Pharmacology & Pharmacy
Vol.06 No.03(2015), Article ID:54783,30 pages
10.4236/pp.2015.63017
Natural Products Modulate the Multifactorial Multidrug Resistance of Cancer
Safaa Yehia Eid1,2, Mahmoud Zaki El-Readi1,2,3*, Sameer Hassan Fatani1, Essam Eldin Mohamed Nour Eldin1, Michael Wink2*
1Department of Biochemistry, Faculty of Medicine, Umm Al-Qura University, Makkah, Kingdom of Saudi Arabia
2Institute of Pharmacy and Molecular Biotechnology, Heidelberg University, Heidelberg, Germany
3Department of Biochemistry, Faculty of Pharmacy, Al-Azhar University, Assiut, Egypt
Email: *mzreadi@uqu.edu.sa, *wink@uni-heidelberg.de
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 4 January 2015; accepted 12 March 2015; published 18 March 2015
ABSTRACT
Multidrug resistance (MDR) is a critical problem in cancer chemotherapy. Cancer cells can develop resistance not only to a single cytotoxic drug, but also to entire classes of structurally and functionally unrelated compounds. Several mechanisms can mediate the development of MDR, including increased drug efflux from the cells by ABC-transporters (ABCT), activation of metabolic enzymes, and defective pathways towards apoptosis. Many plant secondary metabolites (SMs) can potentially increase sensitivity of drug-resistant cancer cells to chemotherapeutical agents. The present thesis investigates the modulation of MDR by certain medicinal plants and their active compounds. The inhibition of ABCTs (P-gp/MDR1, MRP1, BCRP) and metabolic enzymes (GST and CYP3A4), and the induction of apoptosis are useful indicators of the efficacy of a potential medicinal drug. The focus of this study was the possible mechanisms of drug resistance including: expression of resistance proteins, activation of metabolic enzymes, and alteration of the apoptosis and how to overcome their resistance effect on cancer cells. The overall goal of this review was to evaluate how commonly used medicinal plants and their main active secondary metabolites modulate multidrug resistance in cancer cells in order to validate their uses as anticancer drugs, introduce new therapeutic options for resistant cancer, and facilitate the development of their anticancer strategies and/or combination therapies. In conclusion, SMs from medicinal plants exhibit multitarget activity against MDR-related proteins, metabolic enzymes, and apoptotic signaling, this may help to overcome resistance towards chemotherapeutic drugs.
Keywords:
Multidrug Resistance (MDR), ABC-Transporters (ABCT), Metabolic Enzymes, Apoptosis,
CytochromeP3A4 (CYP3A4), Glutathione-S-Transferase (GST)

1. Cancer
Cancer is the second leading cause of death, after heart disease, killing about every fifth or sixth person in western countries. Every year, more than 8.2 million cancer deaths are reported worldwide. Of the 14.1 million new cases each year, more than half occur in developing countries and 32.5 million persons are alive with cancer [1] . WHO predicts 23.6 million cases in 2030, 68% of which to be occurring in developing countries.
Cancer is defined as a disease in which normal tissue is invaded by abnormally dividing cells. Left untreated, it will spread throughout the body and becomes fatal. Chemotherapy (the use of cytotoxic agents to slow the progression of this uncontrolled cell division) is the major treatment when cancer is well established within the patient [2] . Resistance to anticancer drugs is a major problem in chemotherapy with 30% - 80% of cancer patients developing resistance to chemotherapeutical drugs [3] . Thus, counteracting drug resistance is crucial to providing the best treatment.
2. Multifactorial Multidrug Resistance
Cancer cells can not only develop resistance to one drug, but also to entire classes of drugs with similar mechanisms of action. After such resistance is established, some cells even become cross resistant to drugs, which are structurally and mechanistically unrelated; this phenomenon is known as multidrug resistance (MDR). This might explain why the treatment with multiple agent combinations addressing different targets is not effective. The main lines of cellular defense reactions involve:
・ Decreased drug accumulation by enhanced cellular elimination, decreased uptake, and inactivation by intracellular metabolism (Figure 1). One example of the developmental resistance mediated by reduced drug uptake is water-soluble drugs that “piggyback” on transporters and carriers that bring nutrients into the cell [4] [5] .

Figure 1. Cellular causes of drug resistance. Cells may develop resistance to anticancer drugs by increasing efflux pumps activity (like as ATP-dependent transporters) to remove the drug, reduced drug influx (e.g., by “piggyback” on intracellular carriers), activating detoxifying proteins (i.e. cytochrome P450 mixed-function oxidases), repairing their own DNA damage, disrupting apoptotic signalling pathways, or altered cell cycle checkpoints [6] .
・ Impaired drug delivery can result from poor absorption, increased drug metabolism, or increased excretion, resulting in lower levels of the drug in the blood and reduced diffusion of drugs from the blood into the tumor mass [7] [8] . The various causes of drug resistance can work simultaneously, increasing the resistance in a multifactorial manner. For example, the simultaneously induction of CYP3A4 and MDR1 was observed [9] . This type of multidrug resistance can be induced after exposure to any drug. Recent evidence indicates that certain nuclear receptors, such as PXR, might be involved in mediating this response to environmental stress while also acting in regulating metabolic enzymes and ABC transporters [10] .
・ Activation of DNA repair, due to genetic and epigenetic alterations and affecting drug sensitivity.
・ Alteration or modification of the drug targets.
・ Resistance resulting from defective apoptotic pathways. This might occur as a result of malignant transformation; for example, in tumors with mutable nonfunctional p53 [11] .
Alternatively, cells may acquire changes in apoptotic pathways during exposure to chemotherapy, such as an alteration of ceramide levels or changes in the cell-cycle machinery, which activate checkpoints and prevent initiation of apoptosis [6] [12] . In addition, cancer cells, which survive cytotoxic drugs, are likely to be heterogeneous due to their mutated phenotypes. All these causes are involved multifactorial multidrug resistance (Figure 1) [6] .
Current research is centering on how to combat this multifactorial resistance. Thus, we must study multiple means of drug resistance to develop the potent treatment options. Different types of cellular MDR have been identified [13] . They have been broadly classified into, cellular, and noncellular mechanisms, as detailed below.
3. Noncellular Resistance Mechanisms
Noncellular cytotoxic resistance mechanisms, such as in vivo tumor growth [14] , environmental factors [15] , or tumor geometry [16] are also worth further study, but are not the focus of this study.
4. Cellular Mechanisms of Multidrug Resistance
Detailed studies have been devoted to the cellular mechanisms of drug resistance. It is easy to generate in vitro models with cytotoxic drugs. Cellular mechanisms are categorized in terms of alterations in the biochemistry of malignant cells. Such mechanisms can be further classified into two major categories:
1) transport-based classical MDR phenotypes and 2) nonclassical MDR phenotypes.
4.1. Transport-Based Classical MDR Mechanisms
The biological membrane is a lipid bilayer into which numerous proteins are embedded, including transporter proteins. The activities of these transporters are important determinants for the pharmacokinetics and pharmacodynamics of many drugs. Considerable knowledge about these transporters has been gained over the past decade, including their functional characteristics, substrate specificities, and their specialized tissue distribution and subcellular localization [17] . This section will highlight the role of these transporters in MDR.
Classical multidrug resistance exerts increased drug efflux and lowers intracellular drug concentrations. ATP- dependent efflux pumps, belonging to ATP-binding cassette (ABC) transporters (largest family of transmembrane proteins), express broad drug specificity. Today, there are 48 identified human ABC genes. These are divided into seven distinct subfamilies (ABCA-ABCG) based on of their domain organization and sequence homology [18] . ABC transporters are responsible for the ATP-dependent movement of a wide variety of xenobiotics (including cytotoxic drugs), lipids, and metabolic products across the plasma and intracellular membranes [19] [20] . The cytotoxic drugs that are most frequently associated with classical MDR are hydrophobic, amphipathic natural products, such as microtubule-stabilizing taxanes (paclitaxel and docetaxel), vinca alkaloids (vinorelbine, vincristine, and vinblastine), anthracyclines (doxorubicin, daunorubicin, and epirubicin), epipodophyllotoxins (etoposide and teniposide), antimetabolites (methotrexate, fluorouracil, 5-azacytosine, 6-mercap- topurine, and gemcitabine) topotecan, and the RNA transcription inhibitor actinomycin-D [21] .
4.1.1. P-Glycoprotein/Multidrug Resistance 1 (P-gp/MDR1)
Two highly hydrophobic integral membrane domains (each of which spans the membrane six times by alpha helices) and two hydrophilic nucleotide binding domains (NBDs) make up the four distinct parts of MDR1 (Figure 2).

Figure 2. The structures of three categories of ABC transporter known to confer drug resistance. Sample (a) shows ABC transporters (such as MDR1, MRP4, etc), which have 12 transmembrane domains and 2 ATP- binding sites. (b) Depicts those ABC transporters which have 2 ATP-binding domains and a domain consisting of five transmembrane segments at the amino-terminal end; there is a total of 17 transmembrane domains in these transporters (MRP1-6). Finally, (c) shows a “half-transporter” ABCG2. This half transporter, which is believed to function by homodimerizing or heterodimerizing, contains only one ATP-binding region and six transmembrane domains [22] .
A 2.5-nm resolution structure of this broad-spectrum multidrug efflux pump was recently obtained by electron microscopy and single particle image analysis. This shows a large central pore, ~5 nm in diameter, which closes at the inner (cytoplasmic) side of the membrane.A gap may be present in the protein ring, which may allow substrates to access the central pore from the lipid phase [3] [19] (Figure 3).
4.1.2. Mechanism of the Efflux Pump
MDR1 efficiently removes cytotoxic drugs and many commonly used pharmaceuticals from the lipid bilayer. For many years, the model for drug resistance conferred by MDR1 has been a relatively simple one. Cytotoxic drugs are actively transported out of cells that express MDR1 against a concentration gradient, thereby reducing intracellular drug accumulation and inhibiting drug-mediated cell death (Figure 3) [23] .
Initial mechanistic models define efflux of drugs by MDR1 hypothesized that MDR1 forms a hydrophilic pathway, and drugs are transported from the cytosol to the extracellular media through the middle of a pore, thereby shielding the substrate from the hydrophobic lipid phase (Figure 3(a)).
In summary, in order to transport one drug molecule, two ATP hydrolysis events must occur. The hydrophobic substrates that are either neutral or positively charged bind to the transmembrane domains in order to stimulate the ATPase activity of P-gp/MDR1 and cause substrate to be released to either the outer leaflet of the membrane or extracellular space. Then the transporter is required to be “reset” by hydrolysis so that it can bind substrate again. These two events complete in the catalytic cycle [24] .
It appear that P-gp aids in MDR by intercepting a drug as it moves through the lipid membrane then “flipping” the drug so that it effluxes from the cells through the outer leaflet into the extracellular space (Figure 3(b)) [23] . This “flippase” function has also been observed in several related ABC molecules, leading to the assumption
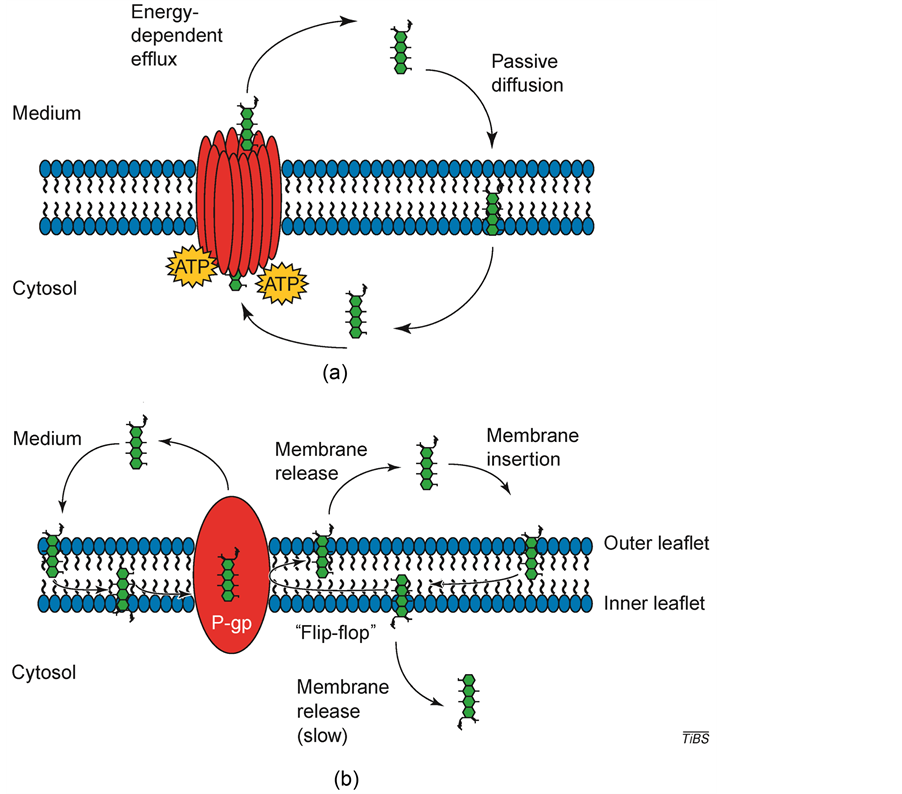
Figure 3. Possible mechanisms of action for drug efflux by P-glycoprotein/MDR1 (P-gp/ MDR1). (a) The “pump” model for drug transport. The three-dimensional structure of MDR1 consists of a single drug pore (shown in red). Chemotherapeutic drugs (green) diffuse through the lipid membrane and are transported out of the cell by P-gp in an ATP-dependent manner. (b) The “flippase” model for drug transport. A drug interacts with lipids of the membrane before it interacts with MDR1. The drug can then interact with MDR1 and is transported from the inner leaflet directly into the extracellular medium. Alternatively, drug intercalated into the inner leaflet of the lipid bilayer is “flipped” into the outer leaflet and released into the extracellular space. Movement of drug from the inner to the outer leaflet is a relatively quick process, whereas drug movement from the inner leaflet to the cytosol is relatively slow [23] .
that they comprise a large, polymorphous drug-binding domain [23] . P-gp/MDR1 also binds with various hydrophobic compounds, making it simple to find potent P-gp/MDR1 inhibitors. Two such inhibitors being studied for their reversal effect on MDR are verapamil (a calcium channel blocker) and cyclosporine A (an immunosuppressant).
4.1.3. Multidrug Resistance Protein (MRP)
Multidrug resistant cells have been found which contain no P-gp/MDR, but instead the efflux pumps protein, multidrug-resistance-associated protein 1 (MRP1, or ABCC1) [25] . MRP1 works similarly to P-gp/MDR1, it transport transports glutathione and its conjugates, cotransports unconjugated glutathione, and recognizes both neutral and anionic hydrophobic natural products. Structurally, it is very similar to P-gp/MDR1 with an added amino terminal containing five-membrane spanning domains attached to the core (Figure 2) [26] . The discovery of MRP1 spurred further research, which led to the discovery of 8 additional members of the ABCC subfamily of transporters [27] . Many of these have a potential in fighting drug resistance [27] .
4.1.4. Breast Cancer Resistance Protein (BCRP)
Mitoxantrone, is a chemotherapeutical agent, but poor substrate for MDR1 and MRP1. It is a selective substrate for ABCG2 protein, which called MXR (mitoxantrone-resistance gene), BCRP (breast cancer resistance protein) [28] . Structurally, BCRP is a homodimer of two half-transporters, each containing an ATP-binding domain at the amino-terminal end of the molecule and six transmembrane segments (Figure 2). The substitution of even a single amino acid can change substrate specificity of P-gp/MDR1 [21] . As observed in ABCG2 gene cloning from resistant cells encoded proteins by substituted arginine at amino acid 482 by threonine or glycine led to increase the ability to transport doxorubicin [29] [30] .
4.1.5. ABC Transporters in Normal Cells and Their Physiological Function
MDR1 is found in both tumor cells (where it contributes to MDR) and normal cells. It is generally found in cell tissues with excretory (kidney, liver, adrenal gland) and barrier functions (intestine, blood brain barrier, pla- centa, testis and ovarian) [31] (Figure 4). These placements allow to suggesting that MDR1 has a role in detoxi- fication and protection of the body against toxic drug and metabolites by secreting these compounds into bile, urine, and the intestinal lumen and by blocking their accumulation in the brain, testis, and fetus. The general function of ABC transporters is the protection of cells from many endogenous or exogenous toxins, as detailed below (Figure 4) [32] :
・ P-gp/MDR1 plays a role in preventing cytotoxines from crossing the endothelium and attacking the brain [33] .
・ MRP proteins are localized to the basolateral membrane of the choroid plexus, and pump the metabolic waste products of CSF into the blood.
・ MDR1 protects the testis by transporting toxins to the capillary lumen.

