Neuroscience & Medicine
Vol. 3 No. 4 (2012) , Article ID: 26184 , 3 pages DOI:10.4236/nm.2012.34042
Pituitary-Thyroid Dysfunction in a Hashimoto’s Encephalopathy Patient
![]()
1Department of Endocrinology, First Affiliated Hospital, General Hospital of Chinese People’s Liberation Army, Beijing, China; 2Department of Radiology, First Affiliated Hospital, General Hospital of Chinese People’s Liberation Army, Beijing, China; 3Telemedicine Center, General Hospital of Chinese People’s Liberation Army, Beijing, China.
Email: hjs304@yahoo.com.cn, *zmk301@126.com
Received June 3rd, 2012; revised July 3rd, 2012; accepted August 13th, 2012
Keywords: Hashimoto’s Thyroiditis; Hashimoto’s Encephalopathy; Pathogenesis; Hypothyroidism; Pituitary Gland
ABSTRACT
The pathogenesis of Hashimoto’s encephalopathy has not been clearly elucidated and involvement of autoimmune damage has been proposed. We report a 23-year-old man who was emergently hospitalized for paroxysmal syncope and involuntary tremor with normal physical examination except for low heart rate. The patient was characterized by neurological symptoms, mild hypothyroidism, enlarged pituitary gland and extremely elevated thyrotropin, which all were reversed with levothyroxine alone. The case indicated that the pituitary-thyroid dysfunction may play a role in the pathogenesis of Hashimoto’s encephalopathy, hence, evaluation of pituitary gland should be recommended in diagnosis and treatment of Hashimoto’s encephalopathy.
1. Introduction
Hashimoto’s encephalopathy (HE) is considered a rare neurologic complication of autoimmune thyroid disease for long-term. Although autoimmune mechanisms are thought to play a major role in HE pathogenesis, the etiology of the disease is still incompletely understood [1]. Due to great variation in clinical manifestations, onset, progression and MRI manifestations, most HE patients were not promptly diagnosed and treated. There is no definite diagnosis criteria for HE so far, and the generally accepted diagnostic criteria are based on the presence of diversied neurologic/neuropsychiatric symptoms, such as cognitive impairment, seizures, stroke-like events, focal neurological deficit or movement disorders with elevated antithyroid antibodies excluding of infectious, metabolic, toxic, vascular, neoplastic and paraneoplastic causes of encephalopathy [2]. Here we report a case manifested as transient acute disturbance of consciousness and myoclonus, characterized by extremely elevated thyrotropin (TSH) and reversible pituitary enlargement, which coincided with high serum titers of antithyroid autoantibodies.
2. Patient Finding
The patient was a 23-year-old man who was emergently hospitalized for paroxysmal syncope and involuntary tremor for 14 h on September 5, 2010. The symptom was immediately followed by stress and fatigue after a sudden death of a colleague of the patient. The patient experienced unconsciousness and recovered after half an hour with whole body fatigue. There was no incontinence of bowel movements and urine, no chest distress or chest pain, no heart palpitations and other discomfort. There was no significant change in body weight recently. Medical history, as well as family history, was negative for other symptoms or diseases. Physical examination: normal body temperature, heart rate 44 beats/min, breath rate 18 beats/min, blood pressure 95/47 mmHg, consciousness clear, the answer to the point, the expression natural without other abnormalities in heart, nerve system normal, no eyelid edema and anterior tibial myxedema. Laboratory examinations revealed extremely high serum titers of TSH (>100 μU/ml, normal range 0.60 to 4.50), elevated thyroglobulin antibody 672 IU/ml (normal range 0 to 4.11) and thyroid peroxidase antibody 124 IU/ml (normal range 0 to 5.61), normal free triiodothyronine (1.78 pg/ml, normal range 1.71 to 3.71) and slightly decreased serum free thyroxine (0.53 ng/dl, normal range 0.7 to 1.48). Further analysis of blood, including differential blood cell count, C-reactive protein, electrolytes, liver enzymes, creatinine, urea, glucose, serum protein were all within the normal ranges. Electrocardiogram showed sinus bradycardia. 24-hour dynamic electrocardiogram and ultrasound echocardiography found no special abnormality. Thyroid ultrasonography showed normal thyroid size, slight rough echo, and slightly increased gland blood flow. Computed tomography of the head showed no abnormality. Electroencephalography (EEG) was slightly abnormal without epileptic form or focal activity.
