Neuroscience & Medicine
Vol.4 No.1(2013), Article ID:28572,7 pages DOI:10.4236/nm.2013.41005
Effects of Methylprednisolone on the Expression and Activity of Calpain Following Ischemia-Reperfusion Spinal Cord Injury in Rats
Zifeng Zhang1,2*, Jinquan Xu2, Yushu Bai3, Tiesheng Hou3
1Guanghua Integrative Medicine Hospital, Changning District, Shanghai, China
2School of Naval Architecture, Ocean and Civil Engineering, Shanghai Jiao Tong University, Shanghai, China
3Department of Orthopedics, Shanghai Changhai Hospital, Shanghai, China
Email: *zzfspine@hotmail.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 30 December 2013; revised 28 January 2014; accepted 18 February 2014
ABSTRACT
The present study was undertaken to examine the effects of methylprednisolone on the expression and activity of calpain in spinal cord tissue following spinal cord ischemia-reperfusion injury in rats. Adult male Sprague-Dawley rats were subjected to sham operations, ischemia-reperfusion and vehicle treated, or ischemia-reperfusion with methylprednisolone administration after injury. The expression of calpain I in the injured segments of the spinal cord as well as the degradation of the 68 kD neurofilament protein (NFP), a calpain-specific substrate, was determined at 3 h, 24 h, 72 h and 7 days after reperfusion using immunohistochemical labeling and western blot analysis, respectively. Three hours after spinal cord reperfusion, calpain I-positive cells and NFP degradation products were evident. The number of positive cells and immunoreactivity increased with time and peaked at 72 h after reperfusion. In addition, the number of calpain I-positive cells and the abundance of NFP degradation products were significantly lower in the methylprednisolone group, compared with vehicle treated animals following ischemia-reperfusion injury. The results of this study suggest that methylprednisolone can inhibit the expression and degradation activity of calpain following ischemia-reperfusion injury, providing further insight into the therapeutic benefits of methylprednisolone treatment for spinal cord injury.
Keywords: Methylprednisolone; Spinal Cord Injury; Ischemia-Reperfusion Injury; Calpain

1. Introduction
Methylprednisolone is a well-established drug for the treatment of spinal cord injury [1] [2] , and has been shown to be capable of reducing secondary lesions, improving secondary functional damage, and exerting a protective effect on the spinal cord following ischemia-reperfusion injury [3] . The mechanisms underlying the therapeutic effects of methylprednisolone are generally considered to include promotion of spinal cord impulse generation, enhancement of signal transmission via monosynaptic or polysynaptic connections, and maintenance of sensory and motor conduction functions of white matter. In addition, methylprednisolone has been purported to scavenge free radicals, reduce lipid peroxidation in the spinal cord, stabilize lysosomal membranes, alleviate tissue degeneration, and prevent neurofilament degradation. Methylprednisolone may also increase blood flow to the spinal cord after injury, preventing progressive ischemia after trauma, as well as suppressing inflammatory responses.
Increases in calpain protein expression are an integral part of the pathological process after spinal cord ischemia-reperfusion injury [4] . Upon binding with calcium ions, calpain causes degradation of cytoskeletal proteins, leading to neuronal necrosis [5] or apoptosis [6] . Calpain inhibitors have been suggested to be capable of protecting neurons [7] and spinal cord function [8] -[12] following injury.
Methylprednisolone has previously been shown to inhibit calpain activity, and this inhibitory effect was hypothesized to be mediated via either interference with the association of calmodulin or Ca2+ with calpain, or by blocking the active sites of calpain [13] . Using a deep hypothermic cardiopulmonary bypass (DHCPB) model in newborn piglets, Schwartz et al. [13] found that methylprednisolone administration (30 mg/kg) six hours before surgery, and at the initiation of surgery, significantly reduced the degradation of the calpain target protein troponin I and preserved levels of the natural calpain inhibitor calpastatin, suggesting that the protective effect of methylprednisolone on the myocardium is mediated by enhancing the activity or increasing the stability of calpain inhibitors. However, few studies have examined the effect of methylprednisolone on calpain expression following spinal cord ischemia-reperfusion injury. The present study was undertaken to assess the effects of methylprednisolone on calpain expression and activity following spinal cord ischemia-reperfusion injury in the rat.
2. Materials and Methods
2.1. Animals
One hundred and eight adult male Sprague-Dawley rats weighing 190 - 220 g were randomly assigned to three groups. The control group, Group A (n = 12), received sham operations only; Group B received an ischemiareperfusion injury with physiological saline administration (n = 48); and Group C received an ischemia-reperfusion injury as well as methylprednisolone administration (n = 48).
