Advances in Microbiology
Vol.05 No.02(2015), Article ID:53996,19 pages
10.4236/aim.2015.52013
A Simulation for Flavivirus Infection Decoy Responses
James K. Peterson1, Alison M. Kesson2, Nicholas J. C. King3
1Department of Mathematical and Biological Sciences, Clemson University, Clemson, USA
2Discipline of Paediatrics and Child Health, Sydney Institute for Emerging Infectious Diseases and Biosecurity Sydney Medical School, The Children’s Hospital at Westmead, Westmead, Australia
3Discipline of Pathology, University of Sydney, Sydney, Australia
Email: petersj@clemson.edu
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 21 January 2015; accepted 9 February 2015; published 12 February 2015
ABSTRACT
In this work, we discuss the development of simulation code for a model of the cross-reactive adaptive immune response seen in flavivirus infections. The model specifically addresses flavivirus pathogen virulence in 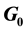 vs.
vs. 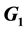 cell states. The MHC-I upregulation of resting cells (
cell states. The MHC-I upregulation of resting cells (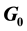 state) allows the T-cells generated for flavivirus peptide antigens to attack healthy cells also. The cells in
state) allows the T-cells generated for flavivirus peptide antigens to attack healthy cells also. The cells in 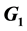 state are not upregulated as much and so virus hides in them and hence is propagated upon rupture. Hence, this type of model is referred to as a decoy model because the immune system is decoyed into preferentially recognizing the upregulated cells while the virus actively propagates in another small, but important, cell population. We show that the generic assumption of upregulation via a model which includes the
state are not upregulated as much and so virus hides in them and hence is propagated upon rupture. Hence, this type of model is referred to as a decoy model because the immune system is decoyed into preferentially recognizing the upregulated cells while the virus actively propagates in another small, but important, cell population. We show that the generic assumption of upregulation via a model which includes the 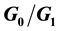 differential upregulation leads to immunopathological consequences. We outline the details behind the simulation code decisions and provide some theoretical justification for our model of collateral damage and upregulation.
differential upregulation leads to immunopathological consequences. We outline the details behind the simulation code decisions and provide some theoretical justification for our model of collateral damage and upregulation.
Keywords:
West Nile Virus, decoy Model, MHC-I Upregulation, 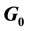 vs
vs 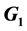 cell State Upregulation differences
cell State Upregulation differences

1. West Nile Virus Infection Models
The family Flaviviridae are single-stranded, plus sense RNA viruses and, with the exception of hepatitis C virus, are all arboviruses, transmitted by mosquitoes and ticks. These viruses cause diseases that include dengue, yellow fever, as well as tick-borne, Japanese and West Nile virus encephalitis. Together these viruses have a
major global impact on human health. They are distributed worldwide with the exception of the polar regions, although a specific flavivirus may be geographically restricted. Notwithstanding, these viruses can expand their geographical distribution, with West Nile virus, for example, emerging in recent years North America, present- ing a significant threat to public and animal health. The most serious manifestation of West Nile virus (WNV) infection is fatal encephalitis in humans and horses, as well as in certain domestic and wild birds. The virus is generally maintained in a zoonotic transmission cycle involving mosquitoes and birds. Most individuals will usually experience an in apparent or mild febrile illness, others, a dengue-like illness, while a minority, in particular including the elderly and immunocompromised, may develop encephalitis which can be fatal. The diagnosis is usually made serologically, although the virus may be detected in the blood by molecular techniques, or in tissue culture. In general, humans are regarded as dead-end hosts since they do not generally produce a high enough viremia to enable transmission by mosquitoes. Currently, no vaccine is available and there is no specific antiviral therapy to modify disease outcomes.
Infection of the host by pathogenic viruses begins an interaction that involves evolutionarily co-developed and counter-developed pathways that confer advantage for both organism and host survival. Infection of cells results in the intracellular processing of virus proteins into peptide fragments that bind to nascent class I and II major histocompatibility complex molecules (MHC-I and II) that are displayed on the infected cell surface. This is additional to the normal processing of self-proteins into peptides that occurs in all cells as part of the main- tenance of tolerance to self. The combined MHC-virus peptide configuration forms the ligand that enables the recognition of infected cells by virus-specific cytotoxic T lymphocytes (CTL) by the cognate T cell receptor (TcR) expressed on the T cell surface. These CTL cause death of the infected cells by lysis, thereby substantially reducing production of progeny virus. This ultimately leads to eradication of virus from the host, enabling host survival (reviewed in [1] ). Flaviviruses such as the neurotropic viruses, West Nile (WNV), Murray Valley encephalitis, Kunjin and Japanese encephalitis viruses, induce an increase in expression of MHC-I and II, as well as other immune recognition molecules, including intracellular adhesion molecule-1 (ICAM-1, CD54), vascular adhesion molecule-1 (VCAM-1, CD106) and E-selectin (CD62E) variously and on a wide variety of cells [2] . WNV-induced increases in cell surface expression of these molecules therefore results in increased efficiency of recognition and killing of infected cells by WNV-specific CTL [3] [4] . Increases in MHC-I concentration on infected cells enable greater numbers of simultaneous TcR-MHC-I-peptide ligand interactions, which enhance the avidity of interaction of virus-specific CTL with the infected target cell. The increased avidity enables the interaction of infected cells with CTL clones that are previously below the recognition threshold by virtue of their low affinity for MHC-virus peptide ligand. Furthermore, a polyclonal anti-viral CTL population may also include low-affinity clones that are self-reactive, i.e., clones that recognize MHC-I-self peptide configurations [4] or even MHC without peptide specificity [5] . Due to the increased avidity of their interaction, these low-affinity, self-reactive clones can now lyse both infected and uninfected target cells that express high cell surface MHC-I concentrations [6] [7] . Furthermore, other non-specific accessory molecules, such as ICAM-1 expressed on the target cell, interacting with its integrin receptor, lymphocyte function- associated antigen-1 (LFA-1; CD11a/CD18), on the T cell, can also increase the avidity of T cell target cell interactions, thus additionally lowering the affinity threshold for T cell recognition and target cell lysis [8] . In addition, interferon-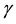 (IFN-
(IFN-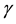 ) also strongly increases the expression of MHC and ICAM-1 on potential target cells in the vicinity, thereby increasing their susceptibility to CTL lysis [9] .
) also strongly increases the expression of MHC and ICAM-1 on potential target cells in the vicinity, thereby increasing their susceptibility to CTL lysis [9] .
Because the avidity of interaction between T cells and their targets can be increased via MHC associated (i.e., MHC-I viral peptide and CD8 MHC-I) and non-MHC-associated (i.e., ICAM-1 LFA-1) interactions, any increases in MHC and/or ICAM-1 expression further enhances CTL recognition of target cells. This in turn increases the chances of low-affinity, self-reactive CTL recognizing these target cells. The position of the cell cycle is also important in flavivirus infection. Cells in G0 (resting) when infected with WNV increase their MHC-I expression 6-10-fold, while infected cycling cells (G1, S, G2 + M) increase MHC-I expression by only 2-3-fold [9] . This results in some 10-fold more lysis of infected G0 target cells than cycling cells by the same CTL [3] . Thus, there is enhanced avidity of interaction between CTL and WNV-infected cells in G0, which express relatively high levels of MHC-I, while infected cycling cells that express much lower levels of MHC-I are less easily recognized. Importantly, WNV replicates to a significantly higher titer in cycling cells than in G0 cells. Thus, virus may be eradicated with relatively poor efficiency in a population of infected cycling cells. In vivo, since most cells are in G0, we hypothesize that it is the small population of infected cycling cells that
support virus replication while maintaining a low immunological profile, thus increasing the probability of virus transmission [4] . As indicated above, the release of IFN-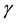 associated with T cell target cell interactions increases MHC and ICAM-1 expression on uninfected cells in the vicinity of virus-infected cells. Thus, high MHC-I expressing (uninfected) targets would be susceptible to lysis by low affinity self-reactive CTL clones. If virus-specific CTL populations included low-affinity self-reactive (i.e., crossreactive) CTL clones, which were able to lyse these uninfected cells, this would result in destruction of uninfected tissue. In the case of an encephalitic virus infection, this collateral damage to uninfected cells would increase the damage to the brain thereby increasing morbidity and mortality. In this way IFN-
associated with T cell target cell interactions increases MHC and ICAM-1 expression on uninfected cells in the vicinity of virus-infected cells. Thus, high MHC-I expressing (uninfected) targets would be susceptible to lysis by low affinity self-reactive CTL clones. If virus-specific CTL populations included low-affinity self-reactive (i.e., crossreactive) CTL clones, which were able to lyse these uninfected cells, this would result in destruction of uninfected tissue. In the case of an encephalitic virus infection, this collateral damage to uninfected cells would increase the damage to the brain thereby increasing morbidity and mortality. In this way IFN-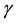 could be responsible for fatal immunopathology in WNV encephalitis in mice [10] .
could be responsible for fatal immunopathology in WNV encephalitis in mice [10] .
This work builds a model of the host-pathogen interactions for flaviviruses in which we focus on WNV. We build a model as closely tied to the biological literature as possible with a mathematical framework and we show that the model exhibits immunopathology that we argue leads to host death in a small but troubling 3% - 8% of the Flavivirus-infected host population.
All modeling of this nature requires many compromises and many levels of abstraction. We model the infection within a single abstract host. We choose initial cell population sizes and immune system components so that our abstract version of a host exhibits reasonable responses closely aligned to what we find in the literature. It is not possible to discuss all of the many design decisions that go into such code development, so we only give some of the details here. The reader can go to [11] for the details for the simulation which is written in C++ and Qt.
Simple T Cell Interaction Algorithms
We begin with a modification of a model of T cell recognition first presented by [12] with changes to handle MHC-I upregulation. Receptors on a given T cell recognize a specific foreign protein snippet, a peptide bound to MHC-I we will call ligand c. This ligand binds to its cognate T cell receptor with high affinity. However, all T cell receptors are also exposed to a variety of possible self-peptide-MHC-I ligands which may bind although with measurably weaker affinity. For clarity, let’s treat these peptides as a single self peptide d complexed with MHC-I, which has a lower affinity for the virus specific T cell receptor. Hence, in an antiviral immune response, at the T cell recognition level there is always a danger of misrecognition in which the virus specific T cell receptor recognizes and responds to a ligand d instead of c. This leads to the possibility of immune-mediated destruction of uninfected cells in the host, i.e. immunopathology. Following [12] , we note the recognition process in T cell receptors includes several steps. Let 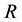 denote the T cell receptor. Then the foreign peptide MHC-I ligand c and R collide at rate
denote the T cell receptor. Then the foreign peptide MHC-I ligand c and R collide at rate 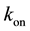 to form a complex
to form a complex 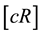 in which the ligand is bound to the receptor. This receptor-ligand complex also disassociates at rate
in which the ligand is bound to the receptor. This receptor-ligand complex also disassociates at rate 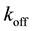 thereby unbinding the ligand c from the receptor R. This leads to the simple dynamics
thereby unbinding the ligand c from the receptor R. This leads to the simple dynamics
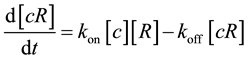 where
where  is the usual notation for concentration of a substance. At steady state,
is the usual notation for concentration of a substance. At steady state,
we then have  leading to
leading to  where
where .