The diagnosis of Hasimoto’s thyroiditis, primary hypothyroidism and bradycardia were made according to clinical manifestations, EEG and laboratory findings. The patient was admitted to the department of cardiology from the emergency department on account of cardiac syncope. The symptoms were alleviated gradually with 25 to 50 μg of levothyroxine daily, but serum TSH was still over 100 μU/ml (136 after 12-fold diluted) 1 month later. Magnetic resonance tomography (MRI) of the butterfly saddle detected an enlarged pituitary gland with the size of 1.27 × 1.32 × 1.17 cm and identical signals at T1 and T2 (Figure 1A). Plasma cortisol level and rhythm were normal. Serum growth hormone and prolactin were normal. With the escalation of levothyroxin dosage to 150 μg daily within 5 months, the patient’s clinical condition was improved completely, and serum free thyroxine and TSH were recovered to normal range. MRI showed that pituitary gland was shrunk to the normal size (0.76 × 0.49 × 0.52 cm, Figure 1B). After being released from the hospital, the patient himself reduced the dose of levothyroxine to 12.5 μg daily. Days after dose reduction, the patient experienced similar symptoms as previously, including rapid onset of paroxysmal left lancinating headache, nausea, and poor sleep. Elevated TSH (>100 μU/ml) and enlarged pituitary gland (Figure 1C) were further confirmed again 3 months later. After restoration of 100 μg daily of levothyroxine, the symptoms disappeared, and serum TSH and pituitary size returned to normal except that a 15 kilograms body-weight gain was noticed in one year.
The patient was admitted to the department of Cardiology on consideration for cardiac syncope and diagnosed of HE after consulting with endocrinologists. HE is an acute/sub-acute, steroid-responsive, recurrent encephalopathy associated with Hashimoto’s thyroiditis. Symptoms such as dementia, alteration of consciousness, stroke-like episodes and epileptic seizures are usually presented. So far there is no established diagnostic criterion for HE, and the diagnosis of HE should be taken into account if infection, tumor or alteration in the cerebral vascular system can be excluded to the patients of encephalopathy with elevated anti-thyroid antibodies [2].
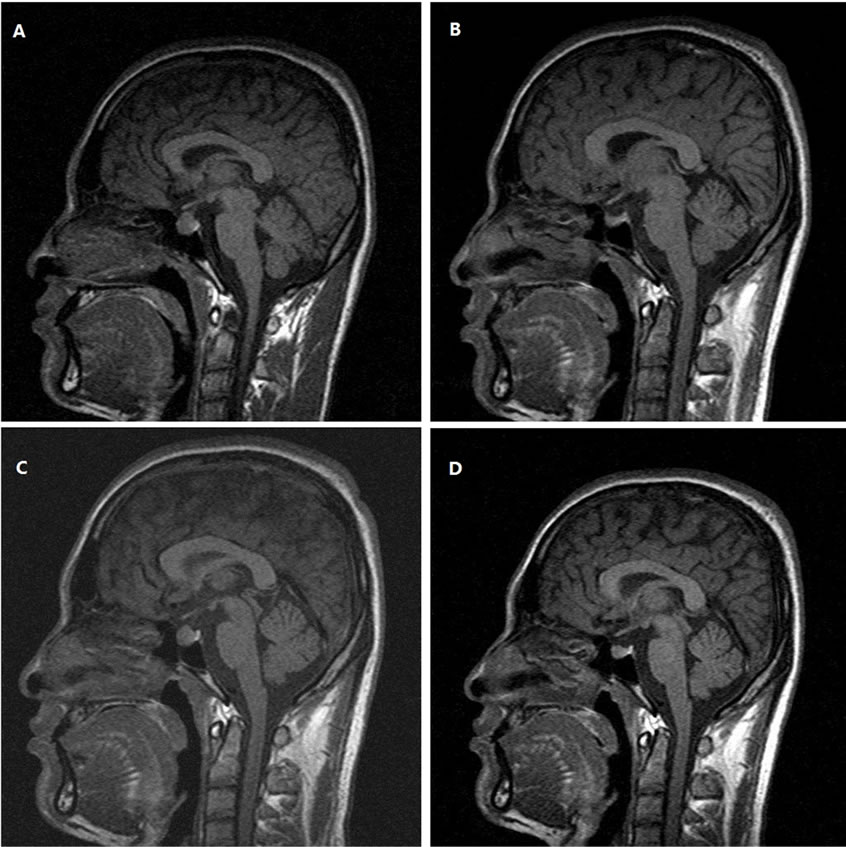
Figure 1. Changes of the pituiary gland MRI scan (plain T1WI, sagittal) following levothyroxine treatment. (A) Pretreatment; (B) 10 months after treatment; (C) 13 months after treatment (reduced dosage by patient for 3 months). (D) 15 months after treatment (increased dosage again for 2 months).
In patients with hypothyroidism and CNS symptoms it is need to rule out hypothyroidism as a cause of the CNS symptoms. It’s well known that thyroid hormone is essential for the development of the central nervous system, but deficiency of thyroid hormone beginning in adult life causes less severe manifestations. In severe cases, all intellectual functions are slowed and psychiatric disorders are common and may induce agitation (myxedema madness). Cerebral hypoxia due to circulatory alterations may predispose to confusional attacks and syncope, which may be prolonged and can lead to stupor or coma. Epileptic seizures have been reported and tend to occur in myxedema coma [3]. The diagnosis of hypothyroidism with central nervous system symptom of the patient was clear, but the patient showed no obvious chronic hypothyroidism symptoms, such as fatigue, lethargy, mental slowing, and myxedema and laboratory tests of thyroid function showed normal levels of free T3. So the patient was not only diagnosed of hypothyroidism but also HE.