Figure 4. Schematic representations of the main sites of localization of P-gp/MDR1 in the body [32] .
・ MDR1 is localized on the apical syncytiotrophoblast surface in the placenta; it can protect the fetus from cationic xenobiotics [34] .
・ MRP and ABCG2 are also localized in the placenta [35] [36] ; they appear have protecting functions for fetal blood [37]
・ MDR1 protects the liver by transporting toxins into the bile [38] .
・ MDR1 is localized in apical membranes of inessential mucosal cells; it has a role in determining oral drug bioavailability [39] .
・ MRP1 is located in the basolateral membrane of mucosal cells; it transports substrates into the blood, rather than across the apical surface into the intestinal lumen [40] .
The protective mechanism of ABC transporter-mediated extrusion of such toxic substances (natural drugs or metabolic waste products) causes cancer cells to be resistant to the toxic effects of many chemotherapeutic agents. ABC transporters not only protect vital tissue from toxins (as in the liver, GIT, and kidneys), they also actively excrete toxins. This function must be considered when determining the bioavailability of oral drugs. In short, ABC transporters effectively protect both normal and cancerous cells. Their protective properties must be circumvented in order to effectively destroy malignant cells.
4.1.6. ABC Transporters in Human Cancers
Early research into ABC transporters focused almost exclusively on P-gp because it is so highly expressed in colon, kidney, adrenocortical, and hepatocellular cancers [41] . Traditionally, research has focused on expression impairing response to chemotherapy, expression levels increasing, as tumor become more drug resistant. The idea was that tumors depending on P-gp expression for survival have the highest rates of MDR1 expression [22] . Initial hopes were that decreasing only P-gp expression could solve the multidrug resistance problem. It became clear that multiple factors (not solely MDR1) are involved when the aforementioned cancers failed to respond to drugs that are not P-gp substrates.
Patient’s studies have shown a correlation between P-gp/MDR1 expression and drug resistance, being strongest in acute myelogenous leukemia (AML). MDR1 expression is present in approximately 30% of newly diagnosed leukemia patients and 50% of those in relapse [42] [43] . In vivo studies have shown that MDR1 expression reduces the intracellular accumulation of doxorubicin and that this can be counteracted by use of a MDR1 inhibitor [44] [45] .
Research has also focused on MRP1 and LRP expression, however, unlike with MDR1 [46] , there has not yet been an indication that these have an effect on prognosis, but observed a correlation between MDR1 expression and prognosis [43] . Interestingly, AML cells have been observed to have low expression levels of BCRP/MXR [47] . Treatment of colorectal cancer, the leading cause of cancer-related death in the western world, has not yet begun to accurate identify subgroups of patients whose differing forms of the disease will respond to varying treatments. The expression of proteins responsible for active processes in Caco-2 cells can also be induced by xenobiotics. This is probably due to the cancerous origin of Caco-2, since e.g. vinca alkaloids induce MDR1 in these cells, according to the concept of multidrug resistance [48] .
Induction of many other phase I and phase II metabolic enzymes has also been reported [49] -[51] . The roles of several active uptake and efflux proteins and metabolic enzymes (as well as the interplay between them) should be studied in order to better understand MDR in Caco-2 cells. Past studies have focused primarily on the easily observable in vitro flavonoids. Pharmacokinetic models of Caco-2 cells have recently been construed [52] [53] .
4.1.7. ABC-Transporter Substrates
MDR1 transports a broad range of substrates, which include several pharmacologically distinct agents used not only for chemotherapy but also for hypertension, allergy relief, infections, immunosupression, neurology, and inflammation.
The structure-activity relationship of these substrates is not clear, though it appears lipophilicity and hydrogen bonds correlate proportionally to the affinity of MDR1 [54] . Since the substrates appear to be freely diffused into the cells, they can be found by looking for MDR1 in the plasma membrane, as it appears from in vitro testing that MDR1 recognizes its substrates before they enter the cytoplasm [55] . Studies find that many substrates of MDR1 are also substrates of drug-metabolizing enzymes, such as CYP3A4. This overlap may be to some extent a result of a coordinated regulation of tissue expression of CYP3A4 and MDR1 (both of which are located in close proximity on the same chromosome) in organs such as the liver and intestine [56] . However, such overlap is not always present; some MDR1 substrates, such as digoxin, fexofonadine, celiprolol, and tarlinolol do not significantly interact with CYP enzymes and others, such as midazolam, are not transported by MDR1 [56] -[58] .
4.1.8. ABC-Transporter Inhibitors
Historically research has focused on the ABC superfamily when searching for the reasons of chemotherapy failure. Only a small number of transporters obtained from patients at diagnosis have been investigated. These studies confirmed that MDR1 is relevant in several cancers; however, the relevance of other transporters is not yet clear [59] . As research in this area continues, developing ABC transport inhibitors appears to be a useful addition to chemotherapy.
Many studies have tried to combat MDR by inhibiting MDR transporters, suppressing MDR mechanisms, or circumventing MDR mechanisms. There are many MDR modulators belonging to several chemical classes, including calcium channel blockers, calmodulin inhibitors, coronary vasodilators, indole alkaloids, quinolines, hormones, cyclosporines, surfactants, and antibodies [60] . These have risks associated with needing very high doses of toxic drugs in order to produce needed results [61] . To minimize these risks, new analogs of these early chemosensitizing agents were tested and developed with the goal of finding patent MDR1 modulators, which require less toxicity. Other avenues to explore are: using anticancer drugs which are not substrates of ABC transporters (such as alkylating drugs, antimetabolites, and anthracycline modified drugs) [62] [63] . Such compounds are called MDR inhibitors, MDR modulators, MDR reversal agents, or chemosensitizers and they may inhibit one or more transporters. Excellent examples of chemosensitizers (e.g. limonin) are included in this study [64] .
Chemosensitization involves the co-administration of an MDR1 inhibitor (MDR modulator) with an anticancer drug in order to cause enhanced intracellular anticancer drug accumulation via impairing the MDR1 function. Many early chemosensitizing agents were themselves substrates for MDR1 and worked by keeping the MDR pump too busy effluxing the inhibitor to fully efflux the cytotoxic drugs [21] . There are advantages of using chemosensitizing agents when they exhibit affinity for the same substrates and interfering with other enzyme systems and transporters [61] [65] .
MDR1 inhibitors, like MDR1 substrates, show no relation between their chemical structures or pharmacological actions and their inhibitory effects. Reports indicate a strong correlation between physicochemical parameters and MDR1 inhibition. An MDR1 modulator is considered a good candidate if it possesses a log p value of 2.92 or higher, a molecular axis of 18 or more atoms in length, one or more tertiary basic nitrogen atoms, and high energy in its highest occupied orbit [66] .
Inhibition of MDR1 could potentially result from the blockage of specific recognition of the substrate, binding to ATP, ATP hydrolysis, or coupling of ATP hydrolysis to translocation of the substrate. Most reversing agents block MDR1 by acting as competitive or noncompetitive inhibitors [67] and by binding either to drug interaction sites, [68] or to other modulator binding sites, leading to allosteric changes. Competitive modulators compete as a substrate with the cytotoxic agent for transport by the pump. This limits the efflux of the cytotoxic agent, increasing its intracellular concentration.
Noncompetitive inhibition of the MDR1 transporter binds with high affinity to the pump but is not itself a substrate. This induces a conformational change in the protein, thereby preventing ATP hydrolysis and transport of the cytotoxic agent out of the cell, resulting in an increased intracellular concentration. MDR modulators such as verapamil are substrates of MDR1, which inhibit the transport function in a competitive manner without interrupting the catalytic cycle of MDR1 [69] . Cyclosporin A, as one of the reversing agents, inhibits MDR1 function by interfering with both substrate recognition and ATP hydrolysis [70] . Because ATP hydrolysis is required for transport, modulators that inhibit ATPase activity are unlikely to be transported by MDR1 [71] . Verapamil inhibits the function of MDR1, making malignant cells more susceptible to cytotoxic drugs [72] . Besides the functional inhibitory effect on MDR1 and down-regulate the MDR1 gene in leukemic cell lines [73] . In this thesis, verapamil was used to inhibit MDR1 as a positive control.
Clinical trials helped to unravel the problems associated with combining together with an MDR inhibitor. The first factor to be determined before a clinical trial is to identify the ABC transporter protein involved in drug resistance. The second factor is to monitor the plasma concentrations and in vivo effectiveness of the tested MDR inhibitor. The pharmacokinetic interaction between the anticancer drug(s) and the MDR inhibitor must be searched and avoided to prevent a reduction in anticancer drug dosage [74] .
4.2. Nonclassical MDR Phenotypes
Mechanisms of drug resistance involving membrane-associated protein pumps, although the most thoroughly characterized, are not the only means by which drug resistance can arise within tumor cells. Clinical studies investigating other drug-resistance mechanisms (called non-classical MDR), are fewer in number, but are no less important. These non-transport mechanisms affect multiple drug classes. This type of resistance can be caused by the altered activity of specific enzyme systems (such as cytochrome P450 (CYP3A4) and glutathione-S- transferase (GST)), which can decrease the cytotoxic activity of drugs in a manner independent of intracellular drug concentrations. In addition, changes in the balance of proteins that control apoptosis can also reduce chemosensitivity since most anticancer drugs are exert their cytotoxicity via apoptotic processes. This section outlines some of these MDR mechanisms and their role in the overall MDR phenomenon.
4.2.1. Cytochrome P450
Since it is estimated that the enzyme CYP (cytochrome P-450) metabolizes (in full or in part) over half of all therapeutic drugs, it is essential that we gain a clear understanding of how CYP3A4 gene expression is regulated. Changes in the hepatic expression of CYP3A4 can significantly impact drug metabolism and thus the pharmacokinetics of medications [75] .
CYP’s main function is catalyzing the metabolic conversion of xenobiotics. Catalyzing helps convert those to polar derivatives (which can be easily excreted) or causes them to become more desirable substrates, which further then biotransformation of phase П enzymes or drug transporters. To clarify, in phase I, a lipophilic molecule is made more hydrophilic by introducing hydroxyl groups by cytochrome P450 oxidases (CYP); other CYPs. They cleave N-methyl, O-methyl, or methylene groups in order to obtain a more hydrophilic or more readily accessible substrate. In phase II, the hydroxylated compounds are conjugated with polar molecules, such as glutathione, sulfate, and glucuronic acid. These conjugates are eliminated via the kidneys and urine.
Several chemomodulating agents inhibit cytochrome P-4503A (CYP3A) activity in addition to inhibiting MDR proteins. Some drugs (such as tamoxifen, methoxymorpholinyl doxorubicin, cyclophosphamide and ifosfamide) requiring activation by CYP3A4 may have a reduced therapeutic effect even when MDR1 inhibition enhances their intracellular accumulation.
On the other hand, some drugs that depend on CYP3A4 for excretion can build up to overly toxic a mounts and the dose may need to be reduced. Therefore, it is essential to understand how CYP3A4 interacts with MDR1 inhibitors when using them in combination with cytotoxic drugs. In addition, reversal agents, which have MDR1 inhibition effects, also inhibit cytochrome P-450 3A (CYP3A) activity [56] . Herbs or drugs may inhibit CYPs by three mechanisms: competitive inhibition, noncompetitive inhibition, and mechanism-based inhibition [76] .
Competitive inhibition may occur between drug and herbal/drug, which are metabolized by the same CYPs enzyme. Noncompetitive inhibition is caused by the binding to the haem portion of the CYPs enzyme this occurs by herbal/drug, which containing electrophilic groups (e.g. hydrazine or imidazole). The formation of a complex between CYPs and herbal/drug metabolites is the main cause of the mechanism-based inhibition of CYPs. Ever since the initial cloning of this receptor, the implications of PXR mediated gene regulation in drug, metabolism and drug-drug interactions were recognized. PXR was first postulated to regulate CYP3A gene expression in both human and rodents [77] (Figure 5).
On the other hand, PXR participates in the modulating metabolic processes and the regulation of drug transporters responsible for both efflux and uptake of endogenous and exogenous chemicals [78] .
Mechanisms involving PXR regulation of efflux transporters tend to be more commonly studied, given that efflux transporters are often postulated as the major blockades for drugs to achieve therapeutic concentrations by crossing barriers such as the blood-brain barrier into the central nervous system, or the placenta during pregnancy [80] (Figure 5).
4.2.2. Glutathione-S-Transferease (GST)
Another component of MDR is the catalytic glutathione-S-transferase (GSTs). Several resistant cell lines have been shown to overexpress GST [81] . GSTs are detoxification enzymes conjugated of glutathione to the electrophilic center of various drugs and resulting in excretion of polar molecules. GSTs are able to metabolized cisplatin, doxorubicin, and melphalan and protecting the cells from environmental or oxidative stress [82] .
There are two intracellular pools of GST, one residing in the cytosol and the other in the microsomal com-

Figure 5. Schematic illustration of the activation mechanisms and target genes of CAR and PXR. Activation of PXR is purely ligand dependent. CAR and PXR shared target genes are grouped in a red box, CAR-specific targets in a blue box, and PXR-specific targets in a purple box [79] .
partment. Cytosolic GSTs are composed of 23-29-kDa subunits which may be homo or hetero-dimers [83] . In contrast, microsomal GSTs are trimeric and composed of identical 17-kDa subunits [84] . Among its various activities, GST plays an important role in protecting cells from reactive epoxides [85] . This is believed to occur via the catalytic addition of GSH to the epoxide moiety, as observed by the metabolism of aflatoxin B1 to an 8,9- epoxide which is detoxified by GST [86] . GST is the predominant enzyme found in ovarian carcinoma and several studies have been performed to determine whether levels of this enzyme have prognostic significance, where the levels of GSTs were correlated to a poor response to chemotherapy.
The first linkage between PXR and GSTs observed in rat GSTA2, GSTA2 expression was suppressed at nanomolar concentration of DEX (GR activation) in cultured hepatocytes, but induced by DEX at micromolar (PXR activation) concentrations or RU486, which is a GR antagonist and PXR agonist [87] (Figure 5). More recently, studies have focused on the inhibition of apoptosis as the so-called de novo mechanism of drug resistance.
4.2.3. Apoptosis
In the last few years, clinical trials using plant-derived drugs for prevention and/or treatment of tumors have become increasingly widespread in cancer therapy. The search for novel agents designed to induce cell cycle arrest and apoptosis in cancer cells is being seriously pursued. The ability of tumor cells to evade apoptosis plays a significant role in their resistance to conventional therapeutic regimens [88] . Apoptosis describes the terminal morphological and biochemical events seen in programmed cell death (PCD). Apoptotic cells are characterized by morphological changes including: cellular shrinkage, plasma and nuclear membrane blebbing, organelle relocalization and compaction, nuclear DNA condensation with or without fragmentation, and hypersegmentation of nuclear chromatin of irregular size. These nuclear structures may then bud from the rapidly blebbing cell surface to form “apoptotic bodies” [89] .
Three mechanisms are known to be involved in the apoptotic process: 1) a receptor-ligand mediated mechanism, 2) a mitochondrial pathway and 3) a mechanism in which the endoplasmic reticulum plays a central role (Figure 6). All three mechanisms activate caspases, which are responsible for the characteristic morphological changes observed during apoptosis. Cell death marked by cellular swelling is called oncosis. Necrosis cell deaths various from apoptosis in several ways. Necrosis results from direct injury, which usually begins at the cell sur- face. Early lysis of the plasma membrane is exhibited by necrotic cells before any significant alterations in