2.2. Spinal Cord Injury Model
Animals were anesthetized with an intraperitoneal injection of 2% sodium pentobarbital (40 mg/kg). A longitudinal incision along the median abdomen was made to open the abdominal cavity. The abdominal aorta was then exposed and the bilateral renal arteries were identified. The abdominal aorta was clipped with an artery clamp approximately 1 cm above the left renal artery, and below the right renal artery. Thirty minutes later, the artery clamp was released to initiate reperfusion. In Group C, intravenous methylprednisolone (165 mg/kg; via tail vein) was immediately administered after the removal of the artery clamp. In Group B, intravenous physiological saline administration (0.9% NaCl, 2 ml; via tail vein) was immediately administered after removal of the artery clamp. Sham operations were performed upon animals in the control group, in which only the abdominal aorta was exposed before closure of the abdominal cavity.
None of the animals had complications related to the surgery.
2.3. Immunohistochemical Staining of Calpain-Positive Cells
At four time points (3 h, 24 h, 72 h and 7 d post-reperfusion), six animals from each group were anesthetized, cannulated in the femoral artery, and perfused with 0.9% saline followed by 4% paraformaldehyde. The L1 - L5 spinal cords were dissected and placed into paraformaldehyde for another 24 h at 4˚C, then sectioned to 4 μm thickness, and processed for immunohistochemistry with goat monoclonal anti-calpain I antibody, developed with HRP chromogen (Santa Cruz Biotechnology, Inc., USA) and counterstained with H&E stain. Cells displaying brownish yellow or brown staining with a granular shape and a purple-blue background were considered to be positive for calpain immunoreactivity. Immunohistochemical images were photographed using an Olympus BH2 microscope and analyzed using the IMS cell image analysis system (Shanghai ShenTeng Information Technology Co., Ltd., China) by randomly selecting three visual fields from each slice (10×). Calpain immunoreactive cells appeared yellowishor dark brown. The optical density of calpain immunoreactivity was measured automatically using the IMS cell image analysis system. The control group at 3 hours was used as the control for all the time points of the experimental groups.
2.4. Western Blot Analysis of NFP Expression
At four time points (3 h, 24 h, 72 h and 7 d post-reperfusion), six animals from each group were anesthetized and the vertebral lamina below the thoracic segment was removed from the posterior median line. The L1 - L5 spinal cords were resected in an ice bath, with care to avoid mechanical injury of the spinal cords during this operation. The L1 - L5 spinal cords were immediately stored at −70˚C.
For western blot analysis, 100 mg of spinal cord tissue from each animal was lysed in RIPA buffer containing PMSF on an ice bath. Protein homogenates were then transferred to fresh tubes and centrifuged at 10,000 rpm for 10 min at 4˚C. Supernatant were collected and preserved at −70˚C until being subjected to SDS-PAGE. Proteins were separated on a 12% SDS-PAGE gel and transferred to a nitrocellulose membrane. After blocking with 5% nonfat milk in TBST, the membrane was incubated with goat monoclonal anti-68 kD NFP primary antibody (1:200, Santa Cruz) at 37˚C for 2 h. Following repeated washes with 0.1 M PBS-Tween, the membrane was incubated with alkaline phosphatase-labeled secondary antibody (1:400, Boster Biotechnology, Inc., China) at 37˚C for 1 h. The signal was developed and photographed (SABC-AP kit, Boster Biotechnology, Inc., China).
2.5. Statistical Analyses
Data are presented as mean ± SD. Differences in calpain and NFP expression were evaluated with t-tests using the SPSS (version 10.0) statistical software. P values less than 0.05 were considered to be statistically significant.
3. Results
3.1. Methylprednisolone Administration Downregulates the Expression of Calpain-I Following Spinal Cord Ischemia-Reperfusion Injury
No obvious immunoreactivity for calpain-I was found in the control group (Figure 1). In contrast, at different time points after reperfusion, a significant number of calpain-I-positive cells were observed in Groups B and C, as evidenced by the presence of quantities of particles with varying intensities of light yellowish or deep brown staining in neurons and glial cells. Figure 2 demonstrates immunohistochemical labeling of calpain-I in the injury-only group (Group B) at 72 h following reperfusion. The red arrow indicates calpain immunoreactive neurons and the black arrow indicates calpain-negative neurons. Figure 3 shows immunohistochemical labeling of calpain-I in the methylprednisolone-treated group (Group C) at 72 h following reperfusion. The red arrow indicates immunoreactive neurons and the black arrow indicates calpain-negative neurons. The number of positive cells per unit area and the staining intensities in Group C were lower than those in Group B at 72 h post-reperfusion. With administration of methylprednisolone (Group C), the number and density of calpain-I positive cells in the spinal cord were significantly lower than those in animals receiving the injury alone (Group B). Quantitative analysis revealed that the intensity of calpain-I immunoreactivity in the two groups was most marked at 72 h post-reperfusion. In addition, the intensity of calpain-I immunoreactivity was significantly lower with methylprednisolone administration (Group C) at all time points compared with the injury-alone group (Group B; Table 1).