Pituitary enlargement with significant hypothyroidism is well described in the literature and is reduced in pituitary size after thyroid hormone administration [4], but there is no report in HE patient with unparalleled thyroid hormone and TSH as such. The markedly elevated TSH accompanied by mildly decreased thyroid hormone in this case, implying the resistance of thyroid to TSH. Such a disparity may be caused by the destruction of thyroid cells in the Hashimoto’s disease. Alternatively, it was reported that the resistance to TSH in Hashimoto’s disease may be caused by abnormalities in thyroid stimulating hormone receptor [5]. Neurological manifestations presented in this case, such as paroxysmal syncope, involuntary tremor, headaches, insomnia and other nervous system symptoms may indicate that the acute hypothyroidism in Hashimoto’s thyroiditis under crisis situation maybe related the onset of the encephalopathy.
Although the pathogenesis of Hashimoto’s encephalopathy is still unclear, involvement of autoimmune damage has been proposed. But there are many facts didn’t support the pathogenesis of autoimmune damage. Firstly, signs of inflammation are usually absent in most cases of HE and mild elevations of erythrocyte sedimentation rate, C-reactive protein, or liver aminotransferase levels have been reported only in few cases [6]. Secondly, examinations of the cerebrospinal fluid were usually normal [7]. Thirdly, the specific anti-brain auto-antibodies has not been found in HE patients so far [7]. Fourthly, therapy effect to glucocorticoids didn’t meet with the other autoimmune diseases, such as many HE patients recovered only with one dosage of steroids and the symptoms can be relieved with thyroid hormone treatment alone in a large proportion of patients [7]. Finally, pathology findings in brain biopsy from HE patients couldn’t explain the autoimmune pathogenesis [8]. As Schiess N., et al. have said that: “In our view, regardless of the role that antithyroid antibodies may play in the pathogenesis of CNS abnormalities, the presence of such antibodies defines a subset of neurological disorders that may be classified under the term HE. The significance of classifying such encephalopathies under the term HE will be determined only in the future once the relevance of antithyroid antibodies is demonstrated or dismissed by more detailed experimental and immunopathological studies” [9].
3. Conclusion
Mild hypothyroidism is insufficient to interpret the neurologic symptoms, enlarged pituitary gland and extremely increased TSH in this case. In a summary, we recommend that thyroid function, thyroid auto-antibodies and evaluation of pituitary should be evaluated for all of unknown cause of nervous system disorders.
REFERENCES
- R. J. Caselli, J. F. Drazkowski and D. M. Wingerchuk, “Autoimmune Encephalopathy,” Mayo Clinic Proceedings, Vol. 85, No. 10, 2010, pp. 878-880.
- G. A. Marshall and J. J. Doyle, “Long-Term Treatment of Hashimoto’s Encephalopathy,” Journal of Neuropsychiatry and Clinical Neurosciences, Vol. 18, No. 1, 2006, pp. 14-20. doi:10.1176/appi.neuropsych.18.1.14
- S. Melmed, K. S. Polonsky, P. R. Larsen and H. M. Kronenberg, “Williams Textbook of Endocrinology, 12th Edition,” In: G. A. Brent and T. Davies, Ed., Hypothyroidism and Thyroiditis, Elsevier Saunders, Inc., Philadelphia, 2011, pp. 410.
- N. J. Sarlis, F. Brucker-Davis, J. L. Doppman and M. C. Skarulis, “MRI-Demonstrable Regression of a Pituitary Mass in a Case of Primary Hypothyroidism after a Week of Acute Thyroid Hormone Therapy,” Journal of Clinical Endocrinology and Metabolism, Vol. 82, No. 3, 1997, pp. 808-811.
- S. Fukata, G. A. Brent and M. Sugawara, “Resistance to Thyroid Hormone in Hashimoto’s Thyroiditis,” New England Journal of Medicine, Vol. 352, No. 5, 2005, pp. 517-518.
- K. W. Nolte, A. Unbehaun, H. Sieker, T. M. Kloss and W. Paulus, “Hashimoto Encephalopathy: A Brainstem Vasculitis?” Neurology, Vol. 54, No. 3, 2000, pp. 769-770. doi:10.1212/WNL.54.3.769
- J. Y. Chong, L. P. Rowland and R. D. Utiger, “Hashimoto Encephalopathy, Syndrome or Myth?” Archives of Neurology, Vol. 60, No. 2, 2003, pp. 164-171.
- P. Striano, M. Pagliuca, V. Andreone, F. Zara, A. Coppola and S. Striano, “Unfavourable Outcome of Hashimoto Encephalopathy Due to Status Epilepticus: One Autopsy Case,” Journal of Neurology, Vol. 253, No. 2, 2006, pp. 248-249. doi:10.1007/s00415-005-0925-6
- N. Schiess and C. A. Pardo, “Hashimoto’s Encephalopa- thy,” Annals of the New York Academy of Sciences, Vol. 1142, 2008, pp. 254-265. doi:10.1196/annals.1444.018
NOTES
*Corresponding author.