International Journal of Otolaryngology and Head & Neck Surgery
Vol.3 No.2(2014), Article ID:44260,7 pages DOI:10.4236/ijohns.2014.32017
Multicentric Cervical Paraganglioma. Rare Case of Five Locations in One Patient. Case Report and Literature Review
José Francisco de Sales Chagas1*, Maria Beatriz Nogueira Pascoal1, José Luis Braga de Aquino1, Luis Antonio Brandi Filho1, Fernanda Fruet1, Douglas Alexandre Rizzanti Pereira1, Amilcar de Castro Matos2, Thalita Ruba3, Fernanda Callegari3
1Head and Neck Service, School of Medicine, Catholic University of Campinas, Campinas, Brazil
2Department of Pathology, School of Medicine, Catholic University of Campinas, Campinas, Brazil
3School of Medicine, Catholic University of Campinas, Campinas, Brazil
Email: *josechagas@ig.com.br, maribibia@ig.com.br, jlaquino@sigmanet.com.br, luisantoniobrandifilho@hotmail.com, ferfruet@yahoo.com.br, med_dpereira@yahoo.com.br, sunsettown@hotmail.com, thalitaruba@gmail.com, fe_callegari@terra.com.br
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 8 December 2013; revised 7 January 2014; accepted 6 February 2014
ABSTRACT
Objective: To report a case of a multiple paraganglioma with 5 concurrent locations and also describe a complication not found in the literature, spontaneous tumor bleeding. Introduction: The paragangliomas are mostly benign and more frequent in women. They can be multiple, and these are considered as the most likely source of familial origin (30% - 35%). The multicentric lesions can occur at different life stages not being necessarily simultaneous, which is a fact that prevents image exam research to be conclusive for tumor presence or absence. In this way the examination of the family members of patients, specially young ones, diagnosed with multicentric tumors may lead to negative results which doesn’t mean that the disease can be developed at a later time period. Therefore routine examination is not justifiable when symptoms relative to tumor presence are not present. Case report: A female patient of 32 years old complaining of tinnitus and diagnosed with five paragangliomas (jugulo-timpanic, bilateral carotid body and bilateral brachiocephalic trunk) through imaging studies. She was reported suffering relatives of paragangliomas. Conclusion: The paper reports a rare case and perhaps unique in the literature, multiple paraganglioma with five locations and discusses difficult choice of treatment and complications.
Keywords:Glomus Tumor; Paraganglioma; Glomus Jugulare; Carotid Body Tumor; Postoperative Complications

1. Introduction
Paragangliomas are tumors of neural crest ectodermal cells that develop in the paravertebral region associated to the cervical vessels, cranial nerves and the autonomic nervous system. There are two cell types: type I, containing catecholamine granules and type II, sustentation cells that circumscribe type I and have similar appearance to Schwann cells. They produce and store vasoactive substances, neurotransmitters (catecholamine, epinephrine and norepinephrine) and hormones, like serotonin, gastrin and somatostatin.
The paraganglionic system can be divided into two groups: 1) branquiomeric group, related to branquial arch development, including the jugulo-tympanic paraganglia, carotid, vagal, orbital, subclavian and laryngeal, corresponding to the non-chromaffin or non-catecholaminergic paraganglia, 2) metameric group, containing the paraganglia situated on spinal metamer, thoracic and abdominal levels; corresponding to the cromafins or catecholaminergic paraganglia. In respect to their starting location, they should be discriminated into two groups, the ones in the head and neck region being the carotid body the highest frequency local and the ones located below the head and neck, having the adrenal medulla as the most frequent location (pheochromocytomas). The head and neck paraganglia are associated to the parasympatic system and locate around arteries and bigger nerves while the adrenal medulla associates itself with the sympathetic nervous system [1] .