Figure 6. Apoptosis can be induced through the activation of death receptors including Fas, TNFαR, DR3, DR4, and DR5 by their respective ligands. Specialized adaptor proteins are recruited as a result. Caspase cascades are also, activated. Fas trimerization is induced by the binding of FasL leading to the initiation (via the adaptor protein FADD) of caspase-8. Then, oligomerzes activated by autocatalysis. This activation stimulates apoptosis in two parallel cascades, which can bind directly and activate caspase-3 or cleave Bid. Next, truncated Bid (tBid) induces cytochrome c release by translocation to mitochondria, which sequentially activating caspase-9 and -3. TNFαR and DR3 (which can deliver pro-or anti-apoptotic signals) encourage apoptosis by use of TRADD/FADD (adapter proteins) and activating caspase-8. TNF-α interacts with TNFαR, which may result in the activation of a NF-κB pathway via NIK/IKK. The expression of prosurvival genes (such as FLIP, which can directly inhibit caspase 8 activation, and NF-κB) is induced by the activation of NF-κB. Additionally FasL and TNF-α, Bcl-2 (by phosphorylation), may activate JNK (via ASKI/MKK7).
nuclear morphology. Necrotic cells also have different surface futures than apoptotic cells. Necrotic cells are characterized by swelling and lysing, in contrast apoptotic cells have intense cell surface zeioticblebbing [90] . Genetically, mutation disturbs apoptosis. It believed that defects in apoptotic pathways contribute to several human diseases, including neurodegenerative disorders to malignancy. A high frequency of apoptosis in spontaneously regressing tumors and in tumors treated with cytotoxic anticancer agents, has been shown in multiple studies [91] .
Thus, we can conclude that anticancer agents may induce apoptotic cell death. It follows that even after the drug-target interaction and cellular responses can have an impact on drug-induced cell death [92] as shown in Figure 6. The caspase-cascade system plays a vital role in the induction, transduction, and amplification of in- tracellular apoptotic signals. Caspases, closely associated with apoptosis, are aspartate-specific cysteine proteases and members of the interleukin-1β-converting enzyme family. The activation and function of caspases, involved in the delicate caspase-cascade system, are regulated by various kinds of molecules, such as the inhibitor of apoptosis protein, Bcl-2 family proteins, Ca2+, and calpain [93] . Not all caspases are involved in apoptosis. The caspases that have been well described are caspase-3,-6,-7,-8, and caspase-9 [94] . Caspase-3 is a key factor in apoptosis execution.
Apoptosis is believed to be started when mitochondrial damage leads to a release of cytochrome C and the activation of Apaf-1, when in turn activates the caspase machinery responsible for executing apoptosis. The chain of events causing this mitochondrial damage begins when procaspase-3 activated (by caspase-3,-8,-9,-10, cysteine protease protein of molecular mass 32 KDa, granzyme B, etc.). Caspase-3 downstream substrates include various components of the cytoskeleton as well as: procaspase-3, procaspase-6, procaspase-9, DNA protein kinase (DNA-PK), protein kinase C (PKCγ), poly(ADP-ribose) polymerase (PARP), D4-GDP-dissociation inhibitor for the Rho family GTPases (D4-GDI) [95] (Figure 6).
It is important that we have this understanding of how apoptosis works because it has effects on tumorgenisis and cancer treatment. Inhibiting apoptosis can make cancer cells more resistant to treatment with chemotherapy and radiotherapy, the cells often increase survival proteins to protect themselves and inhibit apoptosis. On the other hand, apoptosis defects have been shown to increase the number of neoplastic cells.
Two hypotheses can propose the interplay between MDR expression and apoptosis inhibition. First, the expressing of a large molecule (such as MDR1) might disturb the content and context of plasma membrane, thereby interfering with DISC formation. Briefly, the ligation of cell death receptor molecules (e.g. Fas or TNF receptor), recruit the adapter proteins, which followed by the formation of death inducing signaling complex (DISC). This in turn triggers the caspase cascade. It is important to note that interfering with this DISC formation may significantly inhibit subsequent caspase formation.
Second hypothesis is that MDR expression alters intracellular pH (pHi), which contributes to MDR1 gaining resistance to several forms of caspase-dependant cell death stimuli resulting in lower drug concentrations. Briefly, as MDR1-correlates to an increase in pHi, and the transmembrane partitioning or intracellular sequestration is altered. This happens at the intracellular acidification stage, which often precedes apoptosis. Increasing the intracellular pHi can inhibit DNA fragmentation, and decrease the normal sensitivity of cells to inducing the apoptosis as response to Fas cross-linking or serum starvation and cells becoming resistant to caspase-depen- dent-death stimuli [23] [96] [97] .
5. Plant-Derived Natural Products
Throughout history, humankind has used natural products found in plants, animal, microorganisms, etc. to treat disease. Increasingly, scientists are investigating the active ingredients in natural remedies (called secondary metabolites), to further develop effective drug treatments. Much work remains to be done as it is estimated that only 5% - 15% of the approximately 250,000 known plant species have been studied for their bioactive SMS [98] . Interestingly, even as production of and demand for synthetic drugs has been increasingly high, 80% of the world’ s population rely primarily on traditional herbal medicines [99] .
6. Cancer and Natural Products
Increased cancer mortality and the high cost of treatment spur a continued search for better anticancer drugs. In recent decades, natural compounds have attracted considerable attention as cancer chemopreventive agents and as cancer therapeutics [100] . Some of the most effective cancer treatments to date are natural products, or compounds derived from natural products.The first natural product used as an anticancer compound was when podophyllotoxin was islolated from Podophyllumpeltatum in 1947. Later, etoposide and teniposide (chemical derivative), vinca alkaloid (vinblastine and vincristine), andpaclitaxel (Taxol®), were discovered as active principleof Taxusbrevifolia [101] . Natural products and their synthetic derivatives comprise over 77% (63/81) of the approved anticancer drug candidates developed between 1981 and 2006 (Figure 7) [102] . This combined per-

Figure 7. Role of natural products in anticancer.
centage highlights the importance of natural products to drug development.
Our laboratory’s focus is on isolation, identification, and investigation of the biological activities of interests in natural product uses against cancer. There are three main reasons why our laboratory workers believe researches into natural compounds is worthwhile:
First, natural compounds that show anticancer potential fit into the mechanism-based approach as perfectly as a hand fits into a glove. There is solid evidence that these compounds inhibit cancer by interfering with one, or more, of the mechanisms that researchers now feel are central to cancer progression.
Second, although the future does look bright for eventual success in the fight against cancer, we are not there yet. Much work remains to be done. As a science, the field of natural compound research can contribute to a greater understanding of cancer and a faster development of successful therapies.
Third, we must study natural compounds because they are already being used in cancer treatment (and in the treatment of other diseases). For better or for worse, hundreds of thousands if not millions, of patients around the world are experimenting with natural compounds in their efforts to heal themselves of cancer. Researchers estimate that anywhere from 10 to 80 percent of US, European, Australian, and Mexican cancer patients use some form of complementary medicine as part of their overall therapy [103] . For many of these patients, the use of natural compounds is an essential part of the complementary approach. For example, two studies in the United States have reported that roughly 40% - 60% of cancer patients who use some combination of complementary medicine including herbs, vitamins, and/or antioxidants [103] [104] . Most of these patients are using natural compounds without the guidance of their oncologist, or any real guidance from scientific studies. Because the popularity of using natural compounds in cancer treatment appears to be growing rather than declining, we are compelled to study natural compounds so that we can properly guide the public.
On the other hand, the new mechanism-based approach informs us that many different events contribute to the eventual success of a cancer. Any single drug can, at best target a small number of these events, leaving the rest to occur uninterrupted. Moreover, we know that cancer cells have some ability to adapt or resistant to therapy. We can imagine that a cancer cell can adapt better to one, or a few, interrupted events than to many.
To overcome this problem, it is necessary to use multiple compounds in combination. Natural compounds are ideally suited for this type of application; they are active at reasonable concentrations, and yet their mild nature allows a variety of large combinations to be used safely [105] .
Thus, we studied traditional herbs, which are currently in widespread use. We focused on 3 plant families: Chelidoniummajus (Papaveraceae) [106] , Fallopia japonica (Polygonaceae) (in preparation), and Citrus jambhiri (and Citrus pyriformis) (Rutaceae) [64] . Our previous investigations were focused on their SMs such as flavonoids, limonoids, steroids, polyphenols, and alkaloid looking at their possible modulation effect on multidrug resistance in cancer cells in order to validate their traditional use [64] [107] -[110] [111] . The following section highlights the chemistry and anticancer activity (especially the interaction with multidrug resistance proteins) and the resulting effect of these secondary metabolites on cancer.
7. Plant Secondary Metabolites
In the last century, thorough phytochemical investigations were conducted on these herbal drugs using simple and/or modern techniques of isolation and structure elucidation with the aim of finding substances responsible for the claimed medicinal effects.
This has led to a focus on plants secondary metabolites (SMs) [112] -[114] . Secondary metabolites are derived biosynthetically from primary metabolites (e.g., carbohydrates, amino acids, lipids, and acetyl-CoA) and are not directly involved in the growth, development, or reproduction of plants. Thousands of newly isolated secondary metabolites are discovered each year.
The chemical structures of these SMs are quite diverse, but the main classes of these SMs can be easily distinguished. An estimation of the isolated compounds for each class is presented in Table 1.
The screening of anticancer activity of these isolated compounds, and even the parent extracts, has generated as immense body of information and provided many lead compounds that could be modified for better activity. The major classes of secondary metabolites and their anticancer activities are highlighted in the next section.
7.1. Phenolics
Phenolic compounds constitute one of the most numerous groups in the plant kingdom with over 800 identified to date. Phenolics are characterized by the absence of nitrogen atoms and having at least one aromatic ring with one or more hydroxyl groups attached [116] . They range from simple, low molecular weight, single aromatic- ringed compounds to large and complex tannins and can be divided into 10 general classes (based on the number and arrangement of their carbon atoms). They are commonly found conjugated to sugars and organic acids. The most abundantly occurring polyphenols in plants are phenolic acids, flavonoids, stilbenes and lignans, of which flavonoids and phenolic acids account for 60% and 30%, respectively, of dietary polyphenols.
Flavonoids
Over 4000 varieties of flavonoids have been identified to date, making these universally distributed natural plants pigments, one of the most numerous and widespread of all. Additionally, 6500 different flavonoids have been identified from plant sources of which at least 400 appear to be prenylated [117] . Flavonoids are divided

Table 1. Approximate numbers of known secondary metabolites from higher plants [115] .
*Approximate number of known structures, **Total number of all terpenoides exceeds 33,000 at present.
into various classes according to their molecular structure; depend on the oxidation level of the central ring (ring C), however, all share a common carbon skeleton of diphenylpropanes (C6-C3-C6; i.e., two benzene rings joined by a linear three-carbon chain that forms an oxygenated heterocycle). The main groups are flavanols, flavones, flavanones, flavonols, isoflavones and anthocyanidins (Figure 8).
Flavonoid compounds are particularly abundant in fruits (especially in Citrus), vegetables, nuts, stems, flowers, wine, and tea. They constitute an important part of the human diet with an average of 200 mg consumed daily in the Western diet [118] [119] .
The basic flavonoid skeleton can have numerous substituents. Hydroxyl groups are usually present at the 4’, 5, and 7 positions. Sugars are very common with the majority of flavonoids, existing naturally as glycosides. While both sugars and hydroxyl groups increase the water solubility of flavonoids, other substituents, such as methyl groups and isopentyl units, make flavonoids lipophilic [120] . Generally, modifications such as hydroxylation, methylation, and glycosylation are also possible in either of the two aromatic rings. These structural modifications have a great impact on the anticancer activity of these SMs through their effect on the solubility and bioavailability of these compounds.
Anticancer activity of flavonoids: Recently, much research has focused on the anti-cancer properties of flavonoids. Epidemiological studies suggest an association between flavonoid intake and a reduced risk of certain cancers [121] . Flavonoids appear to work with no or little toxicity as large doses of these compounds (up to 500 mg/kg) have been administered to animals, with little or no toxicity reported [122] . There is a long history of human consumption of flavonoids.
Many flavonoids are substrates of the most pharmacologically relevant ABC transporters, P glycoprotein/ MDR1, MRP, and BCRP [123] [124] but so far scientific effort has focused on the modulation of the transporter by flavonoids [125] . Whereas some flavonoids were shown to inhibit MDR1-mediated transport processes by directly interacting with the vicinal ATP- and steroid-binding sites [126] , others (like (?) epicatechin from green tea) were shown to activate MDR1 by a heterotropic allosteric mechanism [127] .
To clarify, MDR1 possesses at least two positively cooperative sites for drug binding, with the H site preferring Hoechst 33,342 to rhodamine 123 and the R site preferring rhodamine 123 to Hoechst 33,342. Binding to one of these sites has been shown to stimulate binding to the other site and transport activity. Therefore, the con- flicting reports regarding flavonoid-MDR1 interactions might be explained by the different binding properties

