Open Journal of Applied Sciences
Vol.4 No.2(2014), Article ID:43240,8 pages DOI:10.4236/ojapps.2014.42008
Novel Inorganic Pyrophosphatase from Soil Metagenomic and Family and Subfamily Prediction
Gisele R. Rodrigues*, Silvana Pompeia Val-Moraes, Eliana G. de Macedo Lemos, João Martins Pizauro*
Technology Department, UNESP—Universidade Estadual Paulista/ College of Agricultural and Veterinarian Sciences, Program of Postgraduate in Agropecuary Microbiology, Jaboticabal, Brazil
Email: *gid_rodrigues@yahoo.com.br, *jpizauro@fcav.unesp.br
Copyright © 2014 Gisele R. Rodrigues et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Gisele R. Rodrigues et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.

Received 14 November 2013; revised 20 December 2013; accepted 30 December 2013
Abstract
Inorganic pyrophosphatase (PPase) is widely studied, to be extremely important for survival of plants and microorganisms. PPases catalyze an essential reaction the hydrolysis of inorganic pyrophosphate (PPi) to inorganic phosphate (Pi). Studies involving the mechanism of PPase were performed in microorganisms culture. We didn’t found reports of PPase derived from soil metagenomic libraries. Soil environment has immense diversity of microorganisms, yet most remains unexplored and the metagenome are the technologies used and investigate uncultured microorganisms potential. The aim is to identify novel genes using the metagenomic approaches from a bioinformatics perspective and hopefully will serve as a useful resource. With this purpose, we used the metagenomic library of Eucalyptus spp. arboretum (EAA). We did a screening to select a positive clone and submitted them to the process of shotgun. The data obtained was submitted to bioinformatics analyses. These analyses identified were the novel MetaPPase gene and were classified according to the predict family and subfamily.
Keywords
Soil Diversity; Bioinformatic; Structure; Membrane Protein; Ion Transport

1. Introduction
Inorganic Pyrophosphatases (PPase, E.C 3.6.1.1), are ubiquitous and is the central enzyme of phosphorus metabolism . This enzyme is an important control of the cellular concentration inorganic pyrophosphate (PPi) and thus controlling biosynthetic reactions like nucleic acid and protein system and is responsible for the pump across the membranes responsible for triplicating ions .
The PPases are made up of two groups, Soluble PPases (Family I and II) and membrane PPase (M-PPase) - . These groups of enzyme are very important for maintaining life. For this reason, we looked for a novel gene of PPase families in a soil metagenome, because we know that soil harbors an immense diversity of microorganisms yet most remains unexplored . For this, the used metagenomic approach has become an indispensable tool for studying the diversity and metabolic potential of environmental microbes, whose bulk is as yet noncultivable, and is a potent method to study this soil demand - . Thus we work with a metagenomic library of a Eucalyptus spp. arboretum (EAA), belonging to Laboratory of Biochemistry and microorganisms of Plants (LBMP), to identify novel gene of PPase families activity, which was submitted to bioinformatics analyses, modeling for testing the biological potential.
2. Methods
We use the metagenomic library of from Eucalyptus spp. arboretum (EAA) . The screening in metagenomic library by PCR for identify the positive clone with degenerate primers, building for this work, using some sequences deposited in NCBI database (DQ182493, DQ916115, DQ916118), Hydro-F- 5’CGTSGGVTAYCGSTAYTTYGA3’; Hydro-R-5’CGMTYDCCYGCSCCDCCYTC3’ . The positive clone A09 plaque 13 was submitted to the process of shotgun sub cloning method . DNAs containing inserts were sequenced by using standard protocols with an ABI PRISM® 3100 Genetic Analyzer. The DNA sequence was determined with the program by the programs Phred , Phrap and Consed . The open reading frames (ORFs) were identified and translated by using the program ORF Finder at NCBI and using BLAST X (http://www.ncbi.nlm.nih.gov/gorf/orfig.cgi), for analysis of similarity with protein. After the genes functional identification the data were analyzed in ProDom is a comprehensive database of protein domain families generated from the global comparison of all available protein sequences. Pfam is a database of protein families, where families are sets of protein regions that share a significant degree of sequence similarity, thereby suggesting homology, similarity is detected using the HMMER3.
