American Journal of Analytical Chemistry
Vol.07 No.05(2016), Article ID:66557,8 pages
10.4236/ajac.2016.75042
Validation of a Capillary Electrophoresis Method for Analyzing Chlorphenamine Maleate-Based Drugs Using the Accuracy Profile Approach
Serigne Omar Sarr1,2*, Eddy Kevin Senan Ouendo1, Thierno Mouhamed Wane1, El Hadji Assane Diop3, Diop Amadou1, Serigne Momar Ndiaye2, Rokhaya Gueye1, Khadidiatou Thiam1, Aminata Sarr1, Ousmane Niass1, Djibril Fall2,4, Bara Ndiaye1, Serge Rudaz3, Yérim Mbagnick Diop1,2
1Laboratory of Analytical Chemistry and Food Sciences, Cheikh Anta Diop University, Dakar, Senegal
2National Medicines Control Laboratory, Ministry of Health, Dakar, Senegal
3Laboratory of Pharmaceutical Analytical Chemistry, School of Pharmacy, Geneva, Switzerland
4Laboratory of Organic and Therapeutic Chemistry, Cheikh Anta Diop University, Dakar, Senegal

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 6 April 2016; accepted 16 May 2016; published 19 May 2016
ABSTRACT
This study aimed at validating an analytical method, using the accuracy profile approach, for the assay of chlorphenamine maleate by capillary electrophoresis. The validation was done using concentrations ranging between 75% and 125% of the target concentration of 600 mg/ml. Validation standards were prepared separately in triplicate for each series. Studied validation criteria were selectivity, linearity, trueness, precision (repeatability and intermediate precision), accuracy and limits of detection and quantification. The method was selective, with recoveries ranging between 99.55% and 99.84%. The relative standard deviations of repeatability and intermediate precision were <5%. The accuracy profile confirmed the performance of the assay method between 75% and 100% of the target concentration of 600 mg/ml. The detection and quantification limits were 5 mg/l and 15 mg/l respectively. This ecological and economical method was applied to identify and quantify chlorphenamine maleate in 3 samples of chlorphenamine maleate-based drugs provided by the Senegalese National Medicines Control Laboratory. All analyzed samples were in accordance with official standards.
Keywords:
Analytical Validation, Chlorphenamine Maleate, Quality, Capillary Electrophoresis, CEB, Senegal

1. Introduction
Counterfeit medicines pose a public health hazard, waste consumer income and endanger stability of health systems particularly in developing countries [1] . It is estimated that this phenomenon reaches about 10% of the global medicines market. In developing countries, the proportion of counterfeit medicines could reach 50%. Indeed, the regular use of counterfeit or substandard medicines can lead to therapeutic failure, drug resistance or death. In some cases, it can cause death. According to WHO South-East Asia region, in 2001, 38% of 104 antimalarial drugs sold in pharmacies did not contain active ingredient [2] .
This deplorable situation can be explained by insufficiency or inadequacy of appropriate technical and human means suitable to monitor the quality of medicines in resource limited countries. Thus it is necessary to train quality control specialists but also to validate new alternative methods for quality control of drugs especially in less developed countries.
In this context, the association for quality control and detection of counterfeit drugs (PHARMELP) developed the CEB (Capillary Electrophoresis Budget) device. It is an apparatus for assessing the quality of medicines using the separative principle of capillary electrophoresis.
The objective of this study was to validate a simple, environmentally friendly and inexpensive method for the determination of chlorphenamine maleate (Figure 1). Then the proposed new method was applied to evaluate the quality of chlorphenamine maleate-based drugs.
The results obtained with this newly proposed method were compared with those given by reference spectrophotometric method described in the American pharmacopeia [3] .
2. Materials and Methods
2.1. Reagents
Sodium phosphate dibasic, potassium phosphate monobasic and HPLC grade methanol were purchased from Sharlau (Barcelona, Spain).
Chlorphenamine maleate standard and diclofenac were provided by the Senegalese National Medicines Control Laboratory (LNCM, 39 Avenue Pasteur, Dakar).
Ultrapure water was prepared using Milli-Q water system (Millipore, Molsheim, France) and used to prepare the buffer and to dissolve standards.
A total of 3 chlorphenamine maleate based-drug samples were taken from the Senegalese National Medicines Control Laboratory (LNCM, Dakar) and used to apply the proposed method.
2.2. Apparatus
In this work, were used:
・ A capillary Electrophoresis (EC Budget) coupled with a UV detector LED 255, type of capillary: TSU UV-Transparent FS Tubing, 50 μm ID, 375 μm OD.
・ A molecular absorption spectrophotometer in the ultraviolet and visible (Jasco model 570) was also used to confirm the assays.