Mostly benign (97%) paragangliomas are rare tumors, corresponding to 0.6% of all head and neck tumors, having chronic hypoxia as one of its possible onset causes. There is a higher incidence in females and the most common symptom is the presence of painless slowly growing cervical mass. Other related symptoms are pulsatile tinnitus, and hearing loss for the tympanic or jugular tumors. In the advanced cases, cranial nerves could also be affected (paralysis). The tumors are isolated or multicentric, either in sporadic or familial cases, the latter following autossomic dominant heritage and having higher multicentric occurrence (30% - 35%). Contrary to pheochromocytomas, the head and neck paragangliomas are rarely (5%) metabolic active and secreting catecholamine [2] [3] .
2. Goals
Report a case of multicentric cervical paranganglioma where tumors were found in five different locations, while literature review shows a maximum of three cases in one patient. We discuss the difficulties on treatment choice and possible complications.
3. Case Report
A 32 years old female patient, presented herself with pulsatile tinnitus on the left ear for six months. Physical exam presented no abnormalities, with normal vital signs. Otoscopy showed reddish mass and secretion in the middle ear and audiometry test resulted in conductive hearing loss. A jugulo-timpanic paraganglioma was confirmed by computer tomography of the temporal region where a high density lesion was found in the middle ear topography with enlargement and erosion of the jugular foramen. Four extra high density lesions were found in the same exam; two in the carotid bulb topography and two in the brachiocephalic trunks, compatible with multiple paraganglioma (Figure 1). The magnetic resonance image confirmed confirmed the findings of computed tomography. The patient was submitted to angiography with carotid balloon test, which showed good perfusion at the contralateral cerebral hemisphere.
The first surgical procedure was performed by means of cranio facial approach through Fish access to remove the jugulo-timpanic paraganglioma. Post operatively, the patient presented complete paralysis of the facial nerve and left vocal cord due to resection of the vagus nerve. The speech impairment was treated, with significant improvement, by compensatory speech therapy. A year later the glomic tumor located at the right carotid body was removed through transverse cervicotomy (Figure 2), with no post operative complications. The tumor in the

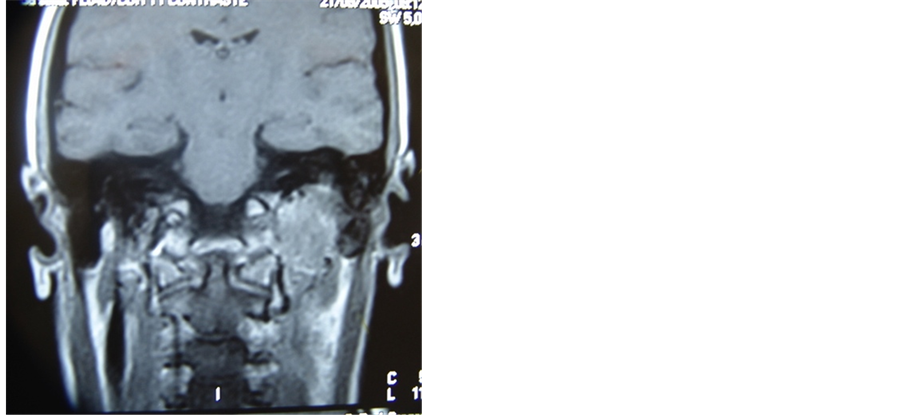

Figure 1. Axial and coronal MRI planes showing left paraganglioma jugulo-tympanic and CT with the remaining 4 paragangliomas (two in carotid bulbs and two in transition to mediastinum).
right parapharyngeal space was removed by cervicotomy. In the third post operative day, left cervical bulginess and respiratory insufficiency were caused by spontaneous bleeding of the tumor located in the contra lateral carotid body, as verified in the emergency surgical procedure performed. After surgery the patient had ischemic vascular accident which led to obit.

Figure 2. Intra-operative view of carotid body tumor.