The determination homology and the I-Tasser server we used are on-line platform for protein structure and function predictions. 3D models are built based on multiple threading alignments by LOMETS and iterative template fragment assembly simulations, functions in slights are derived by matching the 3D models with BioLip protein function database. PyMOL Molecular Graphics System, version 1.5.0.4 is a program user sponsored molecular visualization system on an open-source foundation . We used the MEMSAT3 and MEMSAT-SVM a novel version of a widely used transmembrane topology prediction method and PSIPRED to identify the signature subfamily . The program used for comparison was Basic local alignment search tools (Blast) and the sequences compared with those online at the GenBank. Sequence alignments were first done using Clustal W (version 1.8) , and then adjusted using the BioEdit, version 5.0.9 Program . Phylogenetic relationships were inferred by preferential alignments of the Membrane PPase (MPPase) protein sequences obtained from GenBank. This was done using the program MEGA5 (version 2.1) . Bootstrap analysis was performed with 1000 replicates .
3. Results
The data information of shotgun library was assembled according functional knowledge from genes using OrfFinder, after we analyzed of metagenomic involves functional annotation of the predicted genes by database comparison searches using the and ProDom and Pfam programs . The assembly sequences were submitted to the GenBank, the accession number KF715620.
We identify the novel gene of inorganic pyrophosphatase (PPase) so called MetaPPase. Then we submitted the amino acid sequences on I-Tasser serve, has generated protein structure predictions for thousands of modeling requests from more than 35 countries. A scoring function (C-score) based on the relative clustering structural density and the consensus significance score of multiple threading templates is introduced to estimate the accuracy of the I-TASSER predictions. A large-scale benchmark test demonstrates a strong correlation between the C-score and the TM-score (a structural similarity measurement with values in [0, 1]) of the first models with a correlation coefficient of 0.91. Using a C-score cutoff > −1.5 for the models of correct topology, both false positive and false negative rates are below 0.1. Combining C-score and protein length, the accuracy of the ITASSER models can be predicted with an average error of 0.08 for TM-score and 2 Å for RMSD .
The templates protein of similar folds from PDB (Protein Data Bank) library, with the result: 100.0% confidence by the single highest scoring template is a true homology, code template PDB 4A06, i.d. was of 25%, Fold: H-PPase, Superfamily: H(+)-translocating pyrophosphatase.
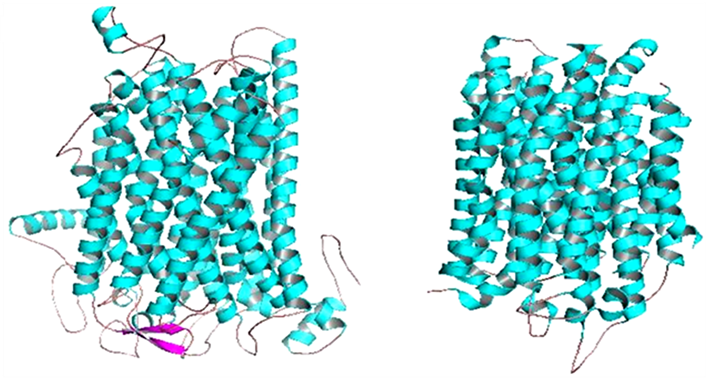
After predictions of structure and function of the MetaPPase gene, we used the PyMol to visualize the image structure (Figure 1). Structure like M-PPase that display particular characteristics in proportions other families, these consist homodimer with 15 - 17 transmembrane (TM) helices . To confirm this characteristic was used the MEMSAT3 and MEMSAT-SVM program , to identify transmembrane helices (Figure 2).
It is possible observed in Figure 2, each yellow segment represents a transmembrane helices and the N-terminal is the periplasm and C-terminal in cytoplasm. Furthermore the M-PPases can represent in H+ Transporting
 (a) (b)
(a) (b)
Figure 1. MetaPPase structure (a) and template conformation PDB accession number 4AV6 (b). The α-helices are in cyan, the β-sheet pink and the loops are salmon, for both structures.
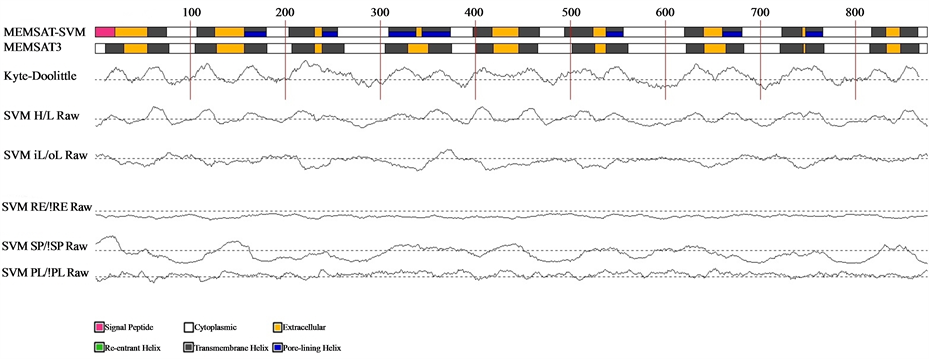
Figure 2. Schematic diagram of the MEMSAT3 and MEMSAT-SVM predictions for the query sequence. Traces indicate the RAW outputs for the prediction SVMs. Dashed lines indicate the prediction threshold. Where PL: Pore lining residue, SP: Signal peptide residue, RE: Re-entrant helix residue and iL/oL&HL: helix prediction.