.
When the ligand binds to the T cell receptor, it triggers a signal transduction pathway inside the T cell which leads to activation of the T cell. Also, after ligand binding, the receptor undergoes a series of modifications such as phosphorylation at several sites. Let’s assume the T cell receptor transduction pathway has five phosphoryla- tion sites. The complex is now renamed  where
where  denotes a site not presently phosphorylated. Phosphorylation can then create complexes of the form
denotes a site not presently phosphorylated. Phosphorylation can then create complexes of the form  (one site is phosphorylated),
(one site is phosphorylated),  (two sites) and so forth to
(two sites) and so forth to  (all sites phosphorylated). Each of these modifications are reversible to the original complex
(all sites phosphorylated). Each of these modifications are reversible to the original complex . Transduction of the T cell signal is not triggered until all the modifications have been made. Hence, this introduces a delay between the ligand binding and the T cell signal generation. This suggests only the ligands that stay bound to the T cell receptor long enough to it to become fully phosphorylated can activate the T cell.
. Transduction of the T cell signal is not triggered until all the modifications have been made. Hence, this introduces a delay between the ligand binding and the T cell signal generation. This suggests only the ligands that stay bound to the T cell receptor long enough to it to become fully phosphorylated can activate the T cell.
From our discussions above, we see the probability that  remains bound for a time
remains bound for a time  after binding is
after binding is
related to the off rate. Hence, letting  denote this probability, we have
denote this probability, we have . Signaling only occurs
. Signaling only occurs
after a delay of  seconds after the complex forms. Therefore, the probability per ligand that the T cell is activated is the same as the probability the ligand stays bound longer than
seconds after the complex forms. Therefore, the probability per ligand that the T cell is activated is the same as the probability the ligand stays bound longer than  seconds. Thus, the rate of T cell activation in the presence of a concentration
seconds. Thus, the rate of T cell activation in the presence of a concentration  of the viral peptide MHC-I T cell receptor complex
of the viral peptide MHC-I T cell receptor complex  is
is
 . And the self peptide
. And the self peptide  gives rise to a rate of T cell activation
gives rise to a rate of T cell activation . where we assume
. where we assume
 is the same for both. The viral ligand
is the same for both. The viral ligand  dissociates from the receptor at a slower rate than the self ligand
dissociates from the receptor at a slower rate than the self ligand
 due to its stronger bond with the receptor. Thus,
due to its stronger bond with the receptor. Thus, . We see the viral ligand spends more time bound
. We see the viral ligand spends more time bound
to the receptor than the self one. The error rate for recognition is thus . The literature
. The literature
suggests a typical  and
and  with
with . Hence,
. Hence, . Therefore,
. Therefore,
long delays can enhance the fidelity of the recognition process. As Alon states, this enhancement comes at a cost: a long signal transduction delay implies that more of the viral ligands can dissociate from the T cell receptor before T cell signaling is triggered, leading to a loss of sensitivity. So loss of sensitivity is acceptable because of the improved specificity, i.e. the discrimination between viral and self ligand recognition.
Now, add the WNV to this model of recognition. What changes with upregulation of the number of MHC-I peptide ligands on the surface of the infected cells? As parameters of affinity of the ligand-receptor interaction, we assume that ,
,  and
and  are intrinsic and not altered by the the MHC-I upregulation caused by the WNV infection. As production of anti-viral interferon by infected cells will also increase MHC-I expression on uninfected cells in the local vicinity, we will assume that if the MHC-I peptide ligands are upregulated by a factor
are intrinsic and not altered by the the MHC-I upregulation caused by the WNV infection. As production of anti-viral interferon by infected cells will also increase MHC-I expression on uninfected cells in the local vicinity, we will assume that if the MHC-I peptide ligands are upregulated by a factor , then the new error rate is scaled by the same factor; i.e., if the upregulation factor is
, then the new error rate is scaled by the same factor; i.e., if the upregulation factor is , we
, we
have . Hence, if upregulation is approximately 6 - 10 fold, we see
. Hence, if upregulation is approximately 6 - 10 fold, we see
 which allows for a significant recognition of self ligands and consequent signal transduction leading to an increase in collateral uninfected cell damage. In other words,
which allows for a significant recognition of self ligands and consequent signal transduction leading to an increase in collateral uninfected cell damage. In other words,  is a measure of the avidity of the T cell-target cell interaction, which is a function of MHC-I peptide ligands expressed on the cell surface. We therefore have theoretical reasons to suspect that WNV infections can lead to healthy cell loss.
is a measure of the avidity of the T cell-target cell interaction, which is a function of MHC-I peptide ligands expressed on the cell surface. We therefore have theoretical reasons to suspect that WNV infections can lead to healthy cell loss.
2. One Host Flavivirus Models
The model is based on a theoretical decoy model of [13] , which simulates the cellular adaptive immune response to flavivirus in vivo. In ( [13] and [14] ), it was argued that natural killer cells did not aid in the recovery of a host from a flavivirus infection. Hence, to investigate this interaction most simply, we choose to model only an adaptive immune response. We now describe the model in detail and justifying the assumptions necessary for its implementation.
The main variables we use are listed in Table 1(a). The cells labeled FIC0 are infected resting cells  and those labeled FIC1 are infected and actively dividing,
and those labeled FIC1 are infected and actively dividing, . The virus level
. The virus level  is a measure of viral load in real data in plaque-forming units (pfu) per cell but its exact definition is not important to the simulation. We initialize the healthy cell populations in our simulations to
is a measure of viral load in real data in plaque-forming units (pfu) per cell but its exact definition is not important to the simulation. We initialize the healthy cell populations in our simulations to  million healthy cells. We could initialize for larger or smaller populations but this is concrete enough to make our points. It is not possible to keep track of detailed information on such a large number of healthy cells individually. Instead, we model our simulation universe as a cube which is 216 on a side. As is common in many simulations, we use a number of auxiliary class objects. In the simulation, addresses of each cell range from 0 to approximately 10 million. These integer addresses correspond to addresses in a
million healthy cells. We could initialize for larger or smaller populations but this is concrete enough to make our points. It is not possible to keep track of detailed information on such a large number of healthy cells individually. Instead, we model our simulation universe as a cube which is 216 on a side. As is common in many simulations, we use a number of auxiliary class objects. In the simulation, addresses of each cell range from 0 to approximately 10 million. These integer addresses correspond to addresses in a  box of size
box of size . This box is then divided into cubes with an edge size of 27 which gives 512 cubes. Each cell is located in one of these cubes
. This box is then divided into cubes with an edge size of 27 which gives 512 cubes. Each cell is located in one of these cubes , where
, where . Hence, we have several different cell addressing choices.
. Hence, we have several different cell addressing choices.
1) A cell has an address  which is an integer from 0 to
which is an integer from 0 to . This address can also be expressed as a triple
. This address can also be expressed as a triple  which corresponds to its location in
which corresponds to its location in  space.
space.
2) A cell lives in a cube whose address is  with
with ,
,  and
and  in the range 0 to 7. These
in the range 0 to 7. These
addresses are determined by the rewriting  as
as ,
,  and
and  where
where ,
,  and
and  are the integer remainders.
are the integer remainders.
3) Inside the cube, the remainders  give the the local cell address
give the the local cell address .
.
(a)  (b)
(b)
Table 1. Variables and probabilities. (a) Variables, (b) Probabilities.
The basic rates and probabilities we use are listed in Table 1(b) and Table 2(a). The nominal values do change with different simulation needs, but they are listed to give a base from which we will diverge as necessary. New viruses are added to the virus population due to lysis of FIC0 and FIC1 cells. In our model,  changes at each iteration and the other probabilities are set randomly around the base values given in Table 1(b). There are also some additional parameters as shown in the bottom panel of the table. The use of these parameters, rates and probabilities are discussed in the following sections.
changes at each iteration and the other probabilities are set randomly around the base values given in Table 1(b). There are also some additional parameters as shown in the bottom panel of the table. The use of these parameters, rates and probabilities are discussed in the following sections.
2.1. Infected Cell Modeling
For dividing infected cells, our model also uses IFN- and ICAM-1 upregulation. Upon recognition of their cognate ligand, MHC-I-peptide, by the T cell receptor (TcR), cytotoxic T cells secrete IFN-
and ICAM-1 upregulation. Upon recognition of their cognate ligand, MHC-I-peptide, by the T cell receptor (TcR), cytotoxic T cells secrete IFN- which upregu- lates the cell surface concentration of MHC-I peptide ligand on the infected cell surface. Thus, MHC-I-peptide upregulation is induced by both the WNV infection and IFN-
which upregu- lates the cell surface concentration of MHC-I peptide ligand on the infected cell surface. Thus, MHC-I-peptide upregulation is induced by both the WNV infection and IFN- . As an accessory adhesion molecule in T cell- target cell interactions, upregulation of ICAM-1 further increases the avidity of the interaction between the TcR and the cognate MHC-I-peptide-ligand. ICAM-1 is also upregulated by IFN-
. As an accessory adhesion molecule in T cell- target cell interactions, upregulation of ICAM-1 further increases the avidity of the interaction between the TcR and the cognate MHC-I-peptide-ligand. ICAM-1 is also upregulated by IFN- . When an infected cell is re- cognized and killed by a T cell, the IFN-
. When an infected cell is re- cognized and killed by a T cell, the IFN- secreted by the T cell upregulates the cell surface concentration of both MHC-I and ICAM-1 on all infected and uninfected cells in a neighborhood of the recognized killed cell. We keep track of this changing upregulation level for the infected cell with the variable
secreted by the T cell upregulates the cell surface concentration of both MHC-I and ICAM-1 on all infected and uninfected cells in a neighborhood of the recognized killed cell. We keep track of this changing upregulation level for the infected cell with the variable .
.
We could handle the neighborhood upregulation of MHC-I simply. Each upregulated cell belongs to a coarse
(a) (b)
Table 2. More probabilities and parameters and a sample upregulation strategy. (a) More probabilities and parameters; (b) A sample upregulation strategy.
cell . We could then upregulate all infected cells that belong to this coarse cell. The difference between the number of infected cells found in a coarse cell and the total number in the coarse cell,
. We could then upregulate all infected cells that belong to this coarse cell. The difference between the number of infected cells found in a coarse cell and the total number in the coarse cell,  , then gives us an estimate of how many uninfected cells have also been upregulated. This upregulation of cell surface MHC-I and ICAM-I concentration increases the probability that an infected cell will be recognized and killed. Unrecognized infected cells continue to move towards a lytic event which releases progeny viruses into the coarse cell. We model the expression of MHC-I-peptide ligands on the surface of an infected cell as follows. Living cells display MHC-I molecules that bind short peptides within a groove. Together these form a ligand recognized by the TcR. These peptides are derived from both self and non-self proteins.