3.2. Methylprednisolone Administration Inhibited the Degradation of NFP Following Spinal Cord Ischemia-Reperfusion Injury
The results of the NFP western blotting following spinal cord ischemia-reperfusion injury are presented in Figure 4. Following ischemia-reperfusion injury alone (Group B), the degradation of the 68 kD NFP species
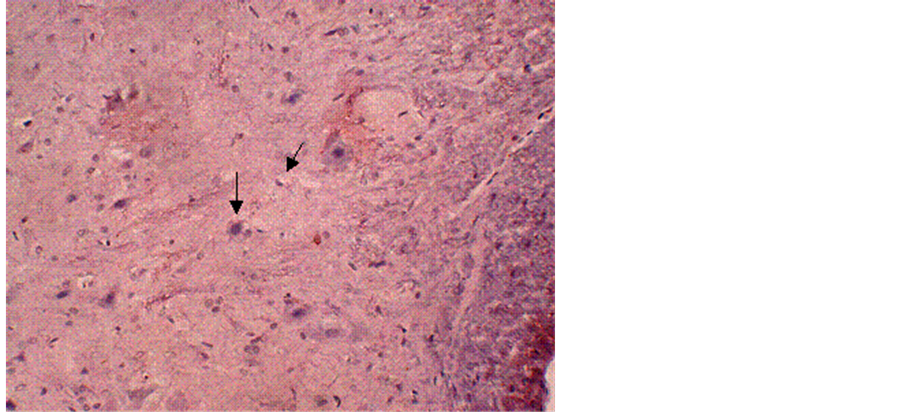
Figure 1. Immunohistochemical labeling of calpain-I in a control tissue section counterstained with H&E. The arrows indicate normal neurons and glial cells. Magnification = 10×.

Figure 2. Immunohistochemical labeling of calpain-I in the injury-only group (Group B) at 72 h following reperfusion. The red arrow indicates positively-stained neurons and the black arrow indicates negativelystained neurons. Magnification = 10×.
could be detected as early as 3 h following ischemia-reperfusion in the spinal cord. The extent of degradation increased with time, reaching a plateau 7 d following the injury. With the administration of methylprednisolone, the degradation of NFP was significantly reduced. The relative levels of the intact 68 kD NFP protein in Group C were 1.4, 1.4, and 2.4 times those of Group B at 3 h, 24 h, and 72 h post reperfusion, respectively (P < 0.01). At 7 days after reperfusion, the levels of NFP in Group C were slightly higher than those in Group B, but this difference was not statistically significant (Table 2).
4. Discussion
Previous studies have established the neuroprotective effects of calpain inhibitors following spinal cord injury [14] -[20] . In addition to specific calpain inhibitors, other molecules such as melatonin have been found to have a
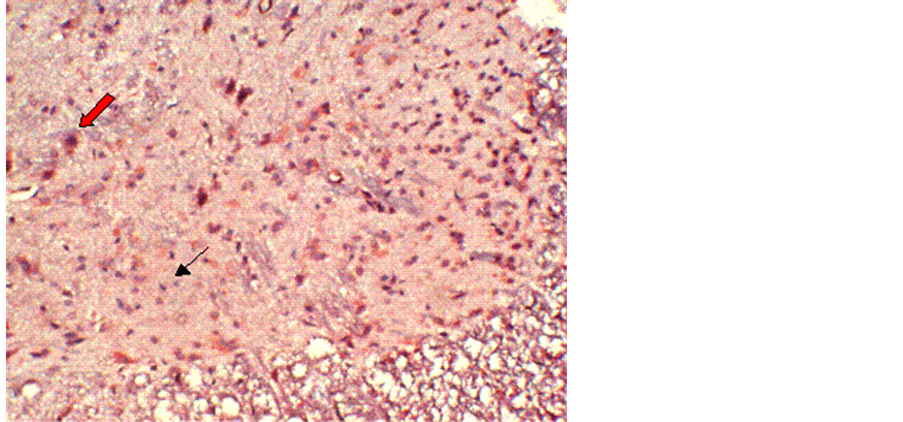
Figure 3. Immunohistochemical labeling of calpain-I in the methylprednisolone-treated group (Group C) at 72 h following reperfusion. The red arrow indicates positively-stained neurons and the black arrow indicates negatively-stained neurons. The number of positive cells per unit area, and the staining intensity, in group C are lower than those in Group B at 72 h post reperfusion. Magnification = 10×.