PPase (H+-PPase), divided in subfamilies the K+ independent and K+ dependent where K+ independent are insensitive to monovalent cations, where K+ dependent enzymes need millimolar concentrations of K+ for activity. These results suggest the possibility of MetaPPase be H+ Transporting PPase (H+-PPase). The signature of MetaPPase and a region contain three conserved aspartates that are involved in the binding of cations (Figure 3).
The analyses performed yet allowed to estimate the family of novel MetaPPase and to obtain more information about the subfamily we led a phylogenetic analysis. The sequences in the tree were selected by redundancy filtering to leave only representative sequences for each group of highly similar sequences . The total number of sequences was found in the NCBI protein sequence database for each PPase subfamily.
The evolutionary history was inferred using the Neighbor-Joining method . The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches . The evolutionary distances were computed using the JTT matrix-based method and are in the units of the number of amino acid substitutions per site. The rate variation among sites was modeled with a gamma distribution (shape parameter = 1). The analysis involved 80 amino acid sequences (Figure 4). All positions containing gaps and missing data were eliminated. There were a total of 367 positions in the final dataset. Evolutionary analyses were conducted in MEGA5 . The tree showed the evolutionary tree of MetaPPase, this tree including plant and protest H+-PPase, consists of independently envolving K+ independent and K+ dependent and families.
4. Discussion
The MetaPPase belongs to membrane integral PPase (M-PPase) family, that can be divided into four subfamilies based on their ion-pumping specificity Na + and/or H+ and the latter they are dependent K+ or not - . We observed the at Figure 1 similarities which represent M-PPase family and couple the hydrolysis of PPi to the transport of cations across membranes - .
This subfamilies of transports are widespread in bacteria, archaea an plants - , but recently identify the Na+ Transporting PPase (Na+PPase) in mesophilic microorganism, and this enzyme are similar to the H+- PPase in many aspects but require both K+ and Na+ for activity .
Our topology model of transmembrane helices Figure 2 has confirmed the 17-transmembrane domain structure predicted by the hydropathy analysis of the primary structure, moreover the N-terminal is the periplasm and C-terminal in cytoplasm, the same characteristics was describe in Streptomyces coelicolor, and H+-PPases subfamily .
The Figure 3 suggests that MetaPPase represents enzymatic function of H+-PPase . According Suzuki , using the alignment of the predicted amino acid sequence for identifies active site and the ligands are highly conserved.
The evolutionary tree of membrane PPases (Figure 4) allowed to identified MetaPPase with H+-PPase and probably belongs the subfamily of K+ independent despite the MetaPPase be in separate clade with low bootstrap

Figure 3. The consensus sequences of membrane PPase identify the conserved signature of subfamily H+-PPase.

Figure 4. The sequence in the tree was selected of verified and putative membrane PPase. The sequences found in the NCBI protein databases for each PPase subfamily (H+-PPase with K+ independent or K+ dependentand and Na+-PPase).
in relation the others microorganisms , demonstrated that all members of the K+ independent family appear to operate as H+ pumps.
5. Conclusions
Our results corroborate with the hypothesis the high unexplored microbial diversity of soil are able to found novel genes. Metagenomic approach has become an indispensable tool, allowing to isolating novel genes and the functional annotation of the predicted genes by database comparison searches and is essential to identify the MetaPPase.
The use of different bioinformatics tools supports the predictions of the family and subfamily of MetaPPase which suppose operations as H+ Transporting PPase (H+-PPase), K+ independent, observed at evolutionary tree.
This suggests a special feature that, our work in situ will be cloning the gene expression vector for subsequent kinetic characterization and crystallization.
Acknowledgements
We thank the Program of Postgraduate in Agropecuary Microbiology (PPMA) and Coordenação de Aperfei- çoamento de Nível Superior (CAPES) for the financial support.