, then gives us an estimate of how many uninfected cells have also been upregulated. This upregulation of cell surface MHC-I and ICAM-I concentration increases the probability that an infected cell will be recognized and killed. Unrecognized infected cells continue to move towards a lytic event which releases progeny viruses into the coarse cell. We model the expression of MHC-I-peptide ligands on the surface of an infected cell as follows. Living cells display MHC-I molecules that bind short peptides within a groove. Together these form a ligand recognized by the TcR. These peptides are derived from both self and non-self proteins.
As a modeling strategy, we make the assumption that there are  peptides that are viral non-self peptides. The model can use any
peptides that are viral non-self peptides. The model can use any  of course, but it is easier to see the thread of our argument with a specific power. This number has been chosen because it is a power of 2 and hence has some advantages in simulations and it was large enough to be pertinent. The self-peptide pool in reality is very large. We will let the self-peptide pool be of the same size as the non-self pool, i.e., 8192. To model peptides, we will number them from 0 to
of course, but it is easier to see the thread of our argument with a specific power. This number has been chosen because it is a power of 2 and hence has some advantages in simulations and it was large enough to be pertinent. The self-peptide pool in reality is very large. We will let the self-peptide pool be of the same size as the non-self pool, i.e., 8192. To model peptides, we will number them from 0 to  (in general
(in general , and divide them in half: viral peptides are labeled from 0 to 8191 and self, from 8192 to 16383. We know that a cell has many MHC-I-peptide ligands on its surface, but we do not want to model all of them. We assume that in a given unit cell surface area, there are
, and divide them in half: viral peptides are labeled from 0 to 8191 and self, from 8192 to 16383. We know that a cell has many MHC-I-peptide ligands on its surface, but we do not want to model all of them. We assume that in a given unit cell surface area, there are  MHC-I-peptide ligands re- cognizable by the TcR, if not upregulated, and a multiple of these if there is upregulation of the MHC-I molecule on the cell surface. Thus, if the upregulation factor is
MHC-I-peptide ligands re- cognizable by the TcR, if not upregulated, and a multiple of these if there is upregulation of the MHC-I molecule on the cell surface. Thus, if the upregulation factor is , then the upregulated cell would have
, then the upregulated cell would have  ligands. In a WNV-infected cell without upregulation, we will assume that the
ligands. In a WNV-infected cell without upregulation, we will assume that the  ligands per unit area pre- sented on the cell surface consist of
ligands per unit area pre- sented on the cell surface consist of  self peptides and
self peptides and  WNV peptide. Then, if an infected cell is upregu- lated by a factor of
WNV peptide. Then, if an infected cell is upregu- lated by a factor of , the cell would present
, the cell would present  self peptides and
self peptides and  WNV peptides. For example if the upregulation factor is
WNV peptides. For example if the upregulation factor is , we would see
, we would see  self peptides and
self peptides and  WNV peptides per unit area on the infected cell. The non-upregulated (
WNV peptides per unit area on the infected cell. The non-upregulated ( ) cells are handled in a similar fashion except that instead of
) cells are handled in a similar fashion except that instead of  displayed ligands each time step, we only use
displayed ligands each time step, we only use . One of them is considered viral and the other three are self. Hence, for an upregulation factor of
. One of them is considered viral and the other three are self. Hence, for an upregulation factor of , the vector
, the vector  in
in  has the form shown in Table 2(b) for resting infected cells and dividing
has the form shown in Table 2(b) for resting infected cells and dividing  infected cells. We would have similar data for upregulation factors other than
infected cells. We would have similar data for upregulation factors other than , of course.
, of course.
We create new FIC0 cells as needed by calling the method SetFIC0() which is discussed in [11] . When we create a new FIC0 cell, we assign it to a randomly chosen coarse cube. We create this coarse cube address by randomly choosing integers ,
,  and
and  in the range
in the range  to
to  (see the cell address discussion earlier). We maintain a list of FIC0 and FIC1 cells throughout our simulations and each such cell has the data vector
(see the cell address discussion earlier). We maintain a list of FIC0 and FIC1 cells throughout our simulations and each such cell has the data vector  associated with it, which has the components shown in Table 3(a).
associated with it, which has the components shown in Table 3(a).
Here  and
and  are as previously described. The variable
are as previously described. The variable  is a counter which is increased for each time unit the infected cell is alive. Once
is a counter which is increased for each time unit the infected cell is alive. Once  exceeds the allowable life time of a typical infected cell, the cell is removed from the simulation. The vector
exceeds the allowable life time of a typical infected cell, the cell is removed from the simulation. The vector  contains
contains  integers where
integers where  is the amount of upregulation the infected cell has had. For example, if the upregulation level is
is the amount of upregulation the infected cell has had. For example, if the upregulation level is , then the vector would be size
, then the vector would be size . The integers refer to the proteins in our simulation which are expressed on the surface of the cell and are therefore available for recognition, as described earlier. Finally, the variables
. The integers refer to the proteins in our simulation which are expressed on the surface of the cell and are therefore available for recognition, as described earlier. Finally, the variables  and
and  are the infected cell’s avidity and upregulation state, respectively. We will discuss how these variables are updated next. Our coding decisions and implementation strategies and choices for these matters are discussed fully in [11] .
are the infected cell’s avidity and upregulation state, respectively. We will discuss how these variables are updated next. Our coding decisions and implementation strategies and choices for these matters are discussed fully in [11] .
(a) (b)
Table 3. Components of  and the coefficient of variation,
and the coefficient of variation, . (a) Components of
. (a) Components of ; (b) The coefficient of variation.
; (b) The coefficient of variation.
2.2. Infected Data Updates
Our model uses both IFN- and ICAM-1 upregulation. IFN-
and ICAM-1 upregulation. IFN- is secreted by cytotoxic T cells upon recogni- tion of their cognate ligand; this upregulates both MHC-I peptide ligand and ICAM-1 on the cell surface. MHC-I and ICAM-1 upregulation are also induced by WNV infection. We discuss the reasoning behind our model of the upregulation issue later in Section 4. This section explains our strategy for IFN-
is secreted by cytotoxic T cells upon recogni- tion of their cognate ligand; this upregulates both MHC-I peptide ligand and ICAM-1 on the cell surface. MHC-I and ICAM-1 upregulation are also induced by WNV infection. We discuss the reasoning behind our model of the upregulation issue later in Section 4. This section explains our strategy for IFN- upregulation as well as the killing of uninfected cells that are susceptible to lysis by low affinity anti-viral T cells as argued by the decoy hypothesis [13] . We will refer to such uninfected cell deaths as collateral damage.
upregulation as well as the killing of uninfected cells that are susceptible to lysis by low affinity anti-viral T cells as argued by the decoy hypothesis [13] . We will refer to such uninfected cell deaths as collateral damage.
At each time point in the simulation, new infected cell’s are created and added to the FIC0 or FIC1 list. For these newly created infected cells, we initialize their age and recognition value to ; and set their initial IFN-
; and set their initial IFN- value to
value to . Hence, as the simulation runs, there is a pool of previously created infected cells and infected cells newly created at this time. As soon as an infected cell is created, we calculate its avidity for the T cells which will interact with it at this time. Once the avidity is calculated, we multiply it by the current IFN-
. Hence, as the simulation runs, there is a pool of previously created infected cells and infected cells newly created at this time. As soon as an infected cell is created, we calculate its avidity for the T cells which will interact with it at this time. Once the avidity is calculated, we multiply it by the current IFN- value of the cell (recall it is
value of the cell (recall it is  at this time). Once a cell is infected, we assume there is a time lag before there is ICAM-I upregulation due to the infection. This time delay is set by the variable ICAMThreshold in the simulation code. Currently, this is set to be
at this time). Once a cell is infected, we assume there is a time lag before there is ICAM-I upregulation due to the infection. This time delay is set by the variable ICAMThreshold in the simulation code. Currently, this is set to be  time steps and since a time step corresponds to
time steps and since a time step corresponds to  minutes, we see ICAM-I is not upregulated until after
minutes, we see ICAM-I is not upregulated until after  hours. This is the only time in the simulation that ICAM-I upregulation is used.
hours. This is the only time in the simulation that ICAM-I upregulation is used.
Hence, at each time in the simulation, we have lists of infected cells whose avidities are known. Infected cells start with their intrinsic avidity which is determined by their displayed MHC-I complexes and the particular mix of T cells that are present to interact with them. Each of these cells also has a life time which is updated each simulation step. However, after these computations are done, we then determine how many of these infected cells are to be recognized and lysed or who have lived long enough to rupture. Hence, there are two events: one is the infected cell recognition which initiates IFN- upregulation which enhances any infected cells avidity which can receive the IFN-
upregulation which enhances any infected cells avidity which can receive the IFN- signal released by the recognized cell and two: the infected cell death event which does not release IFN-
signal released by the recognized cell and two: the infected cell death event which does not release IFN- but does release progeny virus. We keep track of the coarse addresses of the cells targeted for recognition and removal and we increase the avidity of these cells by the IFN-
but does release progeny virus. We keep track of the coarse addresses of the cells targeted for recognition and removal and we increase the avidity of these cells by the IFN- multiplier. We do this up to a maximum multiplier amount which is set by the simulation. Then, at each subsequent simulation step, we check to see if this enhanced avidity exceeds the value needed for recognition. Thus, at each time step, the infected cells removed are any cells whose avidity (newly created or enhanced) allows recognition. Hence, the value of an infected cell’s recognition value increases over the life of the simulation as upregulation of ligand and ICAM-1 spreads through the neighborhood cells.
multiplier. We do this up to a maximum multiplier amount which is set by the simulation. Then, at each subsequent simulation step, we check to see if this enhanced avidity exceeds the value needed for recognition. Thus, at each time step, the infected cells removed are any cells whose avidity (newly created or enhanced) allows recognition. Hence, the value of an infected cell’s recognition value increases over the life of the simulation as upregulation of ligand and ICAM-1 spreads through the neighborhood cells.
We need to look at the details of how we compute the avidity of an infected cell. However, first we will discuss T Cell modeling.
3. T Cell Initialization Code
In this simulation, we create a population of T Cells which can kill WNV-infected cells and if the decoy model is correct, uninfected cells as well. The function we use to do this in is Create T Cells (). The target cell population is divided into big cubes for convenience. At each time step in the simulation, new target cells are infected. New FIC0 (infected, not dividing) and FIC1 (infected, dividing) cells are created.