Figure 8. Generic structures of the major flavonoids subclasses.
of the model substrates used [128] . Thus, polyphenols might be potential agents for modulating the bioavailability of MDR1 substrates at the intestine and the multidrug resistance phenotype associated with expression of this transporter in cancer cells [129] . In any case, the majority of studies have indicated that many flavonoids have an inhibitory activity on MDR1-mediated transport [130] .
Citrus flavonoids are shown to have anti-carcinogenic effects. Nobiletin (5,6,7,8,3’,4’-hexamethoxyflavone), a polymethoxyflavonoids in citrus fruit, showed strong inhibitory effects on tumor promotion in mice and the growth of human prostate carcinoma cells [131] . Therefore, citrus phytochemicals are considered promising chemopreventive agents. Likewise, the ingestion of citrus fruit has been reported to be beneficial for the reduction of certain types of human cancer [132] .
However, further studies of the absorption, distribution, metabolism, and excretion of citrus phytochemicals in the human body are needed to clarify the inhibitory effects of these compounds at the site of drug action, in the tumor tissues and cancer cells. We have recently reported the inhibitory effects of several isolated compounds from citrus on the functions of MDR1 using MDR1-mediated multidrug-resistant human caco-2 and CEM/ADR5000 cells [64] . Recently, reported state to grapefruit juice, compounds in orange juice can inhibit MDR1 [133] . Our results are comparable to this study in that the transport of Rho123, a substrate of MDR1, in Caco-2 cells was inhibited by citrus flavonoids [64] .
Furthermore, increasing in vitro and in vivo evidence has indicated that the pharmacokinetic interaction of drugs with herbal products containing flavonoids may be attributable not only to the modulation of drug transporters such as MDR1 but also to metabolizing enzymes such as several CYP450s, esterases, glucuronidases, oxidases, and other enzymes [134] -[136] . One well-documented food-interaction is the altered oral bioavailability of many marketed drugs from co-administered grapefruit juice and citrus fruit [134] [137] . The citrus appear to inhibit the MDR1 and CYP3A4, thus changing the drug absorption in the small intestine [137] . The enhancement of the detoxification pathway for the elimination of toxic electrophiles by the Phase-II enzymes; glutathione S-transferase (GST), may cause of citrus anticarcinogenic effect. To that end, the effects of citrus flavonoids (hesperidin, naringin, and crude flavonoids mixture) were investigated in various mice tissues. Citrus flavonoids showed the most significant induction of GST in the stomach. The GST induction by the flavonoids appeared to be tissue-specific and related to the structures of the compounds. The enhanced level of phase-II enzymes by citrus flavonoids suggests their importance in chemoprevention.
Finally, the observed antiproliferative properties of flavonoids suggest that these compounds may induce apoptosis by modulating different key targets involved in both apoptotic pathways. However, little is known regarding the precise mechanism of flavonoid-induced apoptosis, and only recently has interest started to focus on flavonoids’ potential to interact with intracellular signaling pathways. Many genes participate in the regulation of the apoptotic process, and activation of caspases is a central effector mechanism. For example in different cell lines, many isoflavonoids increased the activity and levels of caspase-3 and caspase-9, in concentration and time dependent manner [138] - [142] .
7.2. Terpenoids
The term terpenoids is widely used to describe a diverse and widespread class of natural products derived from a common biosynthetic pathway [143] . C5 isopentenoid units constitute the main building block in more than 30,000 reported individual members [144] . Classification of terpenoids is based on the number of the isoprenoid units. Terpenoids with two isoprene units are called monoterpenes, while those containing three to six isoprene units are called sesquiterpenes, diterpenes, sesterterpenes, and triterpenes, respectively [145] .
7.2.1. Limonoids
Limonoids are a group of highly oxygenated triterpenoids. Hundreds of limonoids have been isolated from various plants yet, their occurrence in the plant kingdom is confined to only plant families the Rutales, Meliaceae, and Rutaceae [146] . Limonoids are stereochemically homogenous compounds, with a prototypical structure either containing or derived from a precursor with a 4,4,8-trimethyl-17-furanylsteroid skeleton [146] . All naturally occurring citrus limonoids contain a furan ring attached to the D-ring, at C-17, as well as oxygen containing functional groups at C-3, C-4, C-7, C-16, and C-17 [146] . Citrus fruits and their closely related genera contain about 36 limonoidaglycones and 17 limonoidglucosides [147] . Citrus limonoids and their glucosides, the water- soluble triterpenoid compounds that occur naturally in citrus fruit and citrus juice in amounts comparable to vitamin C, can be reclaimed from citrus processing and citrus seeds as by-products in large quantities [146] . Limonin, the first characterized compound of this group of phytochemicals, has been known as a constituent of Citrus since 1841 (Figure 9) [148] .
Anticancer activity of limonoids: Many studies revealed that the limonoids present in citrus fruits and their juice have cancer chemopreventive property. In vitro limonoids inhibit the growth of estrogen receptor-negative and -positive human breast cancer cells. Limonoids have also been found to target and stop neuroblastoma cells [149] . The decreased colon tumor-genesis associated with ingesting orange juice may be explained by the potential chemopreventive effect of limonin, hesperidin, and other flavonoids [150] . The citrus limonoidsobacunone, limonin, nomilin and their glucosides, and some aglycones inhibit chemically induced carcinogenesis and a series of human cancer cell lines, with remarkable cytotoxicity against lung, colon, oral and skin cancer in animal test system and human breast cancer cells [151] -[153] . Pure limoninglucoside and limonin, (its water insoluble relative lacking glucose) have been found to possess significant antitumor properties in animal tests and with human cells [147] .
Nutritional research on the health benefits of chemicals present in plant foods suggest that citrus limonoids possess substantial anticancer activity and they are also free of any toxic effects in animal models [146] . Moreover, limonoid have shown a modifying effect on the development of aberrant crypt foci. These compounds also have ability to induce specific carcinogen-metabolizing enzymes, glutathione-S-transferase and quinine reductase in the liver and mucosa of the small intestine to detoxify chemical carcinogenesis. Studies show that the activity of phase II enzyme glutathione-S-transferase in the liver of rats, fed diets containing limonin and nomilin, increased significantly in a dose dependent manner. While simultaneously, the limonoidsnomilin and limonin were found to have no significant affect on the phase I enzyme cytochrome P450 [146] . The data from these studies suggest that certain rings in the limonoid nucleus may be critical to antineoplastic activity.
The structure-activity relationships of limonoids, showing that limonoids with an intact apoeuphol skeleton, a 14, 15 b epoxide, and a reactive site such as either a 19 - 28 lactol bridge or a cyclohexanone “A” ring, are biologically very active. In addition, α-,β-unsaturated ketone in ring “A” has been proposed as a common feature that is primarily responsible for their biological activity and absence of these structural features resulting in reduced activity [154] (Figure 9).
7.2.2. Sterols
Phytosterol is a general term widely used to describe a particular subclass of natural products possessing the steroidal nucleus. These steroids represent a large group of natural SMs widely occurring in plants and animals possessing a basic cyclopentanoperhydrophenanthrene skeleton (Figure 10).
Plant steroids comprise sterols, steroid saponins, steroid alkaloids, cardiac glycosides, pregnanes, estranes and ectysteroids [145] . They are all considered as triterpenes which have lost a minimum of three methyl groups during their biosynthesis [155] .
Anticancer activity of sterols: Cholesterol is an essential structural component of animal cell membranes. Plant sterols play analogues roles in plants. The major dietary phytosterols are β-sitosterol and stigmasterol. Their contents are high in edible oils, seeds, and nuts. Steroids, in general have a wide range of biological activities [156] [157] . Epidemiologic and experimental studies also suggest that dietary plant sterols play a role in cancer protection (such as colon, breast, and prostate) [158] . In contrast to cholesterol, β-sitosterol is poorly absorbed from the intestine, and its concentration in the blood and tissues of normal mammals is uniformly low.

Figure 9. Chemical structure of the Citruslimonoid, limonin.

Figure 10. Chemical structures of the major phytosterols.
ABCG5 and ABCG8, which are present almost exclusively in the small intestine and liver, are considered functional transporters for the efflux of plant sterols and stanols as well as cholesterol. In addition to ABCG5 and ABCG8, it has been reported that other ABC transporters, ABCA1 and ABCB4, are implicated in lipid homeostasis. ABCA1 mediates the efflux of cholesterol and phospholipids to form high-density lipoprotein [159] . ABCB4 (MDR2), highly homologous with MDR1 (MDR1), functions in the secretion of phosphatidylcholine into bile ducts from hepatocytes [160] . Therefore, it is conceivable that MDR1 also interacts with membrane lipids.
Foods supplemented with plant sterols, and dietary supplements and/or herbal remedies containing phytosterols have been widely used, often conveniently with prescribed medications. However, little attention has been paid to the interactions between drugs and food and the effects of food components on the function of drug transporters, such as MDR1 and MRP1, have not fully investigated. We reported that the anticancer activity of citrus sterols related to their ability to reverse multidrug resistance (MDR1) in leukemia and human colon carcinoma, which endogenously expresses MDR1. Furthermore, Citrus sterols reversed doxorubicin resistance in both cell lines [64] . Phytosterol glycosides could interact with the cell biomembrane due to their structural similarity with cholesterol, resulting in increasing the fluidity of the cell membrane and leading to leakage of electrolytes and metabolites or even cell death [161] . Additionally, we reported that β-sitosterol-O-glucoside is more effective than stigmasterol in interacting with cell membrane proteins and in reversal of doxorubicin resistance [64] . The pharmacological activity of phytosterols has been shown to be mediated by the antagonism of the nuclear receptor and a ligand for multiple nuclear receptors [162] . Brobst et al. showed that phytosterols activate the estrogen receptor an isoform, progesterone receptor, and pregnane X receptor (PXR, NR1I2) [162] . They also showed that phytosterol-mediated activation of PXR induces the expression of drug-metabolizing enzyme cytochrome P450 (CYP) CYP3A gene. PXR is known to be activated by a variety of drugs, xenobiotics, and bile acids, and is a key regulator of human CYP3A4 and MDR1 genes [78] .
7.3. Alkaloids
Alkaloids, a cyclic compound containing nitrogen in a negative oxidation state which is of limited distribution in living organisms, represent a very extensive group of secondary metabolites [163] . Over 21,000 alkaloids have been identified with diverse structures, distribution in nature, and important biological activities [164] . Alkaloids provide chemical defense against herbivores or predators. Strong physiological effects and the selectivity of some alkaloids present opportunities for utilizing the alkaloids in human medicine. During evolution, the constitution of alkaloids has been modulated so that they usually contain more than one active functional group allowing them to interact with several molecular targets. Therefore, a pleiotropic effect is a common term in alkaloids and other SM [114] [115] [165] (Figure 11).
Isoquinoline Alkaloids
The isoquinolines, one of the largest groups of alkaloids, skeletons are a basic building block of various types of alkaloids including benzylisoquinolines, protopines, benzo[c]phenanthridines, protoberberines, and others. Isoquinolines, alkaloids are biogenetically derived from tyrosine. Sanguinarine (SA), chelidonine (CH) (Figure 11), and chelerythrine (CHE), benzo[c]phen-anthridine alkaloids, have been isolated from Sanguinariacanadensis,

Figure 11. Chemical structures of the isoquinoline alkaloid, chelidonine.
Chelidoniummajus, and Macleyacordata that are known to exert a wide spectrum of biological activities, e.g. from antimicrobial, antifungal, anti-inflammatory, adrenolytic, sympatholytic, and local anaesthetic as well as showing cytotoxicity against various human normal and cancer cell lines [166] .
Anticancer activity of isoquinoline alkaloids: Benzylisoquinoline alkaloids, which are found in several herbal products, such as golden seal (Hydrastiscanadensis), Berberisand Oregon grape root (Mahoniaaquifolia), have been shown to interact with MDR1 and have the potential for drug-diet interactions [167] [168] . For example, Fu et al. (2001) screened potential MDR modifiers from a series of naturally occurring benzylisoquinoline alkaloids that were isolated from natural plants using resistance tumor cells. Many of these natural compounds showed potent activities to decrease the tumor cells resistance to MDR1 substrates, (such as doxorubicin and vincristine) and increased intracellular drug accumulation of [3H] vincristine. Their results suggest that the mechanism of these compounds to reverse MDR was probably linked to increased intracellular drug accumulation by inhibiting the activity of MDR1 [167] .
He and Liu (2002) screened various IQAs to examine their effect on the functional activity of MDR1 in cultured bovine brain capillary endothelial cells (BCEC). All compounds tested increased the intracellular accumulation of MDR1 substrate (Rho123) in a concentration-dependent manner. The rank order of these agents in increasing Rh123 accumulation in endothelial cells was as follows: tetrandrine > vincristine > dl-tetrahydropal- matine > dauricine > azithromycin > berbamine > daurisoline > berberine = doxorubicin > l-te-trahydropalma- tine [169] [170] .
Möller et al. reported that emetine reversed efflux pump function and up regulated the expression of MDR1 in resistance cells. This indicating that emetine acts as competitive substrate for MDR1 but not for MRP1 [171] . This was the first research showing that chelidonine inhibited the ABC transporters in Caco-2 cells, interacted with metabolic enzymes phase I and phase II and induced apoptosis in resistance cells, which indicating that chelidonine may be an active MDR inhibitor.
Extensive studies of the interaction of IQAs (36 compounds) with the most important human CYP enzymes have been performed. Overall, there were clear differences in the ability of these alkaloids to inhibit individual CYP forms [172] [173] .
In addition, IQAs exert cell growth-inhibitory effects via the induction of apoptosis in a variety of cancer cells [106] [174] [175] . IQAs-mediated apoptosis has been found in human cancer through their activation of caspase-3 and depletion of GSH [106] [176] . IQAs were found to result in increased levels of cytochrome c and Apaf-1 and in a significant increase in the active form of caspases 3, 7, 8, and 9 in a dose-dependent manner in many cancer cells [174] [177] -[180] .
8. Molecular Targets of Secondary Metabolites
The modulation of a molecular target will negatively influence its communication with other components of the cellular network, especially proteins (cross-talk of proteins), or elements, or signal transducers. Consequently, the metabolism and function of cells, tissues, organs, and eventually the whole organism will be affected. Although we know the structures of many SM, our knowledge concerning their molecular mode(s) of action is largely fragmentary and incomplete. SM with broad activities interacts mainly with proteins, biomembranes, and DNA/RNA [165] (Figure 12).
Conformational changes, associated with the lessening of protein activity can occur when a covalent or a noncovalent interaction modulates the three-dimensional protein structure. This conformational change is usual-

Figure 12. The main molecular targets of secondary metabolites in mammalian cells [165] .
ly associated with a loss or reduction in the activity of a protein [165] . Covalent bond occur when double bond, epoxide, phenolic radicals, exocyclic or terminal methylene groups are activated by reactive functional groups of polyphenols. When physiological condition cause dissociation for OH groups of flavonoids, and other compounds with phenolic hydroxyl groups, changed it to phenolate anions, which can create hydrogen bonds with proteins or nucleic acids, positively charged amino acid residues (e.g. lysine, arginine, or histidine). Weaker non covalent bonds can be very powerful when they work together. These include: hydrogen bonds, ionic bonds, hydrophobic interactions and Vander Waal forces.
Nitrogen-containing compounds, such as alkaloids, usually have positively charged N-atoms (under physiological conditions) in their molecules that can form ionic bonds with negatively charged amino acid residues of aspartic and glutamic acid in proteins. Alkaloids are able to form covalent bonds with proteins and nucleic acids and noncovalent bonding. IQAs containing also OH groups which are crucial for the biological activity of phenolics. Both the covalent and non-covalent interactions will modulate the three-dimensional protein structure, i.e., the conformation that is so important for their bioactivities. A conformational change is usually associated with a loss or reduction in the activity of a protein [165] [181] .
The activity of IQAs related to their structure, the neutral form (pseudobase) of isoquinoline alkaloids (IQAs) can interact with proteins [182] ; it is involved in the interactions with cellular biomacromolecules and may elicit a cytotoxic response [183] . IQAs interconvert between the cationic vs. neutral form; they penetrate the cell membrane in the form of nonpolar pseudobase [184] . The iminium bond, C6 = N+ in the cationic form is susceptible to nucleophilic attack and plays a key role in inhibition of SH-proteins [185] . The binding of IQAs with human serum albumin and L-cysteine is radically weaker at pH 5.0 than at pH 7.4. In addition, IQAs forms in vitro DNA adducts via modification of the C(11) = C(12) bond detectable by 32P-postlabelling [186] . Although IQAs mediated DNA damage in vitro and in vivo has been published [187] .
The cytotoxic effect of IQAs resulting from their molecular interactions with many important targets in a cancer cell include: DNA, RNA, and the associated enzymes and processes (i.e., replication, repair, transcription, polymerase, topoisomerase, telomerase, protein biosynthesis, protein conformation, biomembranes, and membrane proteins.
In addition, the IQAs are intercalating compounds, it has planar, lipophilic characters, can disturbed the replication and transcription process by interact with base pairs and stabilize the DNA double helix, and inhibition of microtubule [165] [181] . Alkaloids with a planar and polycyclic structure (e.g. isoquinoline, quinoline, and indole) are good candidates for DNA intercalation. Protonable ring nitrogens can stabilize the alkaloid-DNA complex by binding to the negatively charged DNA surface [165] .
Furthermore, terpenoids (lipophilic compounds) have ability to associate with other hydrophobic molecules in a cell (biomembranes or the hydrophobic core of many proteins and of the DNA double helix) [165] [181] . A major target for terpenoids, especially saponins or saponins like structure (e.g. β-sitosterol-O-glucoside) is the biomembrane. This structure can also change the fluidity of biomembranes and reducing their protective function as a permeation barrier [165] [181] (Figure 13).
For example, both saponins and sterol glycosides are amphiphilic molecules, which function as detergents and are active mostly in their mono-desmosidic forms. The monodesmosides are anchored with their lipophilic moiety in the lipophilic membrane bilayer after complexing with cholesterol, while the hydrophilic sugar part remains outside the cell and can interact with other glycoproteins or glycolipids (Figure 13). As a result, loss of the membrane integrity and fluidity occurs with the subsequent leakage of many polar molecules out of the cells or the entry of unwanted molecules into a cell. Such activity of many SMs can explain the cytotoxic effects against a wide range of cells (cancer, bacteria, and fungi) [188] .
In addition, other lipophilic SMs such as mono- and sesquiterpenes can dissolve in biomembranes resulting in a disturbance of the close interaction between membrane lipids and proteins, thereby changing the conformation of membrane proteins leading to loss of function. These membrane proteins include ion channels, transporters for nutrients and intermediates, receptors, and proteins of signal transduction. This type of interaction between

