmacokinetic and Pharmacogenetic Study of Efavirenz in Children: Dosing Guidelines Can Result in Subtherapeutic Concentrations. Antiviral Therapy, 13, 779-787.
- 29. Bienvenu, E., Hoffmann, K.J., Ashton, M. and Kayumba, P.C. (2013) A rapid and Selective HPLC-UV Method for the Quantitation of Efavirenz in Plasma from Patients on Concurrent HIV/AIDS and Tuberculosis Treatments. Biomedical Chromatography, 27, 1554-1559.
http://dx.doi.org/10.1002/bmc.2959 - 30. Gengiah, T.N., Holford, N.H., Botha, J.H., Gray, A.L., Naidoo, K. and Abdool-Karim, S.S. (2012) The Influence of Tuberculosis Treatment on Efavirenz Clearance in Patients Co-Infected with HIV and Tuberculosis. The European Journal of Clinical Pharmacology, 68, 689-695.
http://dx.doi.org/10.1007/s00228-011-1166-5 - 31. Mould, D.R. and Upton, R.N. (2012) Basic Concepts in Population Modeling, Simulation, and Model-Based Drug Development. CPT: Pharmacometrics & Systems Pharmacology, 1, 1-6.
http://dx.doi.org/10.1038/psp.2012.4 - 32. Csajka, C., Marzolini, C., Fattinger, K., Décosterd, L.A., Fellay, J., Telenti, A., Biollaz, J. and Buclin, T. (2003) Population Pharmacokinetics and Effects of Efavirenz in Patients with Human Immunodeficiency Virus Infection. Clinical Pharmacology & Therapeutics, 73, 20-30.
http://dx.doi.org/10.1067/mcp.2003.22 - 33. Kappelhoff, B.S., van Leth, F., MacGregor, T.R., Lange, J., Beijnen, J.H. and Huitema, A.D. (2005) Nevirapine and Efavirenz Pharmacokinetics and Covariate Analysis in the 2NN Study. Antiviral Therapy, 10, 145-155.
- 34. Cohen, K., Grant, A., Dandara, C., McIlleron, H., Pemba, L., Fielding, K., Charalombous, S., Churchyard, G., Smith, P. and Maartens, G. (2009) Effect of Rifampicin-Based Antitubercular Therapy and the Cytochrome P450 2B6 516G>T Polymorphism on Efavirenz Concentrations in Adults in South Africa. Antiviral Therapy, 14, 687-695.
- 35. Ramachandran, G., Hemanth, K.A.K., Rajasekaran, S., Kumar, P., Ramesh, K., Anitha, S., Narendran, G., Menon, P., Gomathi, C. and Swaminathan, S. (2009) CYP2B6 G516T Polymorphism but Not Rifampin Coadministration Influences Steady-State Pharmacokinetics of Efavirenz in Human Immunodeficiency Virus-Infected Patients in South India. Antimicrobial Agents and Chemotherapy, 53, 863-868.
http://dx.doi.org/10.1128/AAC.00899-08 - 36. Li, T. and Chiang, J.Y. (2006) Rifampicin Induction of CYP3A4 Requires Pregnane X Receptor Cross Talk with Hepatocyte Nuclear Factor 4alpha and Coactivators, and Suppression of Small Heterodimer Partner Gene Expression. Drug Metabolism and Disposition, 34, 756-764.
http://dx.doi.org/10.1124/dmd.105.007575 - 37. Bienvenu, E., Swart, M., Dandara, C. and Ashton, M. (2014) The Role of Genetic Polymorphisms in Cytochrome P450 and Effects of Tuberculosis Co-Treatment on the Predictive Value of CYP2B6 SNPs and on Efavirenz Plasma Levels in Adult HIV Patients. Antiviral Research, 102, 44-53.
http://dx.doi.org/10.1016/j.antiviral.2013.11.011 - 38. To, K.W., Liu, S.T., Cheung, S.W., Chan, D.P., Chan, R.C. and Lee, S.S. (2009) Pharmacokinetics of Plasma Efavirenz and CYP2B6 Polymorphism in Southern Chinese. Therapeutic Drug Monitoring, 31, 527-530.