At the time they are created, we calculate the avidity of the T cell-target cell interaction according to the following model. In the simulation, there are  West Nile Virus (WNV) peptides that can be placed in
West Nile Virus (WNV) peptides that can be placed in  MHC-I grooves. The T Cells that can kill an infected cell are then modeled using a distribution of TcR affinities. Each of the T Cells has its highest affinity for one of the
MHC-I grooves. The T Cells that can kill an infected cell are then modeled using a distribution of TcR affinities. Each of the T Cells has its highest affinity for one of the  MHC-I-WNV peptide ligands. The affinity calculation for each TcR-ligand interaction is essentially a normal distribution. There are
MHC-I-WNV peptide ligands. The affinity calculation for each TcR-ligand interaction is essentially a normal distribution. There are  points in the bell curve with the one at the center corresponding to the maximum affinity for given MHC-I-WNV peptide ligand. That leaves
points in the bell curve with the one at the center corresponding to the maximum affinity for given MHC-I-WNV peptide ligand. That leaves  to the right of the center and
to the right of the center and  to the left each with progressively decreasing affinity. This number
to the left each with progressively decreasing affinity. This number  is a simulation parameter. These
is a simulation parameter. These  other affinity values then must correspond to how the TcR interacts with a cell via the MHC-I-peptide ligand. To each of these
other affinity values then must correspond to how the TcR interacts with a cell via the MHC-I-peptide ligand. To each of these  points, we associate self peptides (the ones numbered above
points, we associate self peptides (the ones numbered above ). At the start of each simulation, we pick these
). At the start of each simulation, we pick these  peptides randomly. This gives
peptides randomly. This gives  TCRs which have the potential to interact with
TCRs which have the potential to interact with  MHC-I-self peptide ligands (lower affinity than the center one) and the one WNV peptide. At each time step, we choose to compute the FIC0 or FIC1 affinity/avidity using a fraction of the full T Cell capability. Thus, instead of calculating the affinity using all
MHC-I-self peptide ligands (lower affinity than the center one) and the one WNV peptide. At each time step, we choose to compute the FIC0 or FIC1 affinity/avidity using a fraction of the full T Cell capability. Thus, instead of calculating the affinity using all  affinity calculations for each self peptide in the pool, at each time step, we choose a fraction (now 45%), of these, approximately
affinity calculations for each self peptide in the pool, at each time step, we choose a fraction (now 45%), of these, approximately . Thus, we randomly pick
. Thus, we randomly pick  numbers from
numbers from  to
to . For example, if one of these numbers is
. For example, if one of these numbers is , we think of
, we think of  as
as  and so the number
and so the number  corresponds to the WNV- peptide
corresponds to the WNV- peptide  and the self-peptide
and the self-peptide . The number
. The number  would then be
would then be  corresponding to WNV pep- tide
corresponding to WNV pep- tide  and self peptide
and self peptide . So this collection of
. So this collection of  numbers gives us a mix of T Cells (TcR) correspond- ing to the
numbers gives us a mix of T Cells (TcR) correspond- ing to the  WNV-peptides we selected as well as the other numbers which correspond to the self-peptides.
WNV-peptides we selected as well as the other numbers which correspond to the self-peptides.
We assume the avidity of a T Cell to bind to a WNV-infected cell via MHC-I-peptide ligand depends on the amount of MHC-I and other surface molecule upregulation. If we label this upregulation factor as , we assign
, we assign
the affinity of peptide  using a normal distribution to be
using a normal distribution to be . The parameter
. The parameter  determines the
determines the
coefficient of variation of this bell curve. The coefficient of variation,  , is the width across the bell curve
, is the width across the bell curve
corresponding to an ordinate value of 0.5; this is given by the formula . We want the
. We want the
curve representing the affinity of T Cells which recognize MHC-I-peptide ligand to be very narrow with no upregulation of ligand on the target cells ( ) and to widen as
) and to widen as  increases up to our maximum value of
increases up to our maximum value of . We achieve this by choosing the
. We achieve this by choosing the  values as shown in Table 3(b). The third column in the table shows how the coefficient of variation of the curves varies with the value of
values as shown in Table 3(b). The third column in the table shows how the coefficient of variation of the curves varies with the value of . This behavior is illustrated in Figure 1 where the circles on each of these plots represent the values from
. This behavior is illustrated in Figure 1 where the circles on each of these plots represent the values from  (the affinity of the TcR for MHC-I-WNV-peptide) to
(the affinity of the TcR for MHC-I-WNV-peptide) to , showing the decrease in affinity as it moves away from the center where there is the maximum of 1. In the actual code, we adjust these values so that they scatter randomly around the center value and we also use
, showing the decrease in affinity as it moves away from the center where there is the maximum of 1. In the actual code, we adjust these values so that they scatter randomly around the center value and we also use  where
where  is a random perturbation of
is a random perturbation of . Note that for small values of
. Note that for small values of , the generated affinities are quite small for
, the generated affinities are quite small for . We can easily adjust this affinity model to make the affinity curves much narrower. We can therefore adjust the affinities we use in our avidity calculations so that they depend on the upregulation factor. The simulation would typically generate
. We can easily adjust this affinity model to make the affinity curves much narrower. We can therefore adjust the affinities we use in our avidity calculations so that they depend on the upregulation factor. The simulation would typically generate  such as these for an MHC-I upregulation factor of
such as these for an MHC-I upregulation factor of : WNV-Peptide T Cell
: WNV-Peptide T Cell  would have
would have ; WNV-Peptide T Cell
; WNV-Peptide T Cell ,
,  and so forth. Note all of these are slight perturbations from the base value of
and so forth. Note all of these are slight perturbations from the base value of  shown in Table 3(b) which is the base
shown in Table 3(b) which is the base  for a MHC-I upregulation of
for a MHC-I upregulation of . Then, we compute the actual affinities we will assign to each self-peptide.
. Then, we compute the actual affinities we will assign to each self-peptide.
Infected Cell Avidity Calculations
Now let’s look at a typical infected cell of class FIC0. If the WNV MHC mediated upregulation is  fold, in our model, this FIC0 cell has MHC-I grooves that hold populations of peptides that are comprised propor- tionately from
fold, in our model, this FIC0 cell has MHC-I grooves that hold populations of peptides that are comprised propor- tionately from  self peptides and
self peptides and  WNV peptides. These peptides are chosen randomly from the WNV and self pool, as before. Now at each time point, we use
WNV peptides. These peptides are chosen randomly from the WNV and self pool, as before. Now at each time point, we use  of the T Cells created according to the scheme discussed above. It seems reasonable to model in this way as we get a random selection of a large population of
of the T Cells created according to the scheme discussed above. It seems reasonable to model in this way as we get a random selection of a large population of

Figure 1. The T Cell affinities vary with the amount of MHC-I upregulation.
T Cells with a range of affinities interacting at each time point. For example, at a given time point, the  T Cells we pick could be as follows: the first WNV peptide is
T Cells we pick could be as follows: the first WNV peptide is  and associated with it are the self peptides
and associated with it are the self peptides . We have now selected
. We have now selected  of the
of the  T Cells. The second WNV peptide is
T Cells. The second WNV peptide is  and we select
and we select  of its self peptides. We have thus selected
of its self peptides. We have thus selected  of the
of the  possibilities. Then the third WNV peptide is
possibilities. Then the third WNV peptide is  and for it we select
and for it we select  of its self peptides giving a total of
of its self peptides giving a total of  so far. We finish by selecting WNV peptides
so far. We finish by selecting WNV peptides  and
and  of its self peptides giving
of its self peptides giving  total and WNV peptide
total and WNV peptide  and
and  of its self peptides to finish out the
of its self peptides to finish out the . In a sense, we have selected
. In a sense, we have selected  peptides to match with the peptides on the surface of the FIC0 cell. We can either think of this as
peptides to match with the peptides on the surface of the FIC0 cell. We can either think of this as  separate T Cells or as
separate T Cells or as  T Cells (one for each WNV peptide) with a range of affinities. The T Cells are assembled from these randomly selected numbers. When these numbers are randomly chosen, we do not allow for the same number to be chosen twice. The chances of having the same WNV peptide in a T Cell are small and so we have set up the simulation model to make sure we can not get matches in the random selection process.
T Cells (one for each WNV peptide) with a range of affinities. The T Cells are assembled from these randomly selected numbers. When these numbers are randomly chosen, we do not allow for the same number to be chosen twice. The chances of having the same WNV peptide in a T Cell are small and so we have set up the simulation model to make sure we can not get matches in the random selection process.
We can therefore think of these T Cells as having a response to peptides other than the WNV peptide; we can assemble a T Cell with highest affinity for WNV peptide whose recognition can also include as many as  self peptides. Thus, the model we use can create as many T Cells as there are ways to take
self peptides. Thus, the model we use can create as many T Cells as there are ways to take  peptides out of a set of
peptides out of a set of . For example, if the
. For example, if the  numbers randomly created included
numbers randomly created included ,
,  ,
,  and
and , this would correspond to a T Cell whose WNV peptide is the third one and whose associated self peptides are the ones stored as self peptide
, this would correspond to a T Cell whose WNV peptide is the third one and whose associated self peptides are the ones stored as self peptide ,
,  ,
,  and
and . That T Cell would then be used in the affinity computations at that time step.
. That T Cell would then be used in the affinity computations at that time step.
To calculate the avidity of these  T Cell interactions with this FIC0 cell, we find out which of the
T Cell interactions with this FIC0 cell, we find out which of the  self and
self and  WNV peptides expressed on the surface of this FIC0 cell are recognized. We then sum over all of the affinities that result from the matches. If this sum is
WNV peptides expressed on the surface of this FIC0 cell are recognized. We then sum over all of the affinities that result from the matches. If this sum is  or more, this FIC0 cell will be targeted for lysis at the next time step. This recognition will then trigger the IFN-
or more, this FIC0 cell will be targeted for lysis at the next time step. This recognition will then trigger the IFN- upregulation and the ICAM-1 upregulation on the remaining target cells. This is done simply. The original avidity is multiplied by appropriate multipliers (say
upregulation and the ICAM-1 upregulation on the remaining target cells. This is done simply. The original avidity is multiplied by appropriate multipliers (say  for IFN-
for IFN- and
and  for ICAM-1 upregulation, respectively) to obtain the new avidity. We do this calcula- tion for all the infected FIC0 cells, maintaining them in a list. We add them when they are created and delete them upon T Cell recognition and lysis or upon removal because they have exceeded their lifetime limit.
for ICAM-1 upregulation, respectively) to obtain the new avidity. We do this calcula- tion for all the infected FIC0 cells, maintaining them in a list. We add them when they are created and delete them upon T Cell recognition and lysis or upon removal because they have exceeded their lifetime limit.