Figure 4. Western blot for NFP following spinal cord ischemia-reperfusion injury.

Table 1. Expression of calpain-I in the spinal cord after ischemia-reperfusion injury (values are positive area × optical density) H = hour, D = day.

Note: *Compared with Group B at the same time point, P < 0.01.
Table 2. Degradation of NFP after ischemia-reperfusion injury of the spinal cord (values are optical density). H = hour, D = day.
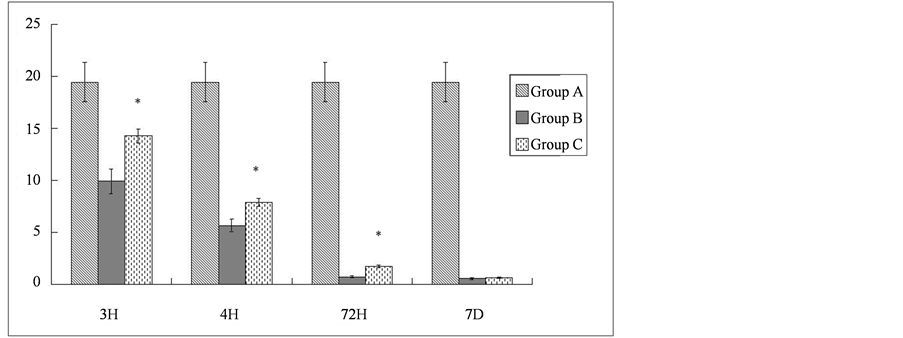
Note: *Compared with Group B at the same time point, P < 0.01.
role in the inhibition of calpain activity [21] . Methylprednisolone is widely acknowledged to have therapeutic effects on spinal cord injury. Free radical scavenging, inhibition of lipid peroxidation [22] and anti-inflammatory [23] functions have been ascribed to methylprednisolone to explain its neuroprotective effects.
Pathological changes in the penumbra, the region adjacent to the damaged spinal cord, have been thought to represent a secondary lesion following the spinal cord injury. The ischemia-reperfusion injury model of spinal cord has been used to study changes in this area of secondary lesion of the damaged spinal cord [24] . This model is able to induce ischemia and reperfusion damage in the targeted region of the spinal cord without any physical injury.
In the present study, we demonstrate that calpain inhibition also underlies the neuroprotection conferred by methylprednisolone following spinal cord injury. The results of the current study showed that, after ischemiareperfusion injury in the rat spinal cord, methylprednisolone administration was able to inhibit both the expression and activity of calpain. Rats with ischemia-reperfusion spinal cord injury showed significantly fewer calpain-positive cells following methylprednisolone administration, compared to saline-treated rats. This suggests that methylprednisolone can directly inhibit the expression of calpain.
An earlier study demonstrated that only calpain can degrade 68 KD NFP [25] ; thus, the degradation of NFP indirectly reflects calpain activity. In our study, methylprednisolone administration preserved NFP from degradation to a significant degree, this effect being most pronounced at 24 h post-administration. Therefore, we consider that the decline in NFP degradation in the spinal cord from methylprednisolone-administered animals (Group C), compared with saline-treated animals (Group B) was due to calpain inhibition. This inhibitory effect may, however, result from several mechanisms, including the inhibition of calpain-calcium binding by methylprednisolone, the direct inhibition of calpain activity, protection of the endogenous calpain inhibitor calpastatin, or the presence of other indirect regulatory pathways associated with additional roles of methylprednisolone. Accordingly, studies are needed to gain further mechanistic insight into the neuroprotective role of methylprednisolone.
The usefulness of methylprednisolone in cases of spinal cord injury has been the subject of recent controversy, largely because of the risk of serious adverse effects and the uncertain efficacy observed in some clinical research [26] . In the current work, we observed the inhibition of calpain activity and the protection of the cytoskeleton after methylprednisolone in the ischemia-reperfusion spinal cord injury was accompanied by the neurological motor function recovery of the animal (data not shown). Whether this effect was from the inhibition of posttraumatic lipid peroxidation [27] or via the direct inhibition of calpain remains to be studied. Methylprednisolone has been shown to depress the activity of calpain in vitro [28] , as well as preserve levels of the endogenous calpain inhibitor, calpastatin, in a model of profound hypothermia cardiopulmonary bypass in the immature pig [13] .