http://dx.doi.org/10.1097/FTD.0b013e3181ad74a4 - 39. Gounden, V., van Niekerk, C., Snyman, T. and George, J.A. (2010) Presence of the CYP2B6 516G> T Polymorphism, Increased Plasma Efavirenz Concentrations and Early Neuropsychiatric Side Effects in South African HIV-Infected Patients. AIDS Research and Therapy, 7, 32.
http://dx.doi.org/10.1186/1742-6405-7-32 - 40. Kwara, A., Lartey, M., Sagoe, K.W., Rzek, N.L. and Court, M.H. (2009) CYP2B6 (c.516G—>T) and CYP2A6 (*9B and/or *17) Polymorphisms Are Independent Predictors of Efavirenz Plasma Concentrations in HIV-Infected Patients. British Journal of Clinical Pharmacology, 67, 427-436.
http://dx.doi.org/10.1111/j.1365-2125.2009.03368.x - 41. Nyakutira, C., Röshammar, D., Chigutsa, E., Chonzi, P., Ashton, M., Nhachi, C. and Masimirembwa, C. (2008) High Prevalence of the CYP2B6 516G—> T(*6) Variant and Effect on the Population Pharmacokinetics of Efavirenz in HIV/AIDS Outpatients in Zimbabwe. European Journal of Clinical Pharmacology, 64, 357-365.
http://dx.doi.org/10.1007/s00228-007-0412-3 - 42. Kwara, A., Lartey, M., Sagoe, K.W., Kenu, E. and Court, M.H. (2009) CYP2B6, CYP2A6 and UGT2B7 Genetic Polymorphisms Are Predictors of Efavirenz Mid-Dose Concentration in HIV-Infected Patients. AIDS, 23, 2101-2106.
http://dx.doi.org/10.1097/QAD.0b013e3283319908 - 43. Kwara, A., Lartey, M., Sagoe, K.W., Xexemeku, F., Kenu, E., Oliver-Commey, J., Boima, V., Sagoe, A., Boamah, I., Greenblatt, D.J. and Court, M.H. (2008) Pharmacokinetics of Efavirenz When Co-Administered with Rifampin in TB/HIV Co-Infected Patients: Pharmacogenetic Effect of CYP2B6 Variation. Journal of Clinical Pharmacology, 48, 1032-1040.
http://dx.doi.org/10.1177/0091270008321790
List of Abbreviation
AIDS = Acquired immunodeficiency syndrome
CD4 = Helper T lymphocyte
CI = Confidence interval
CL/F = Oral clearance
CYP = Cytochrome P450
DDI = Drug-drug interactions
DNA = Deoxyribonucleic acid
DOT = Directly observed therapy
E = Ethambutol
EDTA = Ethylene diamine tetra-acetic acid
EFV = Efavirenz
FOCE = First-order conditional estimation
H = Isoniazid
HAART = Highly active antiretroviral therapy
HAARTPREV = Patients who had previous exposure to HAART
HIV = Human immunodeficiency virus
HWE = Hardy-Weinberg equilibrium
Ka = Absorption rate constant
KASP = Kompetitive Allele Specific PCR*
NNRTI = Non-nucleoside reverse transcriptase inhibitor
NONMEM = Non-linear mixed effects modeling
NRTI = Nucleoside reverse transcriptase inhibitor
OFV = Objective function
PCR* = Polymerase chain reaction
PK = Pharmacokinetics
R = Rifampicin
RBT = Rifampicin-base tuberculosis therapy
RNA = Ribonucleic acid
RNEC = Rwanda National Ethics Committee
SD = Standard deviation
SNP = Single nucleotide polymorphism
TB = Tuberculosis
TBPREV = Patients who had TB treatment prior to HAART
UGT = Uridine diphosphate glucuronosyltransferase
V/F = Volume of distribution
VPC = Visual predictive check
WHO = World Health Organization
Z = Pyrazinamide
NOTES

*Corresponding author.