Our process begins by noting out how many active T Cells and infected cells there already are. Then we find the number of new FIC0 cells that have been created, which indicates where to start the affinity calculations. The previous FIC0 cells in the list have already had their affinity computed. We are now at the place in the list where we need to calculate the affinity for our newly infected cell. To calculate the affinity, we look at the peptides associated with the infected cell and, for the infected cell, we also store their lifetime and IFN- upregulation factor. We have already chosen
upregulation factor. We have already chosen  random T Cells. Each of these corresponds to an integer
random T Cells. Each of these corresponds to an integer 
which can be written as . Each active T Cell,
. Each active T Cell,  , therefore has an associated peptide
, therefore has an associated peptide  and
and
affinity  where
where  is the WNV peptide number and
is the WNV peptide number and  is the number of the self-peptide. To indicate the dependence on the number
is the number of the self-peptide. To indicate the dependence on the number , we will also add a superscript
, we will also add a superscript . Hence, the active T Cell,
. Hence, the active T Cell,  , has an asso-
, has an asso-
ciated peptide  and affinity
and affinity . Then, for each peptide,
. Then, for each peptide,  , on the surface of the infected cell, we check if it matches a peptide that can be recognized by the TcR and add its associated affinity. Let
, on the surface of the infected cell, we check if it matches a peptide that can be recognized by the TcR and add its associated affinity. Let  denote
denote
the check  if V is the same peptide as W and
if V is the same peptide as W and , otherwise. If we let
, otherwise. If we let  denote the
denote the
infected cells avidity, we then have . The
. The  active T Cells at this time step are
active T Cells at this time step are
a mix of the  WNV peptides, so the value of
WNV peptides, so the value of  could be larger than
could be larger than  indicating a recognition event. Then, we multiply
indicating a recognition event. Then, we multiply  by the IFN-
by the IFN- multiplier factor,
multiplier factor,  making sure we don’t exceed the maximum IFN-
making sure we don’t exceed the maximum IFN-
upregulation,  , allowed:
, allowed: . We also allow for ICAM-1 upregulation if the cell has
. We also allow for ICAM-1 upregulation if the cell has
been infected long enough. If the current life of the infected cell exceeds the ICAM threshold time,  , we reset
, we reset
 again by multiplying by the ICAM-1 upregulation factor,
again by multiplying by the ICAM-1 upregulation factor, . Of course, if this value exceeds the maxi-
. Of course, if this value exceeds the maxi-
mum ICAM-1 upregulation amount,  , we set
, we set  to be
to be :
: .
.
4. Handling Collateral Damage
To find the avidity of the uninfected cells in each cube (the approximately  or so), we randomly assign the self peptides for an uninfected cell and using the full
or so), we randomly assign the self peptides for an uninfected cell and using the full  T Cell affinity possibilities, we compute the avidity. To do this, we assume the T Cells are spread uniformly through the cubes and since there are so many uninfected cells compared to infected in general, it would made sense to think all these T Cells could interact with the uninfected cells. When we model an infection, at each time step in a given simulation, we need to estimate collateral damage. We do this by developing an estimate of how much collateral damage occurs during an infection assuming a certain level of upregulation. This estimate is done before we perform any infection simulations. We build these estimates for the upregulation levels from
T Cell affinity possibilities, we compute the avidity. To do this, we assume the T Cells are spread uniformly through the cubes and since there are so many uninfected cells compared to infected in general, it would made sense to think all these T Cells could interact with the uninfected cells. When we model an infection, at each time step in a given simulation, we need to estimate collateral damage. We do this by developing an estimate of how much collateral damage occurs during an infection assuming a certain level of upregulation. This estimate is done before we perform any infection simulations. We build these estimates for the upregulation levels from  to
to  at least
at least  times. The estimates are then averaged over the number of collateral damage computations we have done to provide the collateral damage amounts we use in any of our infection simulations. A typical collateral damage calculation is done as follows. In each cube, we
times. The estimates are then averaged over the number of collateral damage computations we have done to provide the collateral damage amounts we use in any of our infection simulations. A typical collateral damage calculation is done as follows. In each cube, we
・ choose the self peptides for each of these  cells randomly from the self peptide pool.
cells randomly from the self peptide pool.
・ for these  cells, compute their avidities for the
cells, compute their avidities for the  T Cells with all their
T Cells with all their  self peptides. We will assume that each uninfected cell expresses a certain number of self peptides on its surface. This number is arbitrary, but clearly, if the entire universe of self peptides were on the cell, then all uninfected cells would be recognized and potentially lysed. We will assume each uninfected cell expresses
self peptides. We will assume that each uninfected cell expresses a certain number of self peptides on its surface. This number is arbitrary, but clearly, if the entire universe of self peptides were on the cell, then all uninfected cells would be recognized and potentially lysed. We will assume each uninfected cell expresses  or
or  randomly chosen self peptides on their surface which is a small fraction from the self-peptide universe of
randomly chosen self peptides on their surface which is a small fraction from the self-peptide universe of  choices. Our rational for this is as follows. In any cell, peptide snippets are taken from all self-proteins, processed by molecular machinery, placed in MHC-1 cradles and the resulting complex is transported to the cell’s surface. There are a huge number of self-proteins and a limited number of MHC-1 molecules. Hence, at any given moment, a cell displays some fraction of the possible universe of self-peptides as well as a portion of the possible foreign peptides. In addition, MHC-1 molecules are constantly being recycled into component parts for reuse and new MHC-1 molecules with new self-peptides installed in their antigen presenting grooves are being generated. Thus, we can assume only a fraction of the possible self-peptides are displayed at any time and the members of this fraction are probably constantly changing. The immune system generated T Cells that can recognize self-proteins then interact with this fraction of displayed self-peptides generating an affinity. In a typical human, the self-peptide universe is probably on the order of
choices. Our rational for this is as follows. In any cell, peptide snippets are taken from all self-proteins, processed by molecular machinery, placed in MHC-1 cradles and the resulting complex is transported to the cell’s surface. There are a huge number of self-proteins and a limited number of MHC-1 molecules. Hence, at any given moment, a cell displays some fraction of the possible universe of self-peptides as well as a portion of the possible foreign peptides. In addition, MHC-1 molecules are constantly being recycled into component parts for reuse and new MHC-1 molecules with new self-peptides installed in their antigen presenting grooves are being generated. Thus, we can assume only a fraction of the possible self-peptides are displayed at any time and the members of this fraction are probably constantly changing. The immune system generated T Cells that can recognize self-proteins then interact with this fraction of displayed self-peptides generating an affinity. In a typical human, the self-peptide universe is probably on the order of  and
and  of this is
of this is  displayed self-peptides, which is a reasonable approximation to what we would find in a typical cell. Since our universe size is
displayed self-peptides, which is a reasonable approximation to what we would find in a typical cell. Since our universe size is , this amounts to displaying the fraction
, this amounts to displaying the fraction  of the self-peptide universe; approximately
of the self-peptide universe; approximately  of the self-peptides. In our affinity calculations here, therefore we assume each uninfected cell expresses
of the self-peptides. In our affinity calculations here, therefore we assume each uninfected cell expresses  or
or  randomly chosen self peptides on their surface.
randomly chosen self peptides on their surface.
We also know only a fraction of potential T Cells interact with any given uninfected cell at a particular time. Hence, choosing a small number of self peptides to be expressed on each of our uninfected cells and then allowing our T Cell interaction to determine avidity is essentially a more quantitative version of a simple probabilistic law. Such a law might have the form of assuming a certain percentage of uninfected cells are recognized at each time step. However, it is then harder to handle IFN- mediated upregulation. Also here, we don’t use the 45% fraction chosen randomly at each time step. Instead we do the affinity calculation using the full range of T Cells. This gives us avidities for each cell in the cube; i.e.
mediated upregulation. Also here, we don’t use the 45% fraction chosen randomly at each time step. Instead we do the affinity calculation using the full range of T Cells. This gives us avidities for each cell in the cube; i.e.  avidities.
avidities.
Note it is not clear how we should model these avidities. If we simply sum the affinities, it is quite possible that we would have avidities that exceed  or more for a given cell. If the killing threshold is set to
or more for a given cell. If the killing threshold is set to , this could generate an excessive number of uninfected cell deaths. We will divide our summed affinities by
, this could generate an excessive number of uninfected cell deaths. We will divide our summed affinities by  to make it harder to reach the killing threshold. In essence, this is a statistical approach to the calculated avidity. In our calculations, we choose this last approach for our calculated avidities. Our models assume
to make it harder to reach the killing threshold. In essence, this is a statistical approach to the calculated avidity. In our calculations, we choose this last approach for our calculated avidities. Our models assume  WNV pep- tides and hence, we divide all the computed avidities by 5 to generate the avidity of each cell in the cube.
WNV pep- tides and hence, we divide all the computed avidities by 5 to generate the avidity of each cell in the cube.
・ assume when a FIC0 or FIC1 cell is killed, there is IFN- mediated upregulation of these
mediated upregulation of these  cells to some degree. We have the maximum IFN-
cells to some degree. We have the maximum IFN- upregulation set
upregulation set , so if the IFN-
, so if the IFN- multiplier is set to
multiplier is set to , then after
, then after  upregulations,
upregulations,  is approximately
is approximately . Hence, after
. Hence, after  upregulations, the upregulation approaches its maximum value and upregulation of these cells stops. Since we are using the full universe of of available T Cells, if an uninfected cell reaches maximum IFN-
upregulations, the upregulation approaches its maximum value and upregulation of these cells stops. Since we are using the full universe of of available T Cells, if an uninfected cell reaches maximum IFN- mediated upregulation without being lysed, this cell is no longer able to be destroyed by the immune system’s response. The cell will then live out its normal life and be removed following normal cellular metabolic rules. Hence, maximally upregulated cells die y natural causes since the computed avidity for these cells implies they have not been recognized by any of the T Cells.
mediated upregulation without being lysed, this cell is no longer able to be destroyed by the immune system’s response. The cell will then live out its normal life and be removed following normal cellular metabolic rules. Hence, maximally upregulated cells die y natural causes since the computed avidity for these cells implies they have not been recognized by any of the T Cells.
・ next, go through the  avidities just calculated and count how many have a high enough avidity to be killed as they are. Call this number
avidities just calculated and count how many have a high enough avidity to be killed as they are. Call this number  because there is no upregulation yet. Then, go through the re- maining avidities just calculated and count how many have a high enough avidity to be killed with their avidities multiplied by
because there is no upregulation yet. Then, go through the re- maining avidities just calculated and count how many have a high enough avidity to be killed with their avidities multiplied by . Subtract the previous number
. Subtract the previous number  and call this number
and call this number . This gives the number of additional cells killed after
. This gives the number of additional cells killed after  upregulation. We then continue this process: go through the re- maining avidities and count how many have a high enough avidity to be killed with their avidities multiplied by
upregulation. We then continue this process: go through the re- maining avidities and count how many have a high enough avidity to be killed with their avidities multiplied by . Subtract the previous
. Subtract the previous  numbers
numbers  and
and  and call this number
and call this number . This gives the number of additional cells killed after
. This gives the number of additional cells killed after  upregulations. We keep doing this until you have performed
upregulations. We keep doing this until you have performed  upregulations which approximates maximal upregulation. When complete, for each cube we now have the numbers
upregulations which approximates maximal upregulation. When complete, for each cube we now have the numbers ,
,  through
through .
.
・ now in any infection simulation, we model uninfected cell collateral damage in the following way. At each time step in an infection simulation, we know FIC0 and FIC1 cells are killed and their recognition by T Cells generates IFN- mediated upregulation signals to the cube they belong to. It takes several time steps, but eventually, enough upregulation signals occur within a given cube so that
mediated upregulation signals to the cube they belong to. It takes several time steps, but eventually, enough upregulation signals occur within a given cube so that  cells are being killed each time step. This gives us a way to count collateral damage somewhat quantitatively. Note we are assuming, consistent with the acute phase of infection, the upregulation signal is a one way action: there is no mechanism by which this can be down regulated in this model.
cells are being killed each time step. This gives us a way to count collateral damage somewhat quantitatively. Note we are assuming, consistent with the acute phase of infection, the upregulation signal is a one way action: there is no mechanism by which this can be down regulated in this model.