4. Discussion
In the head and neck region the branchiomeric paraganglionic tissue can be found associated to vascular or nervous structures. It also appears, in high concentration, around the carotid and jugular bulbs and in smaller concentrations around cranial nerves. This tissue forms the paraganglionic system, a term used to characterize a structure of the neural crest, composed of chromophilic cells located outside the adrenal gland. Up to 90% of the tumors born in this structure, denominated pheocromocytomas, originate in the adrenal gland. The remaining 10% originate in the abdomen (85%), thorax (12%) and less common originating in the head and neck region (3%) [4] .
The paragangliomas classification into chromophilic and chromophobic was designed to differentiate the catecholamine producing tumors, the ones that react to chromic acid, producing brown polymers, from the ones that do not produce catecholamine [5] . Although all paragangliomas have secreting granules (having, therefore, potential signals of physiological activity), only a small portion (1% to 3%) demonstrates clinical evidences. Patients with clinical indications of exacerbated catecholamine production should be evaluated for the presence of functional or multiple paraganglioma (as for instance in the cases where paragangliomas are associated with other neural crest derived tumors in the multiple endocrine neoplasia). Therefore, paragangliomas are neural crest ectodermic cell tumors and the term “glomic tumor” frequently used to describe these lesions is inappropriate and should be avoided, since the true glomic tumors are located on the skin and superficial tissue, having no relation to paraganglia. The term quimiodectoma (derived from the greek words chemeia, decesthai and oma, meaning respectively infusion, receive and tumor) was introduced in 1950 by Mulligan to describe tumors in the carotid body derived from chemoreceptor cells [6] .
They present equal distribution in between ethinic groups and geographic regions but some authors consider a higher prevalence for populations living in high altitudes. More frequent in women aged between 30 to 60 years, being sporadic or familial with the latter ranging from 10% to 50% of cases, appearing isolated or together with other syndromes e.g., type 2 multiple endocrine neoplasia (MEN 2A and 2B), Von Hippel-Lindau syndrome and type 1 neurofibromatosis. Generally it is assumed 35% of the paraganglioma patients have a hereditary predisposition for developing the disease. Presently 4 cromossomic loci have been associated to the familial forms: 11q23 (PGL1), 11q13 (PGL2), 1q21 (PGL3), and 1p36.1p35 (PGL4). Associative genetic studies have identified the genes involved with the disease in three loci: SDHD (11q23), SDHC (1q21) and SDHB (1p36.1p35), with no gene identified in PGL2. The SDH gene mutations are responsible for 70% of the familial cases and one third of sporadic cases present alterations in the same genes (occult familial forms). In the remaining cases the causing factors are still unknown. The mutations in SDHB and SDHC associated to tumors are expressed in dominant autossomic form, while the ones in SDHD or in PGL2 are also dominant autossomic but ruled by genomic imprinting, i.e., there is no expression when the mutated genes are inherited from the mother, while inheritance from the father will result in disease. Gene identification unfortunately doesn’t correspond to early tumor detection since it would favor early treatment and decrease surgical morbidity [1] [5] .
Originally described in 1743 by Haller Vau, the carotid body is the most common location for the head and neck paragangliomas, followed by the jugulo-tympanic ganglion, initially describe by Vallentin in 1840. Vagal paraganglional bodies were discovered in 1935 by White and represent less than 5% of all head and neck paragangliomas. Those originating from the sympatic chain and superior laryngeal nerve are, most likely, the less frequent and very rare. Even less common are the ones occurring in aberrant locations e.g., nasal cavity, orbit and paranasal sinuses [5] .
Histologically they are well circumscribed and composed of chief cells arranged in nests, with granular cytoplasm and round nuclei with prominent nucleoli. Nuclear pleomorphism may be present, but mitosis is rare, and necrosis is usually present only if the patient underwent preoperative embolisation or if the nests of cells are very large. Compressed sustentacular cells and a rich capillary network surround each nest (Figure 3). Malignant varieties are difficult to distinguish on histologic examination, but generally they have a higher mitotic rate and more necrosis than benign tumours. Vascular invasion may be present in both benign and malignant paragangliomas. The chief cells are positive for neuroendocrine markers such as chromogranin and synaptophysin (Figure 4).