Figure 13. Interactions of some representative secondary metabolites with the cell membrane [161] .
the SM and the biomembranes could explain the cytotoxic, antimicrobial and antiviral effects of many SMs [107] [109] [110] [161] .
Plants extracts are a mixture of SMs, often both phenolics, terpenoids, and alkaloids. Thus, exhibit both covalent and noncovalent interactions. These activities are probably both additive and synergistic [165] [181] .
9. Conclusions
Based on the overall study, it can concluded that certain natural products obtained from medicinal plants may serve as potent drugs for reversing multidrug resistance in cancer cells due to their ability to inhibit ABC- transporters, metabolic enzymes, and to induce apoptosis. In particular, the MDR modulatory activity of the crude extracts such as C. majus may be useful in cancer treatment, which would validate their traditional ethnopharmacological uses in cancer formulas in China and Japan.
Plant extracts contain numerous and diverse secondary metabolites (SMs). Generally, it is difficult to attribute the biological and pharmacological activities of a multicomponent mixture, as plant extracts, to only a single compound of a multicomponent extract. Furthermore, SMs can unselectively affect many molecular targets in the cells.
Active SMs have shown the ideal character of MDR inhibitors or substrates, they have at least two hydrophobic centers containing more than two aromatic rings, which have more than two H-bond acceptors and at least one H-bond donor center. Therefore, under physiological conditions, the OH groups of resveratrol, emodin, and citrus flavonoids (polyphenols) change to phenolate ions and form ionic bonds with positively charged amino acid residues, such as those from lysine, arginine, and histidine in several proteins (e.g. ABCT, CYP3A4, and caspases).
Covalent bonds can be formed also with reactive functional groups of SM, such as epoxides (e.g. limonin), activated double bonds, and exocyclic or terminal methylene groups. Chelidonine has positively charged N- atoms (under physiological conditions) that can form also ionic bonds with negatively charged amino acid residues of glutamic and aspartic acid in proteins.
Chelidonine can be considered as a good candidate as a cytotoxic drug and for induction of apoptosis. One explanation could be that the protonated ring nitrogens with planar and polycyclic structure of chelidonine facilitate its penetration of the cell membrane, forming stable complexes with DNA by binding to the negatively charged DNA surface, leading to DNA intercalation, and inhibiting reverse transcriptase.
Non-covalent bonds, especially hydrogen bonds, ionic bonds, and hydrophobic interactions can be effective if they act cooperatively together. Both the covalent and non-covalent interactions will modulate the three-dimen- sional protein structure, resulting in conformational change, which is usually associated with a loss or reduction in the activity of a protein.
Finally, interference of different SMs/extracts with phase I, CYP3A4, and phase II, GST metabolizing enzymes can cause many serious interactions. Therefore, studies on possible herbal-herbal or herbal-drug interactions and standardization of drugs are very important, especially for integration and acceptance of plant extracts in conventional medicine as anti-cancer drugs.
References
- Stewart, B.W. and Kleihues, P. (2014) International Agency for Research on Cancer. World Cancer Report 351, Lyon.
- Altieri, F., Grillo, C., Maceroni, M. and Chichiarelli, S. (2008) DNA Damage and Repair: From Molecular Mechanisms to Health Implications. Antioxidants & Redox Signaling, 10, 891-938. http://dx.doi.org/10.1089/ars.2007.1830
- Velingkar, V.S. and Dandekar, V.D. (2010) Modulation of P-Glycoprotein Mediated Multidrug Resistance (MDR) in Cancer Using Chemosensitizers. International Journal of Pharma Sciences and Research (IJPSR), 1, 104-111.
- Shen, D., Pastan, I. and Gottesman, M.M. (1998) Cross-Resistance to Methotrexate and Metals in Human Cisplatin- Resistant Cell Lines Results from a Pleiotropic Defect in Accumulation of These Compounds Associated with Reduced Plasma Membrane Binding Proteins. Cancer Research, 58, 268-275.
- Shen, D.W., Goldenberg, S., Pastan, I. and Gottesman, M.M. (2000) Decreased Accumulation of [14C]Carboplatin in Human Cisplatin-Resistant Cells Results from Reduced Energy-Dependent Uptake. Journal of Cellular Physiology, 183, 108-116. http://dx.doi.org/10.1002/(SICI)1097-4652(200004)183:1<108::AID-JCP13>3.0.CO;2-4
- Chai, S., To, K.K. and Lin, G. (2010) Circumvention of Multi-Drug Resistance of Cancer Cells by Chinese Herbal Medicines. Chinese Medicine, 5, 26. http://dx.doi.org/10.1186/1749-8546-5-26
- Jain, R.K. (2001) Delivery of Molecular and Cellular Medicine to Solid Tumors. Advanced Drug Delivery Reviews, 46, 149-168. http://dx.doi.org/10.1016/S0169-409X(00)00131-9
- Pluen, A., Boucher, Y., Ramanujan, S., McKee, T.D., Gohongi, T., di Tomaso, E., et al. (2001) Role of Tumor-Host Interactions in Interstitial Diffusion of Macromolecules: Cranial vs. Subcutaneous Tumors. Proceedings of the National Academy of Sciences of the United States of America, 98, 4628-4633. http://dx.doi.org/10.1073/pnas.081626898
- Schuetz, E.G., Beck, W.T. and Schuetz, J.D. (1996) Modulators and Substrates of P-Glycoprotein and Cytochrome P4503A Coordinately Up-Regulate These Proteins in Human Colon Carcinoma Cells. Molecular Pharmacology, 49, 311-318.
- Jiang, H., Chen, K., He, J., Pan, F., Li, J., Chen, J.F., et al. (2009) Association of Pregnane X Receptor with Multidrug Resistance-Related Protein 3 and Its Role in Human Colon Cancer Chemoresistance. Journal of Gastrointestinal Surgery, 13, 1831-1838. http://dx.doi.org/10.1007/s11605-009-0964-x
- Lowe, S.W., Ruley, H.E., Jacks, T. and Housman, D.E. (1993) P53-Dependent Apoptosis Modulates the Cytotoxicity of Anticancer Agents. Cell, 74, 957-967. http://dx.doi.org/10.1016/0092-8674(93)90719-7
- Liu, Y.Y., Han, T.Y., Giuliano, A.E. and Cabot, M.C. (2001) Ceramide Glycosylation Potentiates Cellular Multidrug Resistance. FASEB Journal, 15, 719-730. http://dx.doi.org/10.1096/fj.00-0223com
- Fan, D., Beltran, P.J. and O’Brien, C.A., Eds. (1994) Reversal of Multidrug Resistance. CRC Press, Boca Raton.
- Huang, G. and Chen, L. (2010) Recombinant Human Endostatin Improves Anti-Tumor Efficacy of Paclitaxel by Normalizing Tumor Vasculature in Lewis Lung Carcinoma. Journal of Cancer Research and Clinical Oncology, 136, 1201-1211. http://dx.doi.org/10.1007/s00432-010-0770-6
- Green, S.K., Frankel, A. and Kerbel, R.S. (1999) Adhesion-Dependent Multicellular Drug Resistance. Anti-Cancer Drug Design, 14, 153-168.
- Durand, R.E. and Olive, P.L. (2001) Resistance of Tumor Cells to Chemo- and Radiotherapy Modulated by the Three-Dimensional Architecture of Solid Tumors and Spheroids. Methods in Cell Biology, 64, 211-233. http://dx.doi.org/10.1016/S0091-679X(01)64015-9
- Litman, T., Druley, T.E., Stein, W.D. and Bates, S.E. (2001) From MDR to MXR: New Understanding of Multidrug Resistance Systems, Their Properties and Clinical Significance. Cellular and Molecular Life Sciences CMLS, 58, 931- 959. http://dx.doi.org/10.1007/PL00000912
- Dean, M., Hamon, Y. and Chimini, G. (2001) The Human ATP-Binding Cassette (ABC) Transporter Superfamily. Journal of Lipid Research, 42, 1007-1017.
- Juliano, R.L. and Ling, V. (1976) A Surface Glycoprotein Modulating Drug Permeability in Chinese Hamster Ovary Cell Mutants. Biochimica et Biophysica Acta, 455, 152-162. http://dx.doi.org/10.1016/0005-2736(76)90160-7
- Ueda, K., Cardarelli, C., Gottesman, M.M. and Pastan, I. (1987) Expression of a Full-Length cDNA for the Human “MDR1” Gene Confers Resistance to Colchicine, Doxorubicin, and Vinblastine. Proceedings of the National Academy of Sciences of the United States of America, 84, 3004-3008. http://dx.doi.org/10.1073/pnas.84.9.3004
- Ambudkar, S.V., Dey, S., Hrycyna, C.A., Ramachandra, M., Pastan, I. and Gottesman, M.M. (1999) Biochemical, Cellular, and Pharmacological Aspects of the Multidrug Transporter. Annual Review of Pharmacology and Toxicology, 39, 361-398. http://dx.doi.org/10.1146/annurev.pharmtox.39.1.361
- Gottesman, M.M., Fojo, T. and Bates, S.E. (2002) Multidrug Resistance in Cancer: Role of ATP-Dependent Transporters. Nature Reviews Cancer, 2, 48-58. http://dx.doi.org/10.1038/nrc706
- Johnstone, R.W., Ruefli, A.A. and Smyth, M.J. (2000) Multiple Physiological Functions for Multidrug Transporter P-Glycoprotein? Trends in Biochemical Sciences, 25, 1-6. http://dx.doi.org/10.1016/S0968-0004(99)01493-0
- Sauna, Z.E. and Ambudkar, S.V. (2000) Evidence for a Requirement for ATP Hydrolysis at Two Distinct Steps during a Single Turnover of the Catalytic Cycle of Human P-Glycoprotein. Proceedings of the National Academy of Sciences of the United States of America, 97, 2515-2520. http://dx.doi.org/10.1073/pnas.97.6.2515
- Cole, S.P., Bhardwaj, G., Gerlach, J.H., Mackie, J.E., Grant, C.E., Almquist, K.C., et al. (1992) Overexpression of a Transporter Gene in a Multidrug-Resistant Human Lung-Cancer Cell-Line. Science, 258, 1650-1654. http://dx.doi.org/10.1126/science.1360704
- Jedlitschky, G., Leier, I., Buchholz, U., Barnouin, K., Kurz, G. and Keppler, D. (1996) Transport of Glutathione, Glucuronate, and Sulfate Conjugates by the MRP Gene-Encoded Conjugate Export Pump. Cancer Research, 56, 988-994.
- Borst, P., Evers, R., Kool, M. and Wijnholds, J. (2000) A Family of Drug Transporters: The Multidrug Resistance- Associated Proteins. Journal of the National Cancer Institute, 92, 1295-1302. http://dx.doi.org/10.1093/jnci/92.16.1295
- Allikmets, R., Schriml, L.M., Hutchinson, A., Romano-Spica, V. and Dean, M. (1998) A Human Placenta-Specific ATP-Binding Cassette Gene (ABCP) on Chromosome 4q22 that Is Involved in Multidrug Resistance. Cancer Research, 58, 5337-5339.
- Honjo, Y., Hrycyna, C.A., Yan, Q.W., Medina-Perez, W.Y., Robey, R.W., van de Laar, A., et al. (2001) Acquired Mutations in the MXR/BCRP/ABCP Gene Alter Substrate Specificity in MXR/BCRP/ABCP-Overexpressing Cells. Cancer Research, 61, 6635-6639.
- Komatani, H., Kotani, H., Hara, Y., Nakagawa, R., Matsumoto, M., Arakawa, H. and Nishimura, S. (2001) Identification of Breast Cancer Resistant Protein/Mitoxantrone Resistance/Placenta-Specific, ATP-Binding Cassette Transporter as a Transporter of NB-506 and J-107088, Topoisomerase I Inhibitors with an Indolocarbazole Structure. Cancer Research, 61, 2827-2832.
- Thiebaut, F., Tsuruo, T., Hamada, H., Gottesman, M.M., Pastan, I. and Willingham, M.C. (1987) Cellular Localization of the Multidrug-Resistance Gene Product P-Glycoprotein in Normal Human Tissues. Proceedings of the National Academy of Sciences of the United States of America, 84, 7735-7738. http://dx.doi.org/10.1073/pnas.84.21.7735
- Fletcher, J.I., Haber, M., Henderson, M.J. and Norris, M.D. (2010) ABC Transporters in Cancer: More than Just Drug Efflux Pumps. Nature Reviews Cancer, 10, 147-156. http://dx.doi.org/10.1038/nrc2789
- Schinkel, A.H., Wagenaar, E., Mol, C.A. and van Deemter, L. (1996) P-Glycoprotein in the Blood-Brain Barrier of Mice Influences the Brain Penetration and Pharmacological Activity of Many Drugs. Journal of Clinical Investigation, 97, 2517-2524. http://dx.doi.org/10.1172/JCI118699
- Cordon-Cardo, C., O’Brien, J.P., Boccia, J., Casals, D., Bertino, J.R. and Melamed, M.R. (1990) Expression of the Multidrug Resistance Gene Product (P-Glycoprotein) in Human Normal and Tumor Tissues. Journal of Histochemistry & Cytochemistry, 38, 1277-1287. http://dx.doi.org/10.1177/38.9.1974900
- Jonker, J.W., Smit, J.W., Brinkhuis, R.F., Maliepaard, M., Beijnen, J.H., Schellens, J.H.M. and Schinkel, A.H. (2000) Role of Breast Cancer Resistance Protein in the Bioavailability and Fetal Penetration of Topotecan. Journal of the National Cancer Institute, 92, 1651-1656. http://dx.doi.org/10.1093/jnci/92.20.1651
- Maliepaard, M., Scheffer, G.L., Faneyte, I.F., van Gastelen, M.A., Pijnenborg, A.C., Schinkel, A.H., et al. (2001) Subcellular Localization and Distribution of the Breast Cancer Resistance Protein Transporter in Normal Human Tissues. Cancer Research, 61, 3458-3464.