The Coarse Update Model
We will assume that our simulation model maintains its level of uninfected cells indefinitely in the absence of infection using a standard logistics growth model. Recall  denotes the uninfected cell level in our model.
denotes the uninfected cell level in our model.
Hence, we assume the uninfected cells follow a logistics growth law:  where the
where the
initial condition  is less than the resource allocation
is less than the resource allocation . For us, we want to maintain the level
. For us, we want to maintain the level
 . This means the uninfected cells grow at the rate
. This means the uninfected cells grow at the rate ,
,  , with decay
, with decay
proportional to the square of their population . If the growth rate of the cell population is on
. If the growth rate of the cell population is on
the order of 1%, we would expect . Hence, a good choice for
. Hence, a good choice for  is
is .
.
We set these values in the simulation as  and
and . Note, in discrete form the logistics growth law
. Note, in discrete form the logistics growth law
gives . Even if the infection has reduced the uninfected cell population to a
. Even if the infection has reduced the uninfected cell population to a
fraction of the initial level, say , the update we see at the next time is
, the update we see at the next time is

Hence, about  cells are created at this time step even though the population is very far below the steady state level of
cells are created at this time step even though the population is very far below the steady state level of . The typical biological response to such a depressed cell level would be relatively small and we want the simulation parameters to be chosen to preserve that level of response.
. The typical biological response to such a depressed cell level would be relatively small and we want the simulation parameters to be chosen to preserve that level of response.
We estimate collateral damage as follows:
・  denotes the number of new infections. In the simulation, we keep track of the number of new FIC0 removals (
denotes the number of new infections. In the simulation, we keep track of the number of new FIC0 removals ( ) and the number of new FIC1 removals (
) and the number of new FIC1 removals ( ) in the
) in the  cube. We then compute the fraction
cube. We then compute the fraction
 where
where  is the current total number of infected cells. The value of
is the current total number of infected cells. The value of  is set to
is set to  if
if
this calculation exceeds . This fraction allows us to estimate what the fraction of cells in a cube are likely to be infected at this time step.
. This fraction allows us to estimate what the fraction of cells in a cube are likely to be infected at this time step.
・ at each time step, we keep track of the number of upregulation signals sent to each coarse cube and store this value in the variable  where
where  is the coarse cube number. Letting
is the coarse cube number. Letting  denote the number of cells in a coarse cube and we see the fraction
denote the number of cells in a coarse cube and we see the fraction  estimates how much upregulation (and hence collateral damage) is being done in a given coarse cube.
estimates how much upregulation (and hence collateral damage) is being done in a given coarse cube.
・ we have stored the average  values for each cube for each possible upregulation value. Denote these
values for each cube for each possible upregulation value. Denote these
values by  where
where  is the upregulation level and
is the upregulation level and  is the index of the coarse cube. We randomly
is the index of the coarse cube. We randomly
perturb the  values around their base values to allow for variation. If we denote this perturbation by
values around their base values to allow for variation. If we denote this perturbation by 
we have the updated value .
.
・ we now have the number

This estimates the collateral damage in the coarse cube .
.
・ we allow for the possibility that there can be a multiplier effect. The number  is the collateral damage
is the collateral damage
estimate for each cell in the cube. Each cell in a cube which is infected sends out upregulation signals which effect additional uninfected cells. This multiplier is the parameter . For example, if
. For example, if , we allow for an eight fold multiplier effect. Hence, the full collateral damage estimate for cube
, we allow for an eight fold multiplier effect. Hence, the full collateral damage estimate for cube  is
is

・ we then sum over all cubes to get the full collateral damage estimate, .
.
5. The Dynamics of the Model
To analyze the dynamics of the model we are building, we will need some auxiliary variables and constants which are listed in Table 4(a) and Table 4(b). In Table 4(b), the variable name IFN_gamma_neighborhood is replaced by IFN_gam_nbhd and ActiveTcellPercentage becomes ActiveTcellPer to make the table less wide.
There are also a number of nonlinear functions which compute various things about the variables and their interactions. These functions are listed below with their mathematical function names as well as the names they have been given in the code we use for the simulation.
 SetFIC0 (
SetFIC0 ( ): The variable
): The variable  denotes the new FIC0 cells. Let the number of these new cells be
denotes the new FIC0 cells. Let the number of these new cells be . This function picks random peptides to populate the MHC complexes of
. This function picks random peptides to populate the MHC complexes of  new FIC0 cells created at this current time step.
new FIC0 cells created at this current time step.
 SetFIC1 (
SetFIC1 ( ): The variable
): The variable  denotes the new FIC1 cells. Let the number of these new cells be
denotes the new FIC1 cells. Let the number of these new cells be . This function picks random peptides to populate the MHC complexes of
. This function picks random peptides to populate the MHC complexes of  new FIC1 cells created at this current time step.
new FIC1 cells created at this current time step.
 ChooseRandomSubset (M,N): This chooses
ChooseRandomSubset (M,N): This chooses  unique elements from a list of
unique elements from a list of  things (i.e. there are no repeats).
things (i.e. there are no repeats).
 CalculateFIC0Avidities (
CalculateFIC0Avidities ( ): This computes the affinities for FIC0 cells given an FIC0 population
): This computes the affinities for FIC0 cells given an FIC0 population .
.
 CalculateFIC1Avidities (
CalculateFIC1Avidities ( ): This computes the affinities for FIC1 cells given an FIC1 population
): This computes the affinities for FIC1 cells given an FIC1 population .
.
 UpdateL
UpdateL : We update the lifetimes of the cells in the FIC0 list
: We update the lifetimes of the cells in the FIC0 list .
.
 UpdateL
UpdateL : We update the lifetimes of the cells in the FIC1 list
: We update the lifetimes of the cells in the FIC1 list .
.
 RemoveFIC0Cells (z): Here, we determine which FIC0 cells in the list
RemoveFIC0Cells (z): Here, we determine which FIC0 cells in the list  have gone past their lifetime and so lyse as well as how many are recognized.
have gone past their lifetime and so lyse as well as how many are recognized.
 RemoveFIC1Cells (w): Here, we determine which FIC1 cells in the list
RemoveFIC1Cells (w): Here, we determine which FIC1 cells in the list  have gone past their lifetime and so lyse as well as how many are recognized.
have gone past their lifetime and so lyse as well as how many are recognized.
 UpregulateIFN
UpregulateIFN : This function IFN-
: This function IFN- upregulates all infected cells in a certain neighborhood of a recognized cell given the current list of cells,
upregulates all infected cells in a certain neighborhood of a recognized cell given the current list of cells,  , to upregulate. It returns an estimate of the number of healthy cells so upregulated.
, to upregulate. It returns an estimate of the number of healthy cells so upregulated.
 UpregulateIFN
UpregulateIFN : This function IFN-
: This function IFN- upregulates all infected cells in a certain neighborhood of a recognized cell given the current list of cells,
upregulates all infected cells in a certain neighborhood of a recognized cell given the current list of cells,  , to upregulate. It returns an estimate of the number of healthy cells so upregulated.
, to upregulate. It returns an estimate of the number of healthy cells so upregulated.
The Time Evolution of the Model
We now discuss the simulation dynamics. In what follows, we will use subscripts  to denote the values of variables at the next time step and
to denote the values of variables at the next time step and  to indicate the value at the current time step. We show the update equations in the table below which are calculated in the order shown. The simulation first must initialize all the T cells we use.
to indicate the value at the current time step. We show the update equations in the table below which are calculated in the order shown. The simulation first must initialize all the T cells we use.
(a) (b)
Table 4. Auxiliary and additional variables. (a) Auxiliary, (b) Additional variables.
・ Free virus,  , is removed by the immune system with a certain probability,
, is removed by the immune system with a certain probability,  and virus is added to the pool by the lysis of FIC0 and FIC1 cells. We assume that FIC1 cell lysis contributes
and virus is added to the pool by the lysis of FIC0 and FIC1 cells. We assume that FIC1 cell lysis contributes  times as much as the lysis of FIC0 cells. From experimental data,
times as much as the lysis of FIC0 cells. From experimental data,  , so if
, so if  free antigen was contributed by the FIC0 lysis, then
free antigen was contributed by the FIC0 lysis, then  would be added by the lysis of FIC1. Thus, the amount of free virus is modeled by new free virus
would be added by the lysis of FIC1. Thus, the amount of free virus is modeled by new free virus  old free virus
old free virus  (virus released due to FIC0 cell lysis)
(virus released due to FIC0 cell lysis)  (virus released due to FIC1 cell lysis)
(virus released due to FIC1 cell lysis)  (virus removed by immune system prior to infection). This is modeled mathema- tically by
(virus removed by immune system prior to infection). This is modeled mathema- tically by  which is Equation (1).
which is Equation (1).
 (1)
(1)
・ Uninfected Cell infections,  , are handled next. We set new uninfected cell HC infections
, are handled next. We set new uninfected cell HC infections  (Pro- bability of uninfected cell infection)
(Pro- bability of uninfected cell infection)  old amount of virus present, or
old amount of virus present, or , Equation (2).
, Equation (2).
・ Infected cells which are not dividing, are the FIC0 cells, . We model their dynamics as new FIC0 cells at the next time step
. We model their dynamics as new FIC0 cells at the next time step  a percentage of new infections,
a percentage of new infections,  , Equation (3).
, Equation (3).
 (2)
(2)
 (3)
(3)
・ Infected cells which are dividing are the FIC1 type, . Their dynamics are modeled as new FIC1 cells at next time step
. Their dynamics are modeled as new FIC1 cells at next time step  percentage of new infections or in mathematical terms
percentage of new infections or in mathematical terms  which is Equation (4).
which is Equation (4).
・ The number of FIC0 cells,  , is given by total FIC0 cells at next time step
, is given by total FIC0 cells at next time step  old FIC0 cells
old FIC0 cells  new
new
FIC0 cells,  , which is Equation (5).
, which is Equation (5).
 (4)
(4)
 (5)
(5)
・ The number of FIC1 cells,  , at the next time is then total FIC1 cells at next time step
, at the next time is then total FIC1 cells at next time step  old FIC1
old FIC1
cells  new FIC1 cells or more quantitatively,
new FIC1 cells or more quantitatively,  , Equation (6).
, Equation (6).
 (6)
(6)
For newly created cells of type FIC0 and FIC1, we assign the MHC-I-peptide ligands that are expressed on their surface by a call to the functions SetFIC0,  , and SetFIC1,
, and SetFIC1, . In the code, this is done via the function calls SetFIC0(y) and SetFIC1(y). The functions
. In the code, this is done via the function calls SetFIC0(y) and SetFIC1(y). The functions  and
and  are used to determine randomly the proteins expressed on the surface of an infected cell as discussed in Section 2.1. As long as there are new FIC1 cells,
are used to determine randomly the proteins expressed on the surface of an infected cell as discussed in Section 2.1. As long as there are new FIC1 cells,  , we build the peptides bound to the MHC-I molecules on the surface. These are infected
, we build the peptides bound to the MHC-I molecules on the surface. These are infected  cells and so they are less likely to be recognized and lysed by T cells, since the MHC-I is upregulated to a lesser extent on these cells. Hence, the call to
cells and so they are less likely to be recognized and lysed by T cells, since the MHC-I is upregulated to a lesser extent on these cells. Hence, the call to  builds
builds  data vectors
data vectors  of the appropriate size for FIC0
of the appropriate size for FIC0
cells. We express this mathematically as  (Equation (7)).