Calpain activation lies downstream of common pathological cascades in tissue injury [29] and affords a long “time window” for therapeutic intervention so that, in the course of the pathological progress, calpain would be activated and cause tissue damage at a later time point than most of the other factors. Kumar et al. studied cerebral ischemia-reperfusion injury and found that massive apoptosis appeared 24 h after injury, peaking at 72 h [30] . In a rat model of spinal cord impact injury induced by heavy drop, Ray [20] et al. found that calpain levels increased at 1, 4, 24, and 72 h after injury, peaking at 24 h, and the same pattern of calpain activation also existed in the ischemic regions surrounding the lesion without direct mechanical injury [31] . The time pattern of calpain activation in our experiment was different from that reported by Ray et al., peaking at 72 h, instead of at 24 h, after spinal cord injury. This discrepancy may be explained by the following reasons: 1) Impact injury predominantly causes cellular structural damage, releasing a large number of calcium ions and rapidly activating a considerable amount of calpain; in our study, the blood supply could be partially compensated and thus the spinal cord experiences incomplete ischemia, suggesting a lesser degree of injury. Consequently, the intensity of Ca2+ influx in our study would differ from that seen after direct mechanical injury; 2) A second stage of calpain activation is present following traumatic injury [32] , the mechanism of which, however, remains elusive. No notable second peak was observed in this experiment, which may be related to the short study time and long observation interval; 3) Calpain is presumed to become rapidly depleted in the impact injury model. In contrast, in spinal cord ischemia reperfusion, calpain may become depleted more slowly, offering a more extended period for the actions of calpain. As a result, calpain may maintain a dynamic and relatively stable level over a longer time period following ischemic injury, thus reaching a peak more gradually than in the impact injury model.
The standard dosing regimen of methylprednisolone for spinal cord injury recommended by the National Acute Spinal Cord Injury Study Group (NASCIS) [2] is: a dose of 30 mg/kg within 15 min after the injury and, after resting for 45 min, infusion of 5.4 mg/kg·h for a total of 23 h, with a total methylprednisolone dosage of 154.2 mg/kg. In the present study, we used a single methylprednisolone dose of 165 mg/kg, as used in Ray et al. [20] , which approximates the one-day dose recommended by NASCIS, instead of the single dose of 30 mg/kg employed in most other animal experiments, which was almost approximated to the one-day dose recommended by NASCIS. The rationale for this dosing regimen was based on the following considerations: 1) we wished to allow the blood concentration of methylprednisolone to rapidly achieve an effective concentration. The serum half-life of methylprednisolone is about 3 hours, and the biological half-life is 12 - 36 hours. A single large dose of 165 mg/kg of methylprednisolone in a given period of time thus has an effective concentration essentially equivalent to the dosage given over 24 hours with the standard regimen; 2) in the spinal cord ischemia-reperfusion injury model, there is theoretically a “no-reflow phenomenon” in the ischemic region after reperfusion meaning that, after the physical obstruction has been removed, blood failed to reperfuse the ischemic area, and that administration of a single large dose can maximize plasma drug concentrations in the injury region.
The protective effects of methylprednisolone in spinal cord injury manifest in several ways. However, methylprednisolone presents a stringent requirement for the proper timing of treatment, and is only effective when administered within eight hours after injury [33] . As calpain activation occurs downstream in the pathway of pathological damage mediated by excitatory toxins and calcium, the targeting of calpain may allow more time for clinical intervention before irreversible or maximum cellular damage has occurred. Thus, the inhibition of calpain activity offers a long “therapeutic window” for the treatment of secondary spinal cord injury. The results from the present study demonstrate that, with the administration of methylprednisolone in the ischemia-reperfusion spinal cord injury rat model, the inhibition of calpain activation and NFP degradation could be observed, suggesting that the modulation of calpain may be a mechanism underlying methylprednisolone-conferred neuroprotection against spinal cord injury.
The combinational use of calpeptin (calpain inhibitor) and methylprednisolone has been examined in a spinal cord impact injury model [20] , in which apoptosis and NFP degradation were substantially suppressed by this treatment. Furthermore, an additional study demonstrated that methylprednisolone in combination with calpeptin exhibited better therapeutic effects than either drug alone [19] .