- St-Pierre, M.V., Serrano, M.A., Macias, R.I., Dubs, U., Hoechli, M., Lauper, U., et al. (2000) Expression of Members of the Multidrug Resistance Protein Family in Human Term Placenta. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 279, R1495-R1503.
- Schinkel, A.H., Mayer, U., Wagenaar, E., Mol, C.A.A.M., van Deemter, L., Smit, J.J.M., et al. (1997) Normal Viability and Altered Pharmacokinetics in Mice Lacking Mdr1-Type (Drug-Transporting) P-Glycoproteins. Proceedings of the National Academy of Sciences of the United States of America, 94, 4028-4033. http://dx.doi.org/10.1073/pnas.94.8.4028
- Zhou, S.F., Lim, L.Y. and Chowbay, B. (2004) Herbal Modulation of P-Glycoprotein. Drug Metabolism Reviews, 36, 57-104. http://dx.doi.org/10.1081/DMR-120028427
- Evers, R., Zaman, G.J., Van Deemter, L., Jansen, H., Calafat, J., Oomen, L.C., et al. (1996) Basolateral Localization and Export Activity of the Human Multidrug Resistance-Associated Protein in Polarized Pig Kidney Cells. Journal of Clinical Investigation, 97, 1211-1218. http://dx.doi.org/10.1172/JCI118535
- Goldstein, L.J., Galski, H., Fojo, A., Willingham, M., Lai, S.L., Gazdar, A., et al. (1989) Expression of Multidrug Resistance Gene in Human Cancers. Journal of the National Cancer Institute, 81, 116-124. http://dx.doi.org/10.1093/jnci/81.2.116
- Dorr, R., Karanes, C., Spier, C., Grogan, T., Greer, J., Moore, J., et al. (2001) Phase I/II Study of the P-Glycoprotein Modulator PSC 833 in Patients with Acute Myeloid Leukemia. Journal of Clinical Oncology, 19, 1589-1599.
- Leith, C.P., Kopecky, K.J., Chen, I.M., et al. (1999) Frequency and Clinical Significance of the Expression of the Multidrug Resistance Proteins MDR1/P-Glycoprotein, MRP1, and LRP in Acute Myeloid Leukemia: A Southwest Oncology Group Study. Blood, 94, 1086-1099.
- Michieli, M., Damiani, D., Ermacora, A., Masolini, P., Raspadori, D., Visani, G., et al. (1999) P-Glycoprotein, Lung Resistance-Related Protein and Multidrug Resistance Associated Protein in de novo Acute Non-Lymphocytic Leukaemias: Biological and Clinical Implications. British Journal of Haematology, 104, 328-335. http://dx.doi.org/10.1046/j.1365-2141.1999.01172.x
- Tidefelt, U., Liliemark, J., Gruber, A., Liliemark, E., Sundman-Engberg, B., Juliusson, G., et al. (2000) P-Glycoprotein Inhibitor Valspodar (PSC 833) Increases the Intracellular Concentrations of Daunorubicin in Vivo in Patients with P-Glycoprotein-Positive Acute Myeloid Leukemia. Journal of Clinical Oncology, 18, 1837-1844.
- El-Sharnouby, J.A., Abou El-Enein, A.M., El Ghannam, D.M., El-Shanshory, M.R., Hagag, A.A., Yahia, S. and Elashry, R. (2010) Expression of Lung Resistance Protein and Multidrug Resistance-Related Protein (MRP1) in Pediatric Acute Lymphoblastic Leukemia. Journal of Oncology Pharmacy Practice, 16, 179-188. http://dx.doi.org/10.1177/1078155209351329
- Van Der Pol, M.A., Broxterman, H.J., Pater, J.M., Feller, N., van der Maas, M., Weijers, G.W., et al. (2003) Function of the ABC Transporters, P-Glycoprotein, Multidrug Resistance Protein and Breast Cancer Resistance Protein, in Minimal Residual Disease in Acute Myeloid Leukemia. Haematologica, 88, 134-147.
- Laska, D.A., Houchins, J.O., Pratt, S.E., Horn, J., Xia, X.L., Hanssen, B.R., et al. (2002) Characterization and Application of a Vinblastine-Selected CACO-2 Cell Line for Evaluation of P-Glycoprotein. In Vitro Cellular & Developmental Biology-Animal, 38, 401-410. http://dx.doi.org/10.1290/1071-2690(2002)038<0401:CAAOAV>2.0.CO;2
- Engman, H.A., Lennernas, H., Taipalensuu, J., Otter, C., Leidvik, B. and Artursson, P. (2001) CYP3A4, CYP3A5, and MDR1 in Human Small and Large Intestinal Cell Lines Suitable for Drug Transport Studies. Journal of Pharmaceutical Sciences, 90, 1736-1751. http://dx.doi.org/10.1002/jps.1123
- Lampen, A., Ebert, B., Stumkat, L., Jacob, J. and Seidel, A. (2004) Induction of Gene Expression of Xenobiotic Metabolism Enzymes and ABC-Transport Proteins by PAH and a Reconstituted PAH Mixture in Human Caco-2 Cells. Biochimica et Biophysica Acta, 1681, 38-46. http://dx.doi.org/10.1016/j.bbaexp.2004.09.010
- Muller, J., Sidler, D., Nachbur, U., Wastling, J., Brunner, T. and Hemphill, A. (2008) Thiazolides Inhibit Growth and Induce Glutathione-S-Transferase Pi (GSTP1)-Dependent Cell Death in Human Colon Cancer Cells. International Journal of Cancer, 123, 1797-1806. http://dx.doi.org/10.1002/ijc.23755
- Dai, J.Y., Yang, J.L. and Li, C. (2008) Transport and Metabolism of Flavonoids from Chinese Herbal Remedy Xiaochaihu-Tang across Human Intestinal Caco-2 Cell Monolayers. Acta Pharmacologica Sinica, 29, 1086-1093. http://dx.doi.org/10.1111/j.1745-7254.2008.00850.x
- Heikkinen, A.T., Monkkonen, J. and Korjamo, T. (2009) Kinetics of Cellular Retention during Caco-2 Permeation Experiments: Role of Lysosomal Sequestration and Impact on Permeability Estimates. Journal of Pharmacology and Experimental Therapeutics, 328, 882-892. http://dx.doi.org/10.1124/jpet.108.145797
- Ecker, G., Huber, M., Schmid, D. and Chiba, P. (1999) The Importance of a Nitrogen Atom in Modulators of Multidrug Resistance. Molecular Pharmacology, 56, 791-796.
- Litman, T., Skovsgaard, T. and Stein, W.D. (2003) Pumping of Drugs by P-Glycoprotein: A Two-Step Process? Journal of Pharmacology and Experimental Therapeutics, 307, 846-853. http://dx.doi.org/10.1124/jpet.103.056960
- Kim, R.B., Wandel, C., Leake, B., Cvetkovic, M., Fromm, M.F., Dempsey, P.J., et al. (1999) Interrelationship between Substrates and Inhibitors of Human CYP3A and P-Glycoprotein. Pharmaceutical Research, 16, 408-414. http://dx.doi.org/10.1023/A:1018877803319
- Marzolini, C., Paus, E., Buclin, T. and Kim, R.B. (2004) Polymorphisms in Human MDR1 (P-Glycoprotein): Recent Advances and Clinical Relevance. Clinical Pharmacology & Therapeutics, 75, 13-33. http://dx.doi.org/10.1016/j.clpt.2003.09.012
- Milne, R.J. and Buckley, M.M.T. (1991) Celiprolol―An Updated Review of Its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Efficacy in Cardiovascular Disease. Drugs, 41, 941-969. http://dx.doi.org/10.2165/00003495-199141060-00009
- Szakacs, G., Paterson, J.K., Ludwig, J.A., Booth-Genthe, C. and Gottesman, M.M. (2006) Targeting Multidrug Resistance in Cancer. Nature Reviews Drug Discovery, 5, 219-234. http://dx.doi.org/10.1038/nrd1984
- Krishna, R. and Mayer, L.D. (2001) Modulation of P-Glycoprotein (PGP) Mediated Multidrug Resistance (MDR) Using Chemosensitizers: Recent Advances in the Design of Selective MDR Modulators. Current Medicinal Chemistry- Anti-Cancer Agents, 1, 163-174. http://dx.doi.org/10.2174/1568011013354705
- Krishna, R. and Mayer, L.D. (2000) Multidrug Resistance (MDR) in Cancer: Mechanisms, Reversal Using Modulators of MDR and the Role of MDR Modulators in Influencing the Pharmacokinetics of Anticancer Drugs. European Journal of Pharmaceutical Sciences, 11, 265-283. http://dx.doi.org/10.1016/S0928-0987(00)00114-7
- Borowski, E., Bontemps-Gracz, M.M. and Piwkowska, A. (2005) Strategies for Overcoming ABC-Transporters-Me- diated Multidrug Resistance (MDR) of Tumor Cells. Acta Biochimica Polonica, 52, 609-627.
- Liscovitch, M. and Lavie, Y. (2002) Cancer Multidrug Resistance: A Review of Recent Drug Discovery Research. Idrugs, 5, 349-355.
- El-Readi, M.Z., Hamdan, D., Farrag, N., El-Shazly, A. and Wink, M. (2010) Inhibition of P-Glycoprotein Activity by Limonin and Other Secondary Metabolites from Citrus Species in Human Colon and Leukaemia Cell Lines. European Journal of Pharmacology, 626, 139-145. http://dx.doi.org/10.1016/j.ejphar.2009.09.040
- Theis, J.G.W., Chan, H.S.L., Greenberg, M.L., Malkin, D., Karaskov, V., Moncica, I., et al. (2000) Assessment of Systemic Toxicity in Children Receiving Chemotherapy with Cyclosporine for Sarcoma. Medical and Pediatric Oncology, 34, 242-249. http://dx.doi.org/10.1002/(SICI)1096-911X(200004)34:4<242::AID-MPO2>3.0.CO;2-U
- Wang, R.B., Kuo, C.L., Lien, L.L. and Lien, E.J. (2003) Structure-Activity Relationship: Analyses of P-Glycoprotein Substrates and Inhibitors. Journal of Clinical Pharmacy and Therapeutics, 28, 203-228. http://dx.doi.org/10.1046/j.1365-2710.2003.00487.x
- Garrigos, M., Mir, L.M. and Orlowski, S. (1997) Competitive and Non-Competitive Inhibition of the Multidrug-Re- sistance-Associated P-Glycoprotein ATPase. Further Experimental Evidence for a Multisite Model. European Journal of Biochemistry, 244, 664-673. http://dx.doi.org/10.1111/j.1432-1033.1997.00664.x
- Dey, S., Ramachandra, M., Pastan, I., Gottesman, M.M. and Ambudkar, S.V. (1997) Evidence for Two Nonidentical Drug-Interaction Sites in the Human P-Glycoprotein. Proceedings of the National Academy of Sciences of the United States of America, 94, 10594-10599. http://dx.doi.org/10.1073/pnas.94.20.10594
- Ford, J.M. (1996) Experimental Reversal of P-Glycoprotein-Mediated Multidrug Resistance by Pharmacological Chemosensitisers. European Journal of Cancer, 32, 991-1001. http://dx.doi.org/10.1016/0959-8049(96)00047-0
- Tamai, I. and Safa, A.R. (1991) Azidopine Noncompetitively Interacts with Vinblastine and Cyclosporin a Binding to P-Glycoprotein in Multidrug Resistant Cells. Journal of Biological Chemistry, 266, 16796-16800.
- Loo, T.W. and Clarke, D.M. (1995) Covalent Modification of Human P-Glycoprotein Mutants Containing a Single Cysteine in Either Nucleotide-Binding Fold Abolishes Drug-Stimulated ATPase Activity. Journal of Biological Chemistry, 270, 22957-22961. http://dx.doi.org/10.1074/jbc.270.39.22957
- Solary, E., Bidan, J.M., Calvo, F., Chauffert, B., Caillot, D., Mugneret, F., et al. (1991) P-Glycoprotein Expression and in Vitro Reversion of Doxorubicin Resistance by Verapamil in Clinical Specimens from Acute Leukaemia and Myeloma. Leukemia, 5, 592-597.
- Muller, C., Goubin, F., Ferrandis, E., Cornil-Scharwtz, I., Bailly, J.D., Bordier, C., et al. (1995) Evidence for Transcriptional Control of Human Mdr1 Gene Expression by Verapamil in Multidrug-Resistant Leukemic Cells. Molecular Pharmacology, 47, 51-56.
- Ozben, T. (2006) Mechanisms and Strategies to Overcome Multiple Drug Resistance in Cancer. FEBS Letters, 580, 2903-2909. http://dx.doi.org/10.1016/j.febslet.2006.02.020
- Guengerich, F.P. (1991) Reactions and Significance of Cytochrome P-450 Enzymes. Journal of Biological Chemistry, 266, 10019-10022.
- Zhou, S.F., Gao, Y.H., Jiang, W.Q., Huang, M., Xu, A.L. and Paxton, J.W. (2003) Interactions of Herbs with Cytochrome P450. Drug Metabolism Reviews, 35, 35-98. http://dx.doi.org/10.1081/DMR-120018248
- Bertilsson, G., Heidrich, J., Svensson, K., Åsman, M., Jendeberg, L., Sydow-Bäckman, M., et al. (1998) Identification of a Human Nuclear Receptor Defines a New Signaling Pathway for CYP3A Induction. Proceedings of the National Academy of Sciences of the United States of America, 95, 12208-12213. http://dx.doi.org/10.1073/pnas.95.21.12208
- Urquhart, B.L., Tirona, R.G. and Kim, R.B. (2007) Nuclear Receptors and the Regulation of Drug-Metabolizing Enzymes and Drug Transporters: Implications for Interindividual Variability in Response to Drugs. Journal of Clinical Pharmacology, 47, 566-578. http://dx.doi.org/10.1177/0091270007299930
- Tolson, A.H. and Wang, H. (2010) Regulation of Drug-Metabolizing Enzymes by Xenobiotic Receptors: PXR and CAR. Advanced Drug Delivery Reviews, 62, 1238-1249. http://dx.doi.org/10.1016/j.addr.2010.08.006
- Kimura, Y., Matsuo, M., Takahashi, K., Saeki, T., Kioka, N., Amachi, T. and Ueda, K. (2004) ATP Hydrolysis-Dependent Multidrug Efflux Transporter: MDR1/P-Glycoprotein. Current Drug Metabolism, 5, 1-10. http://dx.doi.org/10.2174/1389200043489090
- Lewis, A.D., Forrester, L.M., Hayes, J.D., Wareing, C.J., Carmichael, J., Harris, A.L., et al. (1989) Glutathione S-Transferase Isoenzymes in Human Tumours and Tumour Derived Cell Lines. British Journal of Cancer, 60, 327-331. http://dx.doi.org/10.1038/bjc.1989.280
- Green, J.A., Robertson, L.J. and Clark, A.H. (1993) Glutathione S-Transferase Expression in Benign and Malignant Ovarian Tumours. British Journal of Cancer, 68, 235-239. http://dx.doi.org/10.1038/bjc.1993.321