(Equation (7)).
 (7)
(7)
The call to  builds
builds  vectors
vectors  of size appropriate for FIC1 cells,
of size appropriate for FIC1 cells,  , which is listed as Equation (8). The new vectors above are then added to the current list of FIC0 and FIC1 cells as data. Then, we choose a random subset of the WNV-specific T cells to recognize and lyse the infected cells. Recall at each time step we assume there is a heterogeneous population of T cells which can recognize a fraction of the
, which is listed as Equation (8). The new vectors above are then added to the current list of FIC0 and FIC1 cells as data. Then, we choose a random subset of the WNV-specific T cells to recognize and lyse the infected cells. Recall at each time step we assume there is a heterogeneous population of T cells which can recognize a fraction of the  possible peptide ligands. We designate these as active. In our simulation, we let 45% be active at each time step. We store the peptides that can be recognized in the ActiveTcellsIndex and use these peptides to
possible peptide ligands. We designate these as active. In our simulation, we let 45% be active at each time step. We store the peptides that can be recognized in the ActiveTcellsIndex and use these peptides to
compute affinities. The function  chooses
chooses  indices out of the possible list
indices out of the possible list  to use as
to use as
the peptides for which the T cells generated at this time step have an affinity, . This can be expressed
. This can be expressed
quantitatively as  which is Equation (9).
which is Equation (9).
 (8)
(8)
 (9)
(9)
We then calculate affinities for the FIC0 and FIC1 lists. In the simulations, these lists can easily reach  in size, so traversal of these data structures has a serious impact on computational time. So we need to determine the avidity of each new active FIC0 cell clone for each new FIC0 cell. We do this with the Calculate FIC0 Avidities function, labeled
in size, so traversal of these data structures has a serious impact on computational time. So we need to determine the avidity of each new active FIC0 cell clone for each new FIC0 cell. We do this with the Calculate FIC0 Avidities function, labeled . Previous avidities are already stored in our FIC0 data structures. We add the new avidities to the list with the call Calculate FIC0 Avidities (y). Any of the newly infected cells whose avidity exceeds the threshold value that triggers T cell destruction is counted in the variable
. Previous avidities are already stored in our FIC0 data structures. We add the new avidities to the list with the call Calculate FIC0 Avidities (y). Any of the newly infected cells whose avidity exceeds the threshold value that triggers T cell destruction is counted in the variable
new_FIC0_kills. This is stated mathematically as  which is Equation (10). We then
which is Equation (10). We then
do the same sort of thing for new FIC1 cells where the Calculate FIC1 Avidities function is labeled . In the call to Calculate FIC1 Avidities, we also count the number of newly infected cells that are killed and store this value in the variable new_FIC1_kills. The resulting equation has the mathematical form
. In the call to Calculate FIC1 Avidities, we also count the number of newly infected cells that are killed and store this value in the variable new_FIC1_kills. The resulting equation has the mathematical form
 Equation (11). In these equations the argument
Equation (11). In these equations the argument  is the value of the new
is the value of the new
FIC0 cells and  is new value of the FIC1 cells.
is new value of the FIC1 cells.
 (10)
(10)
 (11)
(11)
The avidity calculations are implemented as previously discussed in Section 3.1. Infected cells whose avidity values enable recognition will then generate IFN- which will mediate MHC-I upregulation. We first remove the FIC0 cells and then the FIC1 cells. To remove the FIC0 cells, we
which will mediate MHC-I upregulation. We first remove the FIC0 cells and then the FIC1 cells. To remove the FIC0 cells, we
・ get the current size of the FIC0 list of cells.
・ update the lifetime values of each cell in this list.
・ we remove FIC0 cells whose lifetime has been exceeded.
・ we remove FIC0 cells whose avidity values trigger T Cell destruction.
・ Since  is the number of FIC0 cells, each removal and kill, decreases
is the number of FIC0 cells, each removal and kill, decreases .
.
・ cells dying in the absence of T cell recognition release their viral contents and increase the value of . The amount of free antigen so generated is determined by a random perturbation around the base value
. The amount of free antigen so generated is determined by a random perturbation around the base value  multiplied by the number of dying cells. The resulting amount of free antigen is stored in
multiplied by the number of dying cells. The resulting amount of free antigen is stored in .
.
・ On the other hand, the antigen in the cells destroyed by T Cells is lost and does not increase the  value. However, each destroyed cell has an associated coarse cube address which we keep track of in a list. Some of the cells in this cube will undergo IFN-
value. However, each destroyed cell has an associated coarse cube address which we keep track of in a list. Some of the cells in this cube will undergo IFN- mediated upregulation. We keep track of the number of T Cell mediated lytic events in this time step in
mediated upregulation. We keep track of the number of T Cell mediated lytic events in this time step in .
.
Essentially, we repeat these steps when we remove FIC1 cells. Thus, the FIC0 and FIC1 removal algorithm first sets the elapsed time on all infected cells and an infected cell will die automatically if its life time is
exceeded. This is just a matter of incrementing the life value  in each cell in
in each cell in  and
and . This
. This
amounts to a traversal through each list. The simple functions that do this are called  which
which
returns , the updated list of FIC0 lifetime values. This is written mathematically as
, the updated list of FIC0 lifetime values. This is written mathematically as
 and is shown as Equation (12). The similar equation,
and is shown as Equation (12). The similar equation,  for
for
the updated list of FIC1 lifetime values is listed as Equation (13) where  is the function which
is the function which
does the lifetime updating for the FIC1 list.
 (12)
(12)
 (13)
(13)
As part of this list traversal, recognized infected cells are lysed by a call to the functions  (this computes
(this computes , resets
, resets  and sets the
and sets the  portion due to FIC0 cells) and
portion due to FIC0 cells) and  (this computes
(this computes , resets
, resets  and adds the
and adds the  due to FIC1 cells). We let
due to FIC1 cells). We let  denote the number of FIC0 cells which have exceeded their lifetimes and hence, undergo lysis without T Cell recognition and add released antigen to
denote the number of FIC0 cells which have exceeded their lifetimes and hence, undergo lysis without T Cell recognition and add released antigen to . We let
. We let  be the number of FIC0 cells which are recognized by the immune system and killed (Equation D14).
be the number of FIC0 cells which are recognized by the immune system and killed (Equation D14).
 (14)
(14)
Similarly, the number of FIC1 cells exceeding their lifetimes or recognized by the immune system and killed are denoted by  and
and , respectively (Equation D16).
, respectively (Equation D16).
 (15)
(15)
We then need to upregulate IFN- in a selection of addresses around each FIC0 cell lysed by a T cell. The address
in a selection of addresses around each FIC0 cell lysed by a T cell. The address  in the data
in the data  for each FIC0 cell that has been lysed by a T cell belongs to a corresponding coarse cell in the immune model,
for each FIC0 cell that has been lysed by a T cell belongs to a corresponding coarse cell in the immune model, . We handle the IFN-
. We handle the IFN- in the method Set Neighborhoods as follows:
in the method Set Neighborhoods as follows:
・ the cube addresses associated with destroyed FIC0 and FIC1 cells are stored in a vector which we convert into a list.
・ the list of cube addresses which contain a destroyed infected cell is sorted and duplicates are then removed. This gives us the size of the removed list without duplicates, .
.
・ the FIC0 and FIC1 lists of infected cells are examined and a mapping is set up between the address of the infected cells data and the cube address the infected cell reside in.
・ the number of FIC0 cells that are upregulated at this time step is stored in ; similarly, the number of newly upregulated FIC1 cells is stored in
; similarly, the number of newly upregulated FIC1 cells is stored in .
.
・ FIC0 infected cells are upregulated. The infected cells that live in upregulated cubes must have their avidity values and other parameters reset. As we loop through all the FIC0 cells, if they are in an upregulated cube, we update the avidity value by multiplying the current avidity value by the upregulation factor. If this exceeds the maximum allowable amount, the avidity factor is set to the maximum.
・ FIC1 infected cells are upregulated in the same fashion.
・ the amount of collateral damage is then estimated as . Recall
. Recall  is the number of total new infections. The total number of new T cell triggered deaths is the sum
is the number of total new infections. The total number of new T cell triggered deaths is the sum  new_FIC0_kills
new_FIC0_kills  new_FIC1_kills. We can then find the ratio
new_FIC1_kills. We can then find the ratio  for this coarse cube. If this ratio is larger than
for this coarse cube. If this ratio is larger than , we reset it to
, we reset it to . Here, the MHC-I upregulation level stored as
. Here, the MHC-I upregulation level stored as . We have computed the
. We have computed the ’s for each coarse cube for different choices of
’s for each coarse cube for different choices of . For our damage estimate, choose the
. For our damage estimate, choose the
values  corresponding to the value of
corresponding to the value of  we have. In addition, we randomly perturb the
we have. In addition, we randomly perturb the 
using the parameter  as follows:
as follows:  where
where  is a random number in
is a random number in
 . Finally, the variable, Update Reg [k], stores the number of new IFN-
. Finally, the variable, Update Reg [k], stores the number of new IFN- mediated upregulations for this time interval for the
mediated upregulations for this time interval for the  coarse cube. If
coarse cube. If  is the size of a coarse cube (here
is the size of a coarse cube (here ), then the ratio
), then the ratio
 gives the fraction of cells in each coarse cube which are newly upregulated. For
gives the fraction of cells in each coarse cube which are newly upregulated. For
each coarse cube,  , we can then calculate
, we can then calculate  where,
where,  is the number of
is the number of
cells we estimate are upregulated per IFN- signal (here
signal (here ). This gives an estimate of the number of
). This gives an estimate of the number of
cells which are lost to collateral damage. Then  gives the total collateral damage in this time step.
gives the total collateral damage in this time step.
For typical simulation values, for  (i.e.
(i.e.  % of the newly infected cells are killed) and
% of the newly infected cells are killed) and  (i.e.
(i.e.  % of the cells in a coarse cube are upregulated, we have approximately
% of the cells in a coarse cube are upregulated, we have approximately  or
or  so that
so that . However, this is a dynamic computation which depends on what is happening currently. The collateral damage at this time step is thus stored in
. However, this is a dynamic computation which depends on what is happening currently. The collateral damage at this time step is thus stored in .
.