References
- Fabrichniy, I.P., Lehtiö, L., Tammenkoski, M., Zyryanov, A. B., Oksanen, E., Baykov, A.A., Lahti, R. and Goldman, A. (2007) A trimetal site and substrate distortion in a family II inorganic pyrophosphatase. Journal of Biological Chemistry, 282, 1422-1431. http://dx.doi.org/10.1074/jbc.M513161200
- Kajander, T., Kellosalo, J. and Goldman, A. (2013) Inorganic pyrophosphatases: One substrate, three mechanisms. FEBS Letters, 587, 1863-1869. http://dx.doi.org/10.1016/j.febslet.2013.05.003
- Baykov, A.A., Malinen, A.M., Luoto, H.H. and Lahti, R. (2013) Pyrophosphate-fueled Na+ and H+ transport in prokaryotes. Microbiology and Molecular Biology Reviews, 77, 267-276. http://dx.doi.org/10.1128/MMBR.00003-13
- Kellosalo, J., Kajander, T., Kogan, K., Pokharel, K. and Goldman, A. (2012) The structure and catalytic cycle of a sodium pumping pyrophosphatase. Science, 337, 473-476. http://dx.doi.org/10.1126/science.1222505
- Torsvik, V.L. and Øvreås, L. (2011) DNA reassociation yields broad-scale information on metagenome complexity and microbial diversity. Handbook of Molecular Microbial Ecology I: Molecular Metagenomics and Complementary Approaches, 1, 2.
- Handelsman, J. (2004) Metagenomics: Application of genomics to uncultured microorganisms. Microbiology and Molecular Biology Reviews, 68, 669-685. http://dx.doi.org/10.1128/MMBR.68.4.669-685.2004
- Streit, W.R. and Schmitz, R.A. (2004) Metagenomics—The key to the uncultured microbes. Current Opinion in Microbiology, 7, 492-498. http://dx.doi.org/10.1016/j.mib.2004.08.002
- Yang, D., Weng, H., Wang, M., Xu, W., Li, Y. and Yang, H. (2010) Cloning and expression of a novel thermostable cellulase from newly isolated Bacillus subtilis strain I15. Molecular Biology Reports, 37, 1923-1929. http://dx.doi.org/10.1007/s11033-009-9635-y
- Teeling, H. and Glöckner, F.O. (2012) Current opportunities and challenges in microbial metagenome analysis—A bioinformatic perspective. Briefings in Bioinformatics, 13, 728-742. http://dx.doi.org/10.1093/bib/bbs039
- Huang, Z.X., Xie, Z.R., Ding, J.M., Li, J.J., Yang, Y.J. and Zhang, X.L. (2013) Cloning, expression and characterization of a lipase from Bacillus subtilis strain I4 with potential application in biodiesel production. Applied Mechanics and Materials, 291, 243-248.
- Schuch, V., Gomes, E.S. and Lemos, E.G.M. (2009) Discovery of genes related to antibiotic biosynthesis into a metagenomic library. Novel Biotechnology, 25, S105.
- Sambrook, J., Russell, D.W. and Russell, D.W. (2001) Molecular cloning: A laboratory manual. Cold Spring Harbour Laboratory Press, New York.
- Ewing, B., Hillier, L., Wendl, M.C. and Green, P. (1998) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Research, 8, 175-185. http://dx.doi.org/10.1101/gr.8.3.175
- Gordon, D., Abajian, C. and Green, P. (1998) Consed: A graphical tool for sequence finishing. Genome Research, 8, 195-202. http://dx.doi.org/10.1101/gr.8.3.195
- Servant, F., Abajian, F., Gouzy, J. and Kahn, D. (2000) ProDom and ProDom-CG: Tools for protein domain analysis and whole genome comparisons. Nucleic Acids Research, 28, 267-269. http://dx.doi.org/10.1093/nar/28.1.267
- Punta, M., Coggill, P.C., Eberhardt, R.Y., Mistry, J., Tate, J., Boursnell, P.N., Forslund, K., Ceric, G., Clements, J., Heger, A., Holm, L., Sonnhammer, E.L.L., Eddy, S.R., Bateman, S. and Finn, R.D. (2012) The Pfam protein families database. Nucleic Acids Research, 40, D290-D301. http://dx.doi.org/10.1093/nar/gkr1065
- Zhang ,Y. (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics, 9, 40.
- (2010) The PyMOL molecular graphics system. Version 1. Schrödinger, LLC.