- Mannervik, B. and Jensson, H. (1982) Binary Combinations of Four Protein Subunits with Different Catalytic Specificities Explain the Relationship between Six Basic Glutathione S-Transferases in Rat Liver Cytosol. Journal of Biological Chemistry, 257, 9909-9912.
- Morgenstern, R., Depierre, J.W. and Jornvall, H. (1985) Microsomal Glutathione Transferase. Primary Structure. Journal of Biological Chemistry, 260, 13976-13983.
- Kuzmich, S. and Tew, K.D. (1991) Detoxification Mechanisms and Tumor Cell Resistance to Anticancer Drugs. Medicinal Research Reviews, 11, 185-217.
- Ramsdell, H.S. and Eaton, D.L. (1990) Mouse Liver Glutathione S-Transferase Isoenzyme Activity toward Aflatoxin B1-8,9-Epoxide and Benzo[A]Pyrene-7,8-Dihydrodiol-9,10-Epoxide. Toxicology and Applied Pharmacology, 105, 216- 225. http://dx.doi.org/10.1016/0041-008X(90)90183-U
- Falkner, K.C., Rushmore, T.H., Linder, M.W. and Prough, R.A. (1998) Negative Regulation of the Rat Glutathione S-Transferase A2 Gene by Glucocorticoids Involves a Canonical Glucocorticoid Consensus Sequence. Molecular Pharmacology, 53, 1016-1026.
- Kasibhatla, S. and Tseng, B. (2003) Why Target Apoptosis in Cancer Treatment? Molecular Cancer Therapeutics, 2, 573-580.
- Schultehermann, R., Bursch, W., Graslkraupp, B., Török, L., Ellinger, A. and Müllauer, I. (1995) Role of Active Cell Death (Apoptosis) in Multi-Stage Carcinogenesis. Toxicology Letters, 82-83, 143-148. http://dx.doi.org/10.1016/0378-4274(95)03550-8
- Collins, J.A., Schandl, C.A., Young, K.K., Vesely, J. and Willingham, M.C. (1997) Major DNA Fragmentation Is a Late Event in Apoptosis. Journal of Histochemistry & Cytochemistry, 45, 923-934. http://dx.doi.org/10.1177/002215549704500702
- Bruckheimer, E.M. and Kyprianou, N. (2000) Apoptosis in Prostate Carcinogenesis―A Growth Regulator and a Therapeutic Target. Cell and Tissue Research, 301, 153-162. http://dx.doi.org/10.1007/s004410000196
- Dive, C. and Hickman, J.A. (1991) Drug-Target Interactions: Only the First Step in the Commitment to a Programmed Cell Death? British Journal of Cancer, 64, 192-196. http://dx.doi.org/10.1038/bjc.1991.269
- Launay, S., Hermine, O., Fontenay, M., Kroemer, G., Solary, E. and Garrido, C. (2005) Vital Functions for Lethal Caspases. Oncogene, 24, 5137-5148. http://dx.doi.org/10.1038/sj.onc.1208524
- Thornberry, N.A. and Lazebnik, Y. (1998) Caspases: Enemies Within. Science, 281, 1312-1316. http://dx.doi.org/10.1126/science.281.5381.1312
- Hajra, K.M. and Liu, J.R. (2004) Apoptosome Dysfunction in Human Cancer. Apoptosis, 9, 691-704. http://dx.doi.org/10.1023/B:APPT.0000045786.98031.1d
- Gottlieb, R.A., Nordberg, J., Skowronski, E. and Babior, B.M. (1996) Apoptosis Induced in Jurkat Cells by Several Agents Is Preceded by Intracellular Acidification. Proceedings of the National Academy of Sciences of the United States of America, 93, 654-658. http://dx.doi.org/10.1073/pnas.93.2.654
- Robinson, L.J., Roberts, W.K., Ling, T.T., Lamming, D., Sternberg, S.S. and Roepe, P.D. (1997) Human MDR 1 Protein Overexpression Delays the Apoptotic Cascade in Chinese Hamster Ovary Fibroblasts. Biochemistry, 36, 11169- 11178. http://dx.doi.org/10.1021/bi9627830
- Kinghorn, A.D., Balandrin, M.F., American Chemical Society, Division of Agricultural and Food Chemistry, et al. (1993) Human Medicinal Agents from Plants. American Chemical Society, Washington DC, xii, 356 p.
- Cordell, G.A. (2002) Natural Products in Drug Discovery―Creating a New Vision. Phytochemistry Reviews, 1, 261- 273. http://dx.doi.org/10.1023/A:1026094701495
- Nobili, S., Lippi, D., Witort, E., Donnini, M., Bausi, L., Mini, E. and Capaccioli, S. (2009) Natural Compounds for Cancer Treatment and Prevention. Pharmacological Research, 59, 365-378. http://dx.doi.org/10.1016/j.phrs.2009.01.017
- Colegate, S.M. and Molyneux, R.J. (2008) Bioactive Natural Products: Detection, Isolation, and Structural Determination. 2nd Edition, CRC Press, Boca Raton, xiii, 605 p.
- Newman, D.J. and Cragg, G.M. (2007) Natural Products as Sources of New Drugs over the Last 25 Years. Journal of Natural Products, 70, 461-477. http://dx.doi.org/10.1021/np068054v
- Richardson, M.A., Sanders, T., Palmer, J.L., Greisinger, A. and Singletary, S.E. (2000) Complementary/Alternative Medicine Use in a Comprehensive Cancer Center and the Implications for Oncology. Journal of Clinical Oncology, 18, 2505-2514.
- Sparber, A., Jonas, W., White, J., Derenzo, E., Johnson, E. and Bergerson, S. (2000) Cancer Clinical Trials and Subject Use of Natural Herbal Products. Cancer Investigation, 18, 436-439. http://dx.doi.org/10.3109/07357900009032815
- Boik, J. (2001) Natural Compounds in Cancer Therapy. Oregon Medical Press, Princeton, xiii, 521 p.
- El-Readi, M.Z., Eid, S., Ashour, M.L., Tahrani, A. and Wink, M. (2013) Modulation of Multidrug Resistance in Cancer Cells by Chelidonine and Chelidonium majus Alkaloids. Phytomedicine, 20, 282-294. http://dx.doi.org/10.1016/j.phymed.2012.11.005
- Eid, S.Y., El-Readi, M.Z., Eldin, E.E., Fatani, S.H. and Wink, M. (2013) Influence of Combinations of Digitonin with Selected Phenolics, Terpenoids, and Alkaloids on the Expression and Activity of P-Glycoprotein in Leukaemia and Colon Cancer Cells. Phytomedicine, 21, 47-61. http://dx.doi.org/10.1016/j.phymed.2013.07.019
- Eid, S.Y., El-Readi, M.Z. and Wink, M. (2012) Carotenoids Reverse Multidrug Resistance in Cancer Cells by Interfering with ABC-Transporters. Phytomedicine, 19, 977-987. http://dx.doi.org/10.1016/j.phymed.2012.05.010
- Eid, S.Y., El-Readi, M.Z. and Wink, M. (2012) Digitonin Synergistically Enhances the Cytotoxicity of Plant Secondary Metabolites in Cancer Cells. Phytomedicine, 19, 1307-1314. http://dx.doi.org/10.1016/j.phymed.2012.09.002
- Eid, S.Y., El-Readi, M.Z. and Wink, M. (2012) Synergism of Three-Drug Combinations of Sanguinarine and Other Plant Secondary Metabolites with Digitonin and Doxorubicin in Multi-Drug Resistant Cancer Cells. Phytomedicine, 19, 1288-1297. http://dx.doi.org/10.1016/j.phymed.2012.08.010
- Wink, M., Ashour, M.L. and El-Readi, M.Z. (2012) Secondary Metabolites from Plants Inhibiting ABC Transporters and Reversing Resistance of Cancer Cells and Microbes to Cytotoxic and Antimicrobial Agents. Frontiers in Microbiology, 3, 130. http://dx.doi.org/10.3389/fmicb.2012.00130
- Wink, M. (1999) Introduction: Biochemistry, Role and Biotechnology of Secondary Metabolites. Annual Plant Reviews, 2, 1-16.
- Wink, M. (2003) Evolution of Secondary Metabolites from an Ecological and Molecular Phylogenetic Perspective. Phytochemistry, 64, 3-19. http://dx.doi.org/10.1016/S0031-9422(03)00300-5
- Wink, M. (2008) Plant Secondary Metabolism: Diversity, Function and Its Evolution. Natural Product Communications, 3, 1205-1216.
- Wink M (1999) Introduction: Biochemistry, Role and Biotechnology of Secondary Metabolites. In: Wink, M., Ed., Functions of Plant Secondary Metabolites and Their Exploitation in Biotechnology, Academic, Sheffield, 1-16.
- Mann, J. (1994) Natural Products: Their Chemistry and Biological Significance. 1st Edition, Longman Scientific & Technical, Wiley, Harlow, Essex, England, New York, ix, 455 p.
- Harborne, J.B. and Williams, C.A. (2000) Advances in Flavonoid Research Since 1992. Phytochemistry, 55, 481-504. http://dx.doi.org/10.1016/S0031-9422(00)00235-1
- Manach, C., Morand, C., Gil-Izquierdo, A., Bouteloup-Demange, C. and Rémésy, C. (2003) Bioavailability in Humans of the Flavanones Hesperidin and Narirutin after the Ingestion of Two Doses of Orange Juice. European Journal of Clinical Nutrition, 57, 235-242. http://dx.doi.org/10.1038/sj.ejcn.1601547
- Vallejo, F., Larrosa, M., Escudero, E., Zafrilla, M.P., Cerdá, B., Boza, J., et al. (2010) Concentration and Solubility of Flavanones in Orange Beverages Affect Their Bioavailability in Humans. Journal of Agricultural and Food Chemistry, 58, 6516-6524. http://dx.doi.org/10.1021/jf100752j
- Crozier, A., Clifford, M.N. and Ashihara, H. (2006) Plant Secondary Metabolites: Occurrence, Structure and Role in the Human Diet. Blackwell Pub., Oxford, Ames, Iowa, xii, 372 p.
- Cho, Y.A., Kim, J., Park, K.S., Lim, S.Y., Shin, A., Sung, M.K. and Ro, J. (2010) Effect of Dietary Soy Intake on Breast Cancer Risk According to Menopause and Hormone Receptor Status. European Journal of Clinical Nutrition, 64, 924-932. http://dx.doi.org/10.1038/ejcn.2010.95
- Middleton, E., Kandaswami, C. and Theoharides, T.C. (2000) The Effects of Plant Flavonoids on Mammalian Cells: Implications for Inflammation, Heart Disease, and Cancer. Pharmacological Reviews, 52, 673-751.
- Cao, J., Chen, X., Liang, J., Yu, X.Q., Xu, A.L., Chan, E., et al. (2007) Role of P-Glycoprotein in the Intestinal Absorption of Glabridin, an Active Flavonoid from the Root of Glycyrrhiza glabra. Drug Metabolism and Disposition, 35, 539-553. http://dx.doi.org/10.1124/dmd.106.010801
- Wang, Y., Cao, J. and Zeng, S. (2005) Involvement of P-Glycoprotein in Regulating Cellular Levels of Ginkgo Flavonols: Quercetin, Kaempferol, and Isorhamnetin. Journal of Pharmacy and Pharmacology, 57, 751-758. http://dx.doi.org/10.1211/0022357056299
- Cermak, R. and Wolffram, S. (2006) The Potential of Flavonoids to Influence Drug Metabolism and Pharmacokinetics by Local Gastrointestinal Mechanisms. Current Drug Metabolism, 7, 729-744. http://dx.doi.org/10.2174/138920006778520570
- Conseil, G., Baubichon-Cortay, H., Dayan, G., Jault, J.M., Barron, D. and Di Pietro, A. (1998) Flavonoids: A Class of Modulators with Bifunctional Interactions at Vicinal ATP- and Steroid-Binding Sites on Mouse P-Glycoprotein. Proceedings of the National Academy of Sciences of the United States of America, 95, 9831-9836. http://dx.doi.org/10.1073/pnas.95.17.9831
- Johnson, W.W., Wang, E.J., Barecki-Roach, M., et al. (2002) Allosteric Elevation of P-Glycoprotein Function by a Catechin in Green Tea. Drug Metabolism Reviews, 34, 87.
- Morris, M.E. and Zhang, S. (2006) Flavonoid-Drug Interactions: Effects of Flavonoids on ABC Transporters. Life Sciences, 78, 2116-2130. http://dx.doi.org/10.1016/j.lfs.2005.12.003
- Shapiro, A.B. and Ling, V. (1997) Positively Cooperative Sites for Drug Transport by P-Glycoprotein with Distinct Drug Specificities. European Journal of Biochemistry, 250, 130-137. http://dx.doi.org/10.1111/j.1432-1033.1997.00130.x
- Di Pietro, A., Conseil, G., Perez-Victoria, J.M., Dayan, G., Baubichon-Cortay, H., Trompier, D., et al. (2002) Modulation by Flavonoids of Cell Multidrug Resistance Mediated by P-Glycoprotein and Related ABC Transporters. Cellular and Molecular Life Sciences CMLS, 59, 307-322. http://dx.doi.org/10.1007/s00018-002-8424-8
- Tang, M., Ogawa, K., Asamoto, M., Hokaiwado, N., Seeni, A., Suzuki, S., et al. (2007) Protective Effects of Citrus Nobiletin and Auraptene in Transgenic Rats Developing Adenocarcinoma of the Prostate (TRAP) and Human Prostate Carcinoma Cells. Cancer Science, 98, 471-477. http://dx.doi.org/10.1111/j.1349-7006.2007.00417.x
- Ju-Ichi, M. (2005) Chemical Study of Citrus Plants in the Search for Cancer Chemopreventive Agents. Yakugaku Zasshi, 125, 231-254. http://dx.doi.org/10.1248/yakushi.125.231
- Takanaga, H., Ohnishi, A., Yamada, S., Matsuo, H., Morimoto, S., Shoyama, Y., et al. (2000) Polymethoxylated Flavones in Orange Juice Are Inhibitors of P-Glycoprotein but Not Cytochrome P450 3A4. Journal of Pharmacology and Experimental Therapeutics, 293, 230-236.
- Dresser, G.K. and Bailey, D.G. (2003) The Effects of Fruit Juices on Drug Disposition: A New Model for Drug Interactions. European Journal of Clinical Investigation, 33, 10-16. http://dx.doi.org/10.1046/j.1365-2362.33.s2.2.x
- Evans, A.M. (2000) Influence of Dietary Components on the Gastrointestinal Metabolism and Transport of Drugs. Therapeutic Drug Monitoring, 22, 131-136. http://dx.doi.org/10.1097/00007691-200002000-00028
- Ioannides, C. (2002) Topics in Xenobiochemistry. Pharmacokinetic Interactions between Herbal Remedies and Medicinal Drugs. Xenobiotica, 32, 451-478. http://dx.doi.org/10.1080/00498250210124147
- Dahan, A. and Altman, H. (2004) Food-Drug Interaction: Grapefruit Juice Augments Drug Bioavailability―Mechanism, Extent and Relevance. European Journal of Clinical Nutrition, 58, 1-9. http://dx.doi.org/10.1038/sj.ejcn.1601736