In summary, we scale the IFN- factor for infected cells in upregulated coarse cubes. We can think of these cube addresses as stored in the variables
factor for infected cells in upregulated coarse cubes. We can think of these cube addresses as stored in the variables  and
and . We upregulate by looping over all the addresses in
. We upregulate by looping over all the addresses in  and
and  as described above. The removal of FIC0 cells is done in the function
as described above. The removal of FIC0 cells is done in the function  which returns the number of ruptured FIC0 cells in
which returns the number of ruptured FIC0 cells in , the number of T Cell mediated lytic events in
, the number of T Cell mediated lytic events in  and upregulation addresses in
and upregulation addresses in . This is written mathematically as
. This is written mathematically as
 which is Equation (14). Similar calculations are carried out for
which is Equation (14). Similar calculations are carried out for
the FIC1 case using in the function  which returns the number of ruptured FIC1 cells in
which returns the number of ruptured FIC1 cells in
 , the number of T cell mediated lytic events in
, the number of T cell mediated lytic events in  and upregulation addresses in
and upregulation addresses in ,
,
Equation (15). The upregulation function  then calculates
then calculates  which is written mathematically
which is written mathematically
as , (Equation (16). In a similar fashion, the upregulation function
, (Equation (16). In a similar fashion, the upregulation function 
calculates ,
,  , Equation (17). We also remove cells lysed both by T cells and virus
, Equation (17). We also remove cells lysed both by T cells and virus
infections alone from our master FIC0 list,  , to generate a new
, to generate a new .
.
 (16)
(16)
 (17)
(17)
Cells that are removed due to virus infection only, simply release virus into the extracellular environment of the host. So for these removed cells, virus released from these cells must be added to the total virus present. This amount of new virus added due to the death of FIC0 cells is the variable . We determine how much virus is released at this time step by multiplying by the number of removed cells by a random perturbation of the base amount
. We determine how much virus is released at this time step by multiplying by the number of removed cells by a random perturbation of the base amount . Where infected target cells are lysed by T cell clones before the virus matures, infectious progeny virus is not released. At the start of this time step, the size of the FIC0 list is
. Where infected target cells are lysed by T cell clones before the virus matures, infectious progeny virus is not released. At the start of this time step, the size of the FIC0 list is . So, since
. So, since  is the total number of FIC0 cells, we subtract both the number of cells that die naturally due to virus infection and the cells lysed by T cells to get the current value. The equation needed to do this is
is the total number of FIC0 cells, we subtract both the number of cells that die naturally due to virus infection and the cells lysed by T cells to get the current value. The equation needed to do this is
 which is Equation (19). A similar set of computations is used to
which is Equation (19). A similar set of computations is used to
remove virus and T cell-lysed FIC1 cells. However, there is a difference here. The virus grows in FIC1 cells faster by a factor of . This is done in the
. This is done in the  update equation we discussed earlier using the value computed with the
update equation we discussed earlier using the value computed with the  equation from the previous time step expressed mathematically
equation from the previous time step expressed mathematically
 , which is Equation (20). We then update the FIC1 population using
, which is Equation (20). We then update the FIC1 population using
 Equation (21). Then since
Equation (21). Then since  is an estimate of the number of
is an estimate of the number of
infected cells recognized at this time step, we calculate its updated value using the equation
 as shown in Equation (22).
as shown in Equation (22).
 (18)
(18)
 (19)
(19)
 (20)
(20)
 (21)
(21)
 (22)
(22)
When infected cells are recognized, T cells undergo clonal expansion which improves the T cell killing percentage. Hence, it seems reasonable to allow the parameter  to be reset each time step. To model clonal expansion, we keep track of the fraction of new T cells using the ratio
to be reset each time step. To model clonal expansion, we keep track of the fraction of new T cells using the ratio  defined by
defined by
 (23)
(23)
The parameter  is then reset where we also set up a ceiling threshold which
is then reset where we also set up a ceiling threshold which  can not go past,
can not go past,
 . We update with
. We update with . If the T Cells which have spread avidity have
. If the T Cells which have spread avidity have
been cloned, then there is a concomitant B cell clonal expansion which increases the probability that free virus will be neutralized. This probability is stored in the variable . To implement this idea, we will update
. To implement this idea, we will update  as
as
follows: calculate  and then do the reset
and then do the reset  where we make
where we make
sure we do not exceed a specified maximum. Finally, we look at the Uninfected Cell population dynamics: uninfected cells at next time step  old uninfected cells
old uninfected cells  uninfected cells homeostasis update
uninfected cells homeostasis update  new infected cells
new infected cells  cells lost due to T cell lysis of uninfected cells, i.e. cells lost due to collateral damage. We
cells lost due to T cell lysis of uninfected cells, i.e. cells lost due to collateral damage. We
assume the uninfected cells follow a logistics growth model in the absence of infection,  ,
,
where the values of the growth factor  and the resource limitation
and the resource limitation  are chosen appropriately. We thus generate Equation (25) which is our estimate of healthy cells in the simulation model which uses our collateral damage estimate from Equation (24).
are chosen appropriately. We thus generate Equation (25) which is our estimate of healthy cells in the simulation model which uses our collateral damage estimate from Equation (24).
 (24)
(24)
 (25)
(25)
6. Damage Due to the Decoy Hypothesis
We are now in a position to explain the collateral damage due to the decoy model. In addition, it is known that there have been puzzling results reported in survival experiments for WNV infections. Recall, the amount of virus involved in an infection is measured in plaque forming units (pfu). This determines the concentration of infectious virus by virtue of the number of areas of cell death in a cell monolayer in vitro. Our simulations use a parameter in our simulations for the pfu level and, of course, it is difficult to calibrate artificial pfu levels to measured data levels as the pfu measure itself is a large scale phenomenon and so it is complicated to model. A typical survival simulation generates the data shown in Table 5 for an upregulation level of . We model host infections in
. We model host infections in  groups using initial viral concentrations ranging from
groups using initial viral concentrations ranging from  to
to  pfu. Note there is
pfu. Note there is
Table 5. Sample simulation data.

Figure 2. The percentage of uninfected cells vs. viral infection level.
upward movement in the healthy cell ratio even as the pfu level increases which is an indication that the collateral damage model we have discussed here due to the decoy model is a useful explanatory tool. We can also plot the average uninfected cell percentage vs the logarithm of virus concentration (pfu) in Figure 2. The spikes in the numbers of surviving hosts with increasing inoculating virus concentration indicate that our simulation validates what we would expect to happen if the decoy model holds true.
7. Conclusion
We show that the decoy hypothesis is a reasonable way to explain collateral damage. Our simulations also suggest that the decoy hypothesis might provide an explanation for survival curve data measured in WNV infections which show an increase in survivability even with increasing viral dose. We will explore that further in other work.
Cite this paper
James K.Peterson,Alison M.Kesson,Nicholas J. C.King, (2015) A Simulation for Flavivirus Infection Decoy Responses. Advances in Microbiology,05,123-142. doi: 10.4236/aim.2015.52013
References
- 1. Abbas, A.K., Lichtman, A.H. and Pillai, S. (2010) Cellular and Molecular Immunology. Saunders Elsevier, Philadelphia.
- 2. King, N.J.C. and Kesson, A.M. (1988) Interferon-Independent Increases in Class I Major Histocompatibility Complex Antigen Expression Follow Flavivirus Infection. Journal of General Virology, 69, 2535-2543.
http://dx.doi.org/10.1099/0022-1317-69-10-2535 - 3. Douglas, D.W., Kesson, A.M. and King, N.J.C. (1994) CTL Recognition of West Nile Virus-Infected Fibroblasts Is Cell Cycle Dependent and Is Associated with Virus-Induced Increases in Class I MHC Antigen Expression. Immunology, 82, 561-570.
- 4. Kesson, A.M., Cheng, Y. and King, N.J.C. (2002) Regulation of Immune Recognition Molecules by Flavivirus, West Nile. Viral Immunology, 15, 273-283.
http://dx.doi.org/10.1089/08828240260066224 - 5. Müllbacher, A., Hill, A.B., Blanden, R.V., Cowden, W.B., King, N.J.C. and Hla, R.T. (1991) Alloreactive Cytotoxic T Cells Recognize MHC Class I Antigen without Peptide Specificity. Journal of Immunology, 147, 1765-1772.
- 6. King, N.J.C., Mullbacher, A., Tian, L., Rodger, J.C., Lidbury, B. and Hla, R.T. (1993) West Nile Virus Infection Induces Susceptibility of in Vitro Outgrown Murine Blastocysts to Specific Lysis by Paternally Directed Allo-Immune and Virus-Immune Cytotoxic T Cells. Journal of Reproductive Immunology, 23, 131-144.
http://dx.doi.org/10.1016/0165-0378(93)90003-Z - 7. King, N.J.K., Davison, A., Getts, D., Ping Lu, D., Teague Getts, M., Yeung, A., Peterson, J. and Kesson, A.M. (2008) Enhanced Antigen Processing or Immune Evasion? West Nile Virus and the Induction of Immune Recognition Molecules. In: Diamond, M.S., Ed., West Nile Encephalitis Virus Infection: Viral Pathogenesis and the Host Immune Response, Emerging Infections in the 21st Century, Springer-Verlag, Heidelberg, 309-339.
- 8. Kuhlman, P., Moy, V.T., Lollo, B.A. and Brian, A.A. (1991) The Accessory Function of Murine Intercellular Adhesion Molecule-1 in T Lymphocyte Activation. Contributions of Adhesion and Co-Activation. Journal of Immunology, 146, 1773-1782.
- 9. Shen, J., T-To, S.S., Schrieber, L. and King, N.J.C. (1997) Early E-Selectin and VCAM-1 and ICAM-1 and Late Major Histocompatibility Complex Antigen Induction on Human Endothelial Cells by Flavivirus and Comodulation of Adhesion Molecule Expression by Immune Cytokines. Journal of Virology, 71, 9323-9332.
- 10. Getts, D.R., Matsumoto, I., Muller, M., Getts, M.T., Radford, J., Shrestha, B., Campbell, I.L. and King, N.J.C. (2007) Role of IFN-γ in an Experimental Murine Model of West Nile Virus-Induced Seizures. Journal of Neurochemistry, 103, 1019-1030.
http://dx.doi.org/10.1111/j.1471-4159.2007.04798.x - 11. Peterson, J.K., King, N.J.C. and Kesson, A.M. (2014) Modeling West Nile Virus One Host Infections. Technical Report, Clemson University, Clemson.
http://www.ces.clemson.edu/%7Epetersj/CurrentPapers/WNVSimulationCode-04082014.pdf - 12. Alon, U. (2006) An Introduction to Systems Biology: Design Principles of Biological Circuits. CRC Mathematical and Computational Biology, Chapman Hill.
- 13. King, N.J.C. and Kesson, A.M. (2003) Interaction of Flaviviruses with Cells of the Vertebrate Host and Decoy of the Immune Response. Immunology and Cell Biology, 81, 207-216.
http://dx.doi.org/10.1046/j.1440-1711.2003.01167.x - 14. Lobigs, M., Mullbacher, A. and Regner, M. (2003) MHC Class I Up-Regulation by Flaviviruses: Immune Interaction with Unknown Advantage to Host or Pathogen. Immunology and Cell Biology, 81, 217-223.
http://dx.doi.org/10.1046/j.1440-1711.2003.01161.x