- Nugent, T., Ward, S. and Jones, D.T. (2011) The MEMPACK alpha-helical transmembrane protein structure prediction server. Bioinformatics, 27, 1438-1439. http://dx.doi.org/10.1093/bioinformatics/btr096
- McGuffin, L.J., Bryson, K. and Jones, D.T. (2000) The PSIPRED protein structure prediction server. Bioinformatics, 16, 404-405. http://dx.doi.org/10.1093/bioinformatics/16.4.404
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Hang, J., Hang, Z., Miller, W. and Lepman, D.J. (1997) Gapped BLAST and PSI-BLAST; a novel generation to protein database search programs. Nucleic Acids Research, 25, 3389-3402. http://dx.doi.org/10.1093/nar/25.17.3389
- Thompson, J.D., Higgins, D.G. and Gibson, T.J. (1994) Clustal W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Research, 11, 4673-4680. http://dx.doi.org/10.1093/nar/22.22.4673
- Hall, P. (2001) BioEdit. Version 5.0.6, Department of Microbiology, North Carolina State University, Raleigh.
- Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M. and Kumar, S. (2011) MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 28, 2731-2739. http://dx.doi.org/10.1093/molbev/msr121
- Felsenstein, J. (1985) An approach using the bootstrap. Evolution, 39, 783-791. http://dx.doi.org/10.2307/2408678
- Mimura, H., Nakanishi, Y., Hirono, M. and Maeshima, M. (2004) Membrane topology of the H+-pyrophosphatase of Streptomyces coelicolor determined by cysteine-scanning mutagenesis. The Journal of Biological Chemistry, 279, 35106-35112. http://dx.doi.org/10.1074/jbc.M406264200
- Lin, S.M., Tsai, J.Y., Hsiao, C.D., Huang, Y.T., Chiu, C.L, Liu, M.H., Tung, J.Y., Liu, T.H., Pan, R.L. and Sun, Y.J. (2012) Crystal structure of membrane-embedded H+-translocating pyrophosphatase. Nature, 484, 399-403. http://dx.doi.org/10.1038/nature10963
- Luoto, H.H., Belogurov, G.A., Baykov, A.A., Lahti, R. and Malinen, A.M. (2011) Na+-translocating membrane pyrophosphatases are widespread in the microbial world and evolutionarily precede H+-translocating pyrophosphatases. Journal of Biological Chemistry, 286, 21633-21642. http://dx.doi.org/10.1074/jbc.M111.244483
- Jones, D.T., Taylor, W.R. and Thornton, J.M. (1992) The rapid generation of mutation data matrices from protein sequences. Computer Applications in the Biosciences, 8, 275-282.
- Suzuki, J., Mutton, M.A., Ferro, M.I.T., Lemos, M.V.F., Pizauro, J.M., Mutton, M.J.R. and Di Mauro, S.M.Z. (2003) Putative pyrophosphate phosphofructose 1-kinase genes identified in sugar cane may be getting energy from pyrophosphate. Genetics and Molecular Research, 2, 376-382.
Abbreviations List
ABI: PRISM® 3100 Genetic Analyzer
BioEdit: Sequence Alignment Editor for Windows 95/98/NT/XP/Vista/7
BLAST: Basic Local Alignment Search Tool
ClustalW: Multiple Sequence Alignment
Consed: Sequence assembly editor companion to Phrap
DNA: Deoxyribonucleic acid
EAA: Eucalyptus spp.Arboretum
GenBank: Sequence database provided by the National Center for Biotechnology Information (NCBI)
H-PPase: Pyrophosphate-energised proton pump
H+-PPase:Hydrogen ions transporting PPase
HMMER3: Databases for homologs of protein sequences
iL/oL&HL:Helix prediction
I-Tasser: Server for protein structure and function prediction
K+:potassium ions
LBMP: Laboratory of Biochemistry and Microorganisms of Plants
LOMETS:Local Meta-Threading-Server
MEGA5: Molecular Evolutionary Genetics Analysis
MEMSAT3 & MEMSAT-SVM: Membrane Helix Prediction
MetaPPase: inorganic pyrophosphatase metagenomic
Na+-PPase: Sodium ions transporting PPase
NCBI: National Center for Biotechnology Information
OrfFinder: Searches for open reading frames
ORFs: Open reading frames
PCR: Polymerase Chain Reaction
PDB: Protein Data Bank
Pfam: Database is a large collection of protein families
Phrap: Program for shotgun sequence assembly
Phred: Base calling software with quality estimation
PL: Pore lining residue
PPases: inorganic pyrophosphatase
PPi: inorganic phosphatase
ProDom: Protein Domain Prediction
PSIPRED: Protein Sequence Analysis Workbench
PyMOL: Molecular Graphics System
RAW: Traces indicate outputs for the prediction SVMs
RE: Re-entrant helix
SP: Signal peptide
SVMs: Support Vector Machine
TM: Transmembrane.
NOTES

*Corresponding authors.