Complex anatomical location and high vascularization make the surgical handling difficult and controversial in relation to the best treatment course. One of the main parameters considered to define proper therapy is the
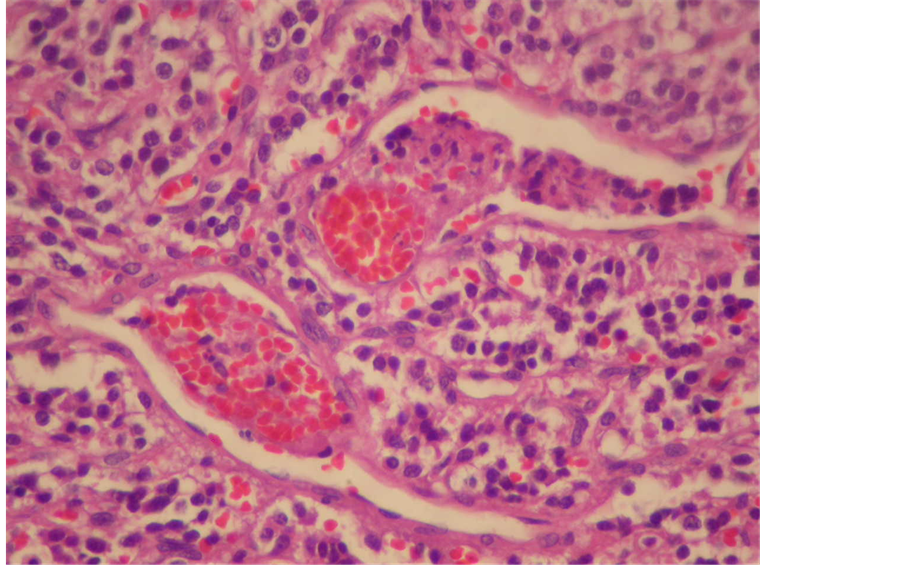
Figure 3. Tumor cells containing abundant cytoplasm, nuclear regularity and organoid appearance.
Figure 4. Presence of cells strongly marked by positivity for granules of chromogranin.
patient’s age, since the tumor’s insidious growth profile allows elderly patients to be initially treated through more conservative methods, such as radiotherapy or observation of the tumor development, due to the potential surgical morbidity caused by the anatomical structures involved. Such approach is not interesting for young patients since the tumoral growth could affect important structures such as the medial ear, the central nervous system and cranial nerves. These factors affect the therapeutic approach choice, because they increase the surgical difficulty and consequently the post operative morbidity due to the increased risk of stroke, dysphonia, dysphagia or aspiration. Inversely, in advanced disease situations, the cranial pairs are paralyzed, with permanent nerve functional loss surgical ablation is the chosen therapeutic approach, corroborated by the fact that radiotherapy only slows disease progress, even for early cases. The symptoms (tinnitus and paralysis of the cranial nerves) persisted in all patients and there was no regression in the post treatment evaluation. For that reason the surgical treatment begun through resection of the jugulo-tympanic lesion that, according to Fisch’s classification, presented itself as D1 i.e., invading the central nervous sistem. Guiding the therapeutical choices were the patient’s age, global clinical condition, the tumor’s biological behavior, its size and the treatment’s risks [7] -[9] .
Bocian and Tuschka, in 1958, described a multicentric cervical paragangioma with three locations [10] , since then there were several reports about paraganglioma multicentricity in the literature, with familial cases ranging from 30% to 35% of cases and 10% to 20% of sporadic cases, all of them showing maximally 3 locations in the same patient. Our report describes 5 locations occurring at the same time with first symptoms appearing due to the jugulo-tympanic lesion which led to a tomography that revealed the remaining four tumors. Spontaneous bleeding from the tumor as seen in this patient after cervical exploratory surgery was not found in literature review. The multicentric lesions can occur at different life stages not being necessarily simultaneous as proposed by Isik et al. [3] , a fact that prevents image exam research to be conclusive for tumor presence or absence. The reported familial cases were diagnosed through image exams only after the symptoms justified the use of the former [3] [5] . In this way the examination of the family members of patients, specially young ones, diagnosed with multicentric tumors may lead to negative results which doesn’t meant that the relatives are free from developing the same disease at a later time period. Therefore routine examination is not justifiable when symptoms relative to tumor presence are not present.