5. Conclusion
The results of this study suggest that methylprednisolone can inhibit the expression and degradation activity of calpain following ischemia-reperfusion injury, providing further insight into the therapeutic benefits of methylprednisolone treatment for spinal cord injury.
Acknowledgements
We thank Medjaden Bioscience Limited for assisting in the preparation of this manuscript.
References
- S. Ozawa, H. Kamiya and K. Tsuzuki K, “Glutamate Receptors in the Mammalian Central Nervous System,” Progress in Neurobiology, Vol. 54, No. 5, 1998, pp. 581-618. doi:10.1016/S0301-0082(97)00085-3
- B. Bettler and C. Mulle, “Review: Neurotransmitter Receptors II: AMPA and Kainate Receptors,” Neuropharmacology, Vol. 34, No. 2, 1995, pp. 123-139. doi:10.1016/0028-3908(94)00141-E
- M. Jorgensen, C. K. Tygesen and P. H. Andersen, “Ionotropic Glutamate Receptors-Focus on Non-NMDA Receptors,” Pharmacology & Toxicology, Vol. 76, No. 5, 1995, pp. 312-319. doi:10.1111/j.1600-0773.1995.tb00153.x
- D. Bleakman and D. Lodge, “Neuropharmacology of AMPA and Kainate Receptors,” Neuropharmacology, Vol. 37, No. 10-11, 1998, pp. 1187-1204. doi:10.1016/S0028-3908(98)00139-7
- J. W. McDonald and M. V. Johnston, “Physiological and Pathophysiological Roles of Excitatory Amino Acids during Central Nervous System Development,” Brain Research, Brain Research Reviews, Vol. 15, No. 1, 1990, pp. 41-70. doi:10.1016/0165-0173(90)90011-C
- C. G. Parsons, W. Danysz and G. Quack G, “Glutamate in CNS Disorders as a Target for Drug Development: An Update,” Drug News & Perspectivew, Vol. 11, No. 9, 1998, pp. 523-569. doi:10.1358/dnp.1998.11.9.863689
- S. I. Deutsch, R. B. Rosse, B. L. Schwartz and J. Mastropaolo, “A Revised Excitotoxic Hypothesis of Schizophrenia: Therapeutic Implications,” Clinical Neuropharmacology, Vol. 24, No. 1, 2001, pp. 43-49. doi:10.1097/00002826-200101000-00008
- V. Bubeníková-Valesová, J. Horácek, M. Vrajová and C. H?schl, “Models of Schizophrenia in Humans and Animals Based on Inhibition of NMDA Receptors,” Neuroscience and Biobehavioral Reviews, Vol. 32, No. 5, 2008, pp. 1014-1023. doi:10.1016/j.neubiorev.2008.03.012�
- K. Y. Tseng, R. A. Chambers and B. K. Lipska, “The Neonatal Ventral Hippocampal Lesion as a Heuristic Neurodevelopmental Model of Schizophrenia,” Behavioural Brain Research, Vol. 204, No. 2, 2009, pp. 295-305. doi:10.1016/j.bbr.2008.11.039
- H. P. Jedema and B. Moghddam, “Characterization of Excitatory Amino Acid Modulation of Dopamine Release in the Prefrontal Cortex of Conscious Rats,” Journal of Neurochemistry, Vol. 66, No. 4, 1996, pp. 1448-1453. doi:10.1046/j.1471-4159.1996.66041448.x
- W. R. Wu, N. Li and B. A. Sorg, “Regulation of Medial Prefrontal Cortex Dopamine by Alphaamino-3-Hydroxy5-Methylisoxazole-4-Propionate/Kainate Receptors,” Neuroscience, Vol. 114, No. 2, 2002, pp. 507-516. doi:10.1016/S0306-4522(02)00276-2
- C. L. Ryan, M. A. Robbins, M. T. Smith, I. C. Gallant, A. L. Adams-Marriott and T. A. Doucette, “Altered Social Interaction in Adult Rats Following Neonatal Treatment with Domoic Acid,” Physiology & Behavior, Vol. 102, No. 3-4, 2011, pp. 291-295. doi:10.1016/j.physbeh.2010.11.020
- A. L. Adams, T. A. Doucette and C. L. Ryan, “Altered Pre-Pulse Inhibition in Adult Rats Treated Neonatally with Domoic Acid,” Amino Acids, Vol. 35, No. 1, 2008, pp. 157-160. doi:10.1007/s00726-007-0603-3
- A. L. Marriott, C. L. Ryan and T. A. Doucette, “Neonatal Domoic Acid Treatment Produces Alterations to Prepulse Inhibition and Latent Inhibition in Adult Rats,” Pharmacology, Biochemistry and Behavior, Vol. 103, No. 2, 2012, pp. 338-344. doi:10.1016/j.pbb.2012.08.022