- Chen, A.C. and Donovan, S.M. (2004) Genistein at a Concentration Present in Soy Infant Formula Inhibits Caco-2BBe Cell Proliferation by Causing G2/M Cell Cycle Arrest. Journal of Nutrition, 134, 1303-1308.
- Horie, N., Hirabayashi, N., Takahashi, Y., Miyauchi, Y., Taguchi, H. and Takeishi, K. (2005) Synergistic Effect of Green Tea Catechins on Cell Growth and Apoptosis Induction in Gastric Carcinoma Cells. Biological and Pharmaceutical Bulletin, 28, 574-579. http://dx.doi.org/10.1248/bpb.28.574
- Kaneuchi, M., Sasaki, M., Tanaka, Y., Sakuragi, N., Fujimoto, S. and Dahiya, R. (2003) Quercetin Regulates Growth of Ishikawa Cells through the Suppression of EGF and Cyclin D1. International Journal of Oncology, 22, 159-164.
- Kumi-Diaka, J., Sanderson, N.A. and Hall, A. (2000) The Mediating Role of Caspase-3 Protease in the Intracellular Mechanism of Genistein-Induced Apoptosis in Human Prostatic Carcinoma Cell Lines, DU145 and LNCaP. Biology of the Cell, 92, 595-604. http://dx.doi.org/10.1016/S0248-4900(00)01109-6
- Nguyen, T.T., Tran, E., Nguyen, T.H., Do, P.T., Huynh, T.H. and Huynh, H. (2004) The Role of Activated MEK-ERK Pathway in Quercetin-Induced Growth Inhibition and Apoptosis in A549 Lung Cancer Cells. Carcinogenesis, 25, 647- 659. http://dx.doi.org/10.1093/carcin/bgh052
- Banthorpe, D. (1991) Classification of Terpenoids and General Procedures for Their Characterization. In: Charlwood, B. and Banthorpe, D., Eds., Methods in Plant Biochemistry: Terpenoids, Academic Press, London, 1-41.
- Davis, E. and Croteau, R. (2000) Cyclization Enzymes in the Biosynthesis of Monoterpenes, Sesquiterpenes, and Diterpenes. In: Leeper, F. and Vederas, J., Eds., Topics in Current Chemistry: Biosynthesis: Aromatic Polyketides, Isoprenoids, Alkaloids, Springer, Berlin, New York, 53-95.
- Gershenzon, J. and Kreis, W. (1999) Biochemistry of Terpenoids. In: Wink, M., Ed., Biochemistry of Plant Secondary Metabolism, Annual Plant Reviews, Sheffield Academic Press, CRC Press, Sheffield, Boca Raton, 222-299.
- Roy, A. and Saraf, S. (2006) Limonoids: Overview of Significant Bioactive Triterpenes Distributed in Plants Kingdom. Biological & Pharmaceutical Bulletin, 29, 191-201. http://dx.doi.org/10.1248/bpb.29.191
- Manners, G.D., Jacob, R.A., Breksa III, A.P., Schoch, T.K. and Hasegawa, S. (2003) Bioavailability of Citrus Limonoids in Humans. Journal of Agricultural and Food Chemistry, 51, 4156-4161. http://dx.doi.org/10.1021/jf0300691
- Berhow, M.A., Hasegawa, S., Manners, G.D., et al. (2000) Citrus Limonoids: Functional Chemicals in Agriculture and Food. American Chemical Society, Washington DC, xiii, 253 p.
- Poulose, S.M., Harris, E.D. and Patil, B.S. (2005) Citrus Limonoids Induce Apoptosis in Human Neuroblastoma Cells and Have Radical Scavenging Activity. Journal of Nutrition, 135, 870-877.
- Miyagi, Y., Om, A.S., Chee, K.M. and Bennink, M.R. (2000) Inhibition of Azoxymethane-Induced Colon Cancer by Orange Juice. Nutrition and Cancer―An International Journal, 36, 224-229. http://dx.doi.org/10.1207/S15327914NC3602_12
- Berhow, M.A., Omura, M., Ohta, H., Ozaki, Y. and Hasegawa, S. (1994) Limonoids in Seeds of 3 Citrus Hybrids Related to Citrus ichangensis. Phytochemistry, 36, 923-925. http://dx.doi.org/10.1016/S0031-9422(00)90464-3
- Silalahi, J. (2002) Anticancer and Health Protective Properties of Citrus Fruit Components. Asia Pacific Journal of Clinical Nutrition, 11, 79-84. http://dx.doi.org/10.1046/j.1440-6047.2002.00271.x
- Tanaka, T., Kohno, H., Tsukio, Y., Honjo, S., Tanino, M., Miyake, M. and Wada, K. (2000) Citrus Limonoids Obacunone and Limonin Inhibit Azoxymethane-Induced Colon Carcinogenesis in Rats. BioFactors, 13, 213-218. http://dx.doi.org/10.1002/biof.5520130133
- Madyastha, K.M. and Venkatakrishnan, K. (2000) Structural Flexibility in the Biocatalyst-Mediated Functionalization of Ring “A” in Salannin, a Tetranortriterpene from Azadirachta indica. Journal of the Chemical Society, Perkin Transactions, 1, 3055-3062. http://dx.doi.org/10.1039/b004260i
- Dewick, P.M. (2009) Medicinal Natural Products: A Biosynthetic Approach. 3rd Edition, Wiley, Chichester, x, 539 p.
- Katan, M.B., Grundy, S.M., Jones, P., Law, M., Miettinen, T. and Paoletti, R. (2003) Efficacy and Safety of Plant Stanols and Sterols in the Management of Blood Cholesterol Levels. Mayo Clinic Proceedings, 78, 965-978. http://dx.doi.org/10.1016/S0025-6196(11)63144-3
- Lichtenstein, A.H. and Deckelbaum, R.J. (2001) AHA Science Advisory. Stanol/Sterol Ester-Containing Foods and Blood Cholesterol Levels. A Statement for Healthcare Professionals from the Nutrition Committee of the Council on Nutrition, Physical Activity, and Metabolism of the American Heart Association. Circulation, 103, 1177-1179. http://dx.doi.org/10.1161/01.CIR.103.8.1177
- Awad, A.B. and Fink, C.S. (2002) Phytosterols as Anticancer Dietary Components: Evidence and Mechanism of Action (Reprinted from Vol 130, Pg 2127, 2000). Journal of Nutrition, 132, 2127-2130.
- Oram, J.F. and Vaughan, A.M. (2006) ATP-Binding Cassette Cholesterol Transporters and Cardiovascular Disease. Circulation Research, 99, 1031-1043. http://dx.doi.org/10.1161/01.RES.0000250171.54048.5c
- Sarkadi, B., Homolya, L., Szakacs, G. and Váradi, A. (2006) Human Multidrug Resistance ABCB and ABCG Transporters: Participation in a Chemoimmunity Defense System. Physiological Reviews, 86, 1179-1236. http://dx.doi.org/10.1152/physrev.00037.2005
- Wink, M. (2008) Evolutionary Advantage and Molecular Modes of Action of Multi-Component Mixtures Used in Phytomedicine. Current Drug Metabolism, 9, 996-1009. http://dx.doi.org/10.2174/138920008786927794
- Brobst, D.E., Ding, X., Creech, K.L., Goodwin, B., Kelley, B. and Staudinger, J.L. (2004) Guggulsterone Activates Multiple Nuclear Receptors and Induces CYP3A Gene Expression through the Pregnane X Receptor. Journal of Pharmacology and Experimental Therapeutics, 310, 528-535. http://dx.doi.org/10.1124/jpet.103.064329
- Roberts, M.F. and Wink, M. (1998) Alkaloids: Biochemistry, Ecology, and Medicinal Applications. Springer, New York, 1-7.
- Wink, M. (1998) A Short History of Alkaloids. In: Alkaloids: Biochemistry, Ecology, and Medicinal Applications, Springer, New York, 11-43.
- Wink, M. (2007) Molecular Modes of Action of Cytotoxic Alkaloids: From DNA Intercalation, Spindle Poisoning, Topoisomerase Inhibition to Apoptosis and Multiple Drug Resistance. Alkaloids: Chemistry and Biology, 64, 1-47. http://dx.doi.org/10.1016/S1099-4831(07)64001-2
- Zdarilova, A., Malikova, J., Dvorak, Z., Ulrichová, J. and Šimánek, V. (2006) Quaternary Isoquinoline Alkaloids Sanguinarine and Chelerythrine. In Vitro and in Vivo Effects. Chemické Listy, 100, 30-41.
- Fu, L.W., Deng, Z.A., Pan, Q.C. and Fan, W. (2001) Screening and Discovery of Novel MDR Modifiers from Naturally Occurring Bisbenzylisoquinoline Alkaloids. Anticancer Research, 21, 2273-2280.
- Jakubikova, J., Duraj, J., Hunakova, L., Chorvath, B. and Sedlak, J. (2002) PK11195, an Isoquinoline Carboxamide Ligand of the Mitochondrial Benzodiazepine Receptor, Increased Drug Uptake and Facilitated Drug-Induced Apoptosis in Human Multidrug-Resistant Leukemia Cells in Vitro. Neoplasma, 49, 231-236.
- He, L. and Liu, G.Q. (2002) Effects of Various Principles from Chinese Herbal Medicine on Rhodamine123 Accumulation in Brain Capillary Endothelial Cells. Acta Pharmacologica Sinica, 23, 591-596.
- Wakusawa, S., Nakamura, S., Tajima, K., Miyamoto, K., Hagiwara, M. and Hidaka, H. (1992) Overcoming of Vin- blastine Resistance by Isoquinolinesulfonamide Compounds in Adriamycin-Resistant Leukemia Cells. Molecular Phar- macology, 41, 1034-1038.
- Moller, M., Weiss, J. and Wink, M. (2006) Reduction of Cytotoxicity of the Alkaloid Emetine through P-Glycoprotein (MDR1/ABCB1) in Human Caco-2 Cells and Leukemia Cell Lines. Planta Medica, 72, 1121-1126. http://dx.doi.org/10.1055/s-2006-941546
- Belyaeva, T., Leontieva, E., Shpakov, A., Mozhenok, T. and Faddejeva, M. (2003) Sensitivity of Lysosomal Enzymes to the Plant Alkaloid Sanguinarine: Comparison with Other SH-Specific Agents. Cell Biology International, 27, 887- 895. http://dx.doi.org/10.1016/S1065-6995(03)00161-6
- Salminen, K.A., Meyer, A., Jerabkova, L., Korhonen, L.E., Rahnasto, M., Juvonen, R.O., et al. (2010) Inhibition of Human Drug Metabolizing Cytochrome P450 Enzymes by Plant Isoquinoline Alkaloids. Phytomedicine, 18, 533-538.
- Adhami, V.M., Aziz, M.H., Mukhtar, H. and Ahmad, N. (2003) Activation of Prodeath Bcl-2 Family Proteins and Mitochondrial Apoptosis Pathway by Sanguinarine in Immortalized Human HaCaT Keratinocytes. Clinical Cancer Research, 9, 3176-3182.
- Adhami, V.M., Aziz, M.H., Reagan-Shaw, S.R., Nihal, M., Mukhtar, H. and Ahmad, N. (2004) Sanguinarine Causes Cell Cycle Blockade and Apoptosis of Human Prostate Carcinoma Cells via Modulation of Cyclin Kinase Inhibitor- Cyclin-Cyclin-Dependent Kinase Machinery. Molecular Cancer Therapeutics, 3, 933-940.
- Malikova, J., Zdarilova, A. and Hlobilkova, A. (2006) Effects of Sanguinarine and Chelerythrine on the Cell Cycle and Apoptosis. Biomedical Papers of the Medical Faculty of Palacký University, 150, 5-12. http://dx.doi.org/10.5507/bp.2006.001
- Ding, Z.H., Tang, S.C., Weerasinghe, P., Yang, X., Pater, A. and Liepins, A. (2002) The Alkaloid Sanguinarine Is Effective against Multidrug Resistance in Human Cervical Cells via Bimodal Cell Death. Biochemical Pharmacology, 63, 1415-1421. http://dx.doi.org/10.1016/S0006-2952(02)00902-4
- Moller, M., Herzer, K., Wenger, T., Herr, I. and Wink, M. (2007) The Alkaloid Emetine as a Promising Agent for the Induction and Enhancement of Drug-Induced Apoptosis in Leukemia Cells. Oncology Reports, 18, 737-744.
- Moller, M. and Wink, M. (2007) Characteristics of Apoptosis Induction by the Alkaloid Emetine in Human Tumour Cell Lines. Planta Medica, 73, 1389-1396. http://dx.doi.org/10.1055/s-2007-990229
- Rosenkranz, V. and Wink, M. (2008) Alkaloids Induce Programmed Cell Death in Bloodstream Forms of Trypanosomes (Trypanosoma b. brucei). Molecules, 13, 2462-2473. http://dx.doi.org/10.3390/molecules13102462
- Van Wyk, B.E. and Wink, M. (2004) Medicinal Plants of the World: An Illustrated Scientific Guide to Important Medicinal Plants and Their Uses. 1st Edition, Timber Press, Portland, 480 p.
- Bartak, P., Simanek, V., Vlckova, M., Ulrichová, J. and Vespalec, R. (2003) Interactions of Sanguinarine and Chelerythrine with Molecules Containing a Mercapto Group. Journal of Physical Organic Chemistry, 16, 803-810. http://dx.doi.org/10.1002/poc.659
- Ulrichova, J., Dvorak, Z., Vicar, J., Lata, J., Smržová, J., Šedo, A. and Šimánek, V. (2001) Cytotoxicity of Natural Compounds in Hepatocyte Cell Culture Models: The Case of Quaternary Benzo[C]phenanthridine Alkaloids. Toxicology Letters, 125, 125-132. http://dx.doi.org/10.1016/S0378-4274(01)00430-1
- Slaninova, I., Taborska, E., Bochorakova, H. and Slanina, J. (2001) Interaction of Benzo[C]phenanthridine and Protoberberine Alkaloids with Animal and Yeast Cells. Cell Biology and Toxicology, 17, 51-63. http://dx.doi.org/10.1023/A:1010907231602
- Walterova, D., Ulrichova, J., Preininger, V., Simanek, V., Lenfeld, J. and Lasovsky, J. (1981) Inhibition of Liver Alanine Aminotransferase Activity by Some Benzophenanthridine Alkaloids. Journal of Medicinal Chemistry, 24, 1100- 1103. http://dx.doi.org/10.1021/jm00141a019
- Stiborova, M., Simanek, V., Frei, E., Hobza, P. and Ulrichová, J. (2002) DNA Adduct Formation from Quaternary Benzo[C]phenanthridine Alkaloids Sanguinarine and Chelerythrine as Revealed by the P-32-Postlabeling Technique. Chemico-Biological Interactions, 140, 231-242. http://dx.doi.org/10.1016/S0009-2797(02)00038-8
- Das, M., Ansari, K.M., Dhawan, A., Shukla, Y. and Khanna, S.K. (2005) Correlation of DNA Damage in Epidemic Dropsy Patients to Carcinogenic Potential of Argemone Oil and Isolated Sanguinarine Alkaloid in Mice. International Journal of Cancer, 117, 709-717. http://dx.doi.org/10.1002/ijc.21234
- Wink, M. (2006) Chapter 11 Importance of Plant Secondary Metabolites for Protection against Insects and Microbial Infections. In: Mahendra, R. and María Cecilia, C., Eds., Advances in Phytomedicine, Vol. 3, Elsevier, Amsterdam, 251-268.
NOTES

*Corresponding authors.