Pre operative embolization is recommended by several authors, but the procedure should not be mandatory due to the risk of cerebral embolism and the lack of proof in the operative bleeding reduction, even in the jugular gulf tumors. Endocrine activity can be observed through taquicardia, hypertension and tremor, although paragangliomas are normally not functional which leads to controversy as to use routine investigation used for secreting tumors. Patients with familial or multiple tumors may have higher incidence of catecholamine producing lesions, generally feocromocitomas [3] .
5. Conclusion
This article demonstrates rare aspects of the paragangliomas which were not found previously in the literature: multicentric through five sinchronic locations in one patient, spontaneous bleeding of the tumor and higher treatment complexity as compared to non-multicentric cases.
References
- Suárez Nieto, C., Llorente Pendás, J.L. and Martínez, J.G. (2009) Tratamiento Quirúrgico de los Paragangliomas Vagales. Acta Otorrinolaringológica Española, 60, 97-105.
- Köhler, H.F., Carvalho, A.L., Nishinari, K. and Kowalski, L.P. (2010) Internal Carotid Artery Reconstruction after Paraganglioma Resection: Report of Six Cases and Analysis of Available Techniques. Revista Brasileira de Cirurgia de Cabeça e Pescoço, 39, 210-213.
- Isik, A.C.U., Imamoglu, A.C.E.M., Cinel, A. and Maral, A.S.G. (2006) Familial Paraganglioma. European Archives of Oto-Rhino-Laryngology, 263, 23-31. http://dx.doi.org/10.1007/s00405-004-0885-y
- Havas, T.E. and Gullane, P.J. (1989) Multicentric Chemodectoma in the Head and Neck. ANZ Journal of Surgery, 59, 641-645. http://dx.doi.org/10.1111/j.1445-2197.1989.tb01648.x
- Sobol, S.M. and Dailey, J.C. (1990) Familial Multiple Cervical Paragangliomas: Report of a Kindred and Review of the Literature. Otolaryngology—Head and Neck Surgery, 102, 382-390.
- Pellitteri, P.K., Rinaldo, A., Myssiorek, D., Jackson, C.G., Bradley, P.J., Devaney, K.O., Shaha, A.R., Netterville, J.L., Manni, J.L. and Ferlito, A. (2004) Paragangliomas of the Head and Neck. Oral Oncology, 40, 563-575.http://dx.doi.org/10.1016/j.oraloncology.2003.09.004
- Dall′Igna, C., Antunes, M.B. and Dall′Igna, D.P. (2005) Radiation Therapy for Glomus Tumors of the Temporal Bone. Brazilian Journal of Otorhinolaryngology, 71, 752-757.
- Borba, L.A.B., Araújo, J.C., Oliveira, J.G., Giudicissi Filho, M., Moro, M.S., Tirapelli, L.F. and Colli, B.O. (2009) Surgical Management of Glomus Jugulare Tumors: A Proposal for Approach Selection Based on Tumor Relationships with the Facial Nerve. The Journal of Neuroscience, 8, 1-11.
- Chung, S.M., Kim, H.S., Jung, J., Lee, H.K. and Lee, W.S. (200) Clinical Presentation and Management of Jugular Foramen Paraganglioma. Clinical and Experimental Otorhinolaryngology, 9, 28-32.http://dx.doi.org/10.3342/ceo.2009.2.1.28
- Bocian, J.J. and Tushka, O.J. (1958) Multicentric Paragangliomas. Vagal and Carotid Body Tumors. California Medicine, 88, 51-54.
NOTES

*Corresponding author.