- M. A. Burt, C. L. Ryan and T. A. Doucette, “Altered Responses to Novelty and Drug Reinforcement in Adult Rats Treated Neonatally with Domoic Acid,” Physiology & Behavior, Vol. 93, No. 1-2, 2008, pp. 327-336. doi:10.1016/j.physbeh.2007.09.003
- M. A. Burt, C. L. Ryan and T. A. Doucette, “Low Dose Domoic Acid in Neonatal Rats Abolishes Nicotine Induced Conditioned Place Preference during Late Adolescence,” Amino Acids, Vol. 35, No. 1, 2008, pp. 247-249. doi:10.1007/s00726-007-0584-2
- A. L. Adams, T. A. Doucette, R. James and C. L. Ryan, “Persistent Changes in Learning and Memory in Rats Following Neonatal Treatment with Domoic Acid,” Physiology & Behavior, Vol. 96, No. 4-5, 2009, pp. 505-512. doi:10.1016/j.physbeh.2008.11.019
- J. M. Gold, C. Carpenter, C. Randolph, T. E. Goldberg, and D. R. Weinberger, “Auditory Working Memory and Wisconsin Card Sorting Test Performance in Schizophrenia,” Archives of General Psychiatry, Vol. 54, No. 2, 1997, pp. 159-165. doi:10.1001/archpsyc.1997.01830140071013
- R. S. E. Keefe, S. E. Lees-Roitman and R. L. Dupre, “Performance of Patients with Schizophrenia on a Pen and Paper Visuospatial Working Memory Task with Short Delay,” Schizophrenia Research, Vol. 26, No. 1, 1997, pp. 9-14. doi:10.1016/S0920-9964(97)00037-6
- G. W. Dalack, D. J. Healy and J. H. Meador-Woodruff, “Nicotine Dependence in Schizophrenia: Clinical Phenomena and Laboratory Findings,” The American Journal of Psychiatry, Vol., 155, No. 11, 1998, pp. 1490-1501.
- R. A. Chambers and D. W. Self, “Motivational Responses to Natural and Drug Rewards in Rats with Neonatal Ventral Hippocampal Lesions: An Animal Model of Dual Diagnosis Schizophrenia,” Neuropsychopharmacology, Vol. 27, No. 6, 2002, pp. 889-905. doi:10.1016/S0893-133X(02)00365-2
- B. K. Lipska, J. M. Aultman, A. Verma, D. R. Weinberger and B. Moghaddam, “Neonatal Damage of the Ventral Hippocampus Impairs Working Memory in the Rat,” Neuropsychopharmacology, Vol. 27, No. 1, 2002, pp. 47-54. doi:10.1016/S0893-133X(02)00282-8
- B. Eleveg, G. D. A. Brown, T. McCormack, J. I. Vousden and T. E. Goldberg, “Identification of Tone Duration, Line Length, and Letter Position: An Experimental Approach to Timing and Working Memory Deficits in Schizophrenia,” Journal of Abnormal Psychology, Vol. 113, No. 4, 2004, pp. 509-521. doi:10.1037/0021-843X.113.4.509
- B. L. Schwartz, L. H. Deutsch, C. Cohen, D. Warden and S. I. Deutsch, “Memory for Temporal Order in Schizophrenia,” Biological Psychiatry, Vol. 29, No. 4, 1991, pp. 329-339. doi:10.1016/0006-3223(91)90218-B
- F. A. V. Waters, M. T. Maybery, J. C. Badcock and P. T. Michie, “Context Memory and Binding in Schizophrenia,” Schizophrenia Research, Vol. 68, No. 2-3, 2004, pp. 119-125. doi:10.1016/S0920-9964(03)00221-4
- T. A. Doucette, S. M. Strain, G. V. Allen, C. L. Ryan and R. A. R. Tasker, “Comparative Behavioural Toxicity of Domoic Acid and Kainic Acid in Neonatal Rats,” Neurotoxicology and Teratology, Vol. 22, No. 6, 2000, pp. 863-869. doi:10.1016/S0892-0362(00)00110-0
- T. A. Doucette, P. B. Bernard, P. C. Yuill, R. A. Tasker and C. L. Ryan, “Low Doses of Non-NMDA Glutamate Receptor agonists Alter Neurobehavioural Development in the Rat,” Neurotoxicology and Teratology, Vol. 25, No. 4, 2003, pp. 473-479. doi:10.1016/S0892-0362(03)00034-5
- J. B. Mitchell and J. Laiacona, “The Medial Frontal Cortex and Temporal Memory: Tests Using Spontaneous Exploratory Behaviour in the Rat,” Behavioural Brain Research, Vol. 97, No. 1-2, 1998, pp. 107-113. doi:10.1016/S0166-4328(98)00032-1
- G. Paxinos and C. Watson C, “The Rat Brain in Stereotaxic Coodinates,” 4th Editon, Academic Press, Toronto, 1998.
- L. M. DeVito, R. Konigsberg, C. Lykken, M. Sauvage, W. S. Young and H. Eichenbaum, “Vasopressin 1b Receptor Knock-Out Impairs Memory for Temporal Order,” The Journal of Neuroscience, Vol. 29, No. 9, 2009, pp. 2676-2683. doi:10.1523/JNEUROSCI.5488-08.2009
- J. G. Howland, R. A. Harrison, D. K. Hannesson and A. G. Phillips, “Ventral Hippocampal Involvement in Temporal Order, but Not Recognition, Memory for Spatial Information,” Hippocampus, Vol. 18, No. 3, 2008, pp. 251-257. doi:10.1002/hipo.20396
- M. Akil, C. L. Edgar, J. N. Pierri, S. Casali and D. A. Lewis, “Decreased Density of Tyrosine HydroxylaseImmunoreactive Axons in the Entorhinal-Cortex of Schizophrenic Subjects,” Biological Psychiatry, Vol. 47, No. 5, 2000, pp. 361-370. doi:10.1016/S0006-3223(99)00282-6
- M. Akil, J. N. Pierri, R. E. Whitehead, C. L. Edgar, C. Mohila, A. R. Sampson and D. A. Lewis, “Lamina-Specific Alterations in the Dopamine Innervation of the Prefrontal Cortex in Schizophrenic Subjects,” The American Journal of Psychiatry, Vol. 156, No. 10, 1999, pp. 1580-1589.
- K. Wedzony, K. Fijal and A. Chocyk, “Blockade of NMDA Receptors in Postnatal Period Decreased Density of Tyrosine Hydroxylase Immunoreactive Axonal Arbors in the Medial Prefrontal Cortex of Adult Rats,” Journal of Physiology and Pharmacology, Vol., 56, No. 2, 2005, pp. 205-221.
- A. Aguilar-Valles, C. Flores and G. N. Luheshi, “Prenatal Inflammation-Induced Hypoferremia Alters Dopamine Function in Adult Offspring in Rat: Relevance for Schizophrenia,” PloS One, Vol. 5, No. 6, 2010, e10967. doi:10.1371/journal.pone.0010967
- D. J. Balfour, “Neuroplasticity within the Mesoaccumbens Dopamine System and Its Role in Tobacco Dependence,” Current Drug Targets, CNS and Neurological Disorders, Vol. 1, No. 4, 2002, pp. 413-421. doi:10.2174/1568007023339076
- T. A. Doucette, C. L. Ryan and R. A. Tasker, “Genderbased Changes in Cognition and Emotionality in a New Rat Model of Epilepsy,” Amino Acids, Vol. 32, No. 3, 2007, pp. 317-322. doi:10.1007/s00726-006-0418-7
- J. L. Nunez and M. M. McCarthy, “Evidence for an Extended Duration of GABA-Mediated Excitation in the Developing Male versus Female Hippocampus,” Developmental Neurobiology, Vol. 67, No. 14, 2007, pp. 1879-1890. doi:10.1002/dneu.20567
- A. Kyrozis, O. Chudomel, S. L. Moshe and A. S. Galanopoulou, “Sex-Dependent Maturation of GABAA Receptor-Mediated Synaptic Events in Rat Substantia Nigra Reticulate,” Neuroscience Letters, Vol. 398, No. 1-2, 2006, pp. 1-5. doi:10.1016/j.neulet.2005.12.018
- R. K. Lenroot, N. Gogtay, D. K. Greenstein, E. MolloyWells, G. L. Wallace, L. S. Clasen, J. D. Blumenthal, J. Lerch, A. P. Zijdebnos, A. C. Evans, P. M. Thompson and J. N. Giedd, “Sexual Dimorphism of Brain Developmental Trajectories during Childhood and Adolescence,” NeuroImage, Vol. 36, No. 4, 2007, pp. 1065-1073. doi:10.1016/j.neuroimage.2007.03.053
NOTES

*Corresponding author.