Figure 1. Chemical structure of chlorphenamine maleate.
・ A pH Meter Mettler Toledo Ion Analyzer 365.
・ A Sartorius balance LA230S model.
2.3. Analytical Parameters
For electrophoretic analysis, a separation voltage of 20 kV was applied to the electrophoretic system. The injection of solutions (<1 μl) was done at 50 mbar during 10 s for an acquisition frequency of 9.5 Hz. A phosphate buffer (pH = 6.8) was used and UV detection at 255 nm was conducted for an analysis time of 5 min.
The spectrophotometric UV-VISIBLE analysis was conducted using the method described in the United States Pharmacopeia [3] .
2.4. Preparation of Solutions
Standard solutions of chlorphenamine maleate and diclofenac were prepared for recovery studies (accuracy). Three levels of chlorphenamine maleate concentrations were prepared at 450 mg/ml, 600 mg/ml, and 750 mg/ml. In each case, the concentration of diclofenac standard was 200 mg/l.
For sample preparation, 4 tablets each containing 4 mg of chlorphenamine maleate drugs were grounded in a mortar. Then quantity of powder equivalent to 12 mg of chlorphenamine maleate was weighed, and placed in a 10 ml flask and filled up to the mark with ultrapure water. After complete dissolution, this solution was sonicated for 10 minutes and filtered through a 0.45 μm Whatman cellulose filter grade 42® (GE Healthcare, France).
The chlorphenamine maleate content in samples was calculated using the following formula:
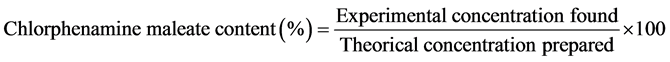
2.5. Fisher Test
The Fischer-test was used to compare the results of the assay by capillary electrophoresis with those obtained by spectrophotometric method [3] . The F-test is a statistical test comparing the precision of two analytical methods from the formula F = Sa2/Sb2; where Sa is the largest value of standard deviation and Sb the smallest standard deviation value for the same sample.
2.6. Validation Criteria
The protocol consisted of several criteria that were evaluated as follows [4] - [7] :
2.6.1. Selectivity
The chlorphenamine maleate standard and diclofenac internal standard solutions were injected individually, then together to appreciate the separation of the different peaks of substances.
2.6.2. Linearity
It is measured over 3 days from 3 chlorphenamine maleate standard solutions at the following concentrations: 450 mg/l; 600 mg/l and 750 mg/l of chlorphenamine maleate.
2.6.3. Trueness
It is expressed in recovery, by the ratio of the concentration found with the proposed method and actually prepared theoretical concentration. It is evaluated for each of the concentration levels used in the study of the linearity, which corresponds in value at 75%, 100%, 125% of the nominal concentration of 600 mg/l.
2.6.4. Precision
Precision is expressed as relative standard deviation (RSD) for the repeatability and intermediate precision. It is evaluated for each of the levels (75%, 100%, and 125% of the target value of the assay).
Repeatability is determined by injecting 3 times solutions containing chlorphenamine maleate and internal standard to the standard 3 different concentrations of chlorphenamine maleate for 3 consecutive days, this to study “intra-day” variations of the method.
Intermediate precision is evaluated by injecting solutions containing chlorphenamine maleate standard and internal standard for the 3 different concentrations of chlorphenamine maleate, 3 times during 3 consecutive days to study the “inter-day” variations of the method by concentration level.
2.6.5. Accuracy
Accuracy is the total error after the sum of systematic error (trueness) and random errors (precision). It is represented by an accuracy profile for the area of measures (75% - 125%). Accuracy of the method is established through the accuracy profile described by Feinberg [6] .
2.6.6. Limits of Detection (LOD) and Quantification (LOQ)
They were determined as specified in the ICH protocol [8] .
3. Results and Discussion
3.1. Validation Method
3.1.1. Selectivity
Chlorphenamine maleate has a pKa of 9.13. After testing citrate and phosphate buffers at different pH of 6.8, 5.5 and 2.5 with different internal standards (quinine sulfate, procaine, diclofenac) t better results were obtained with diclofenac as internal standard and phosphate buffer at pH = 6.8. In these conditions, the elution time was around 5 minutes. The solvent used was ultrapure water. The influence of the voltage and current was also studied and optimized. Results were a separation voltage of 20 kV, a pressure of 50 mbar, an acquisition frequency of 9.5 Hz and UV detection at 255 nm. These parameters are different to those described by other authors who used a 10 mM sodium dihydrogen-phosphate-sodium tetraborate buffer containing 50 mM sodium dodecyl sulfate (SDS) and 5% methanol (MeOH), pH 9.0 [9] .
Electrophoregrams obtained after analyzing chlorphenamine maleate standard, internal standard (diclofenac), mixture of chlorphenamine maleate sample and diclofenac in the described conditions are presented respectively in Figures 2-4. These figures show distinct peaks at specific migration times corresponding specifically to the injected products.
Figure 2 shows the chlorphenamine maleate standard’s electrophoregram with a migration time of 1.32 min and Figure 3 shows the diclofenac standard’s electrophoregram with a migration time of 4.13 min.
Figure 4 shows the mixture of chlorphenamine maleate and diclofenac standard’s electrophoregrams with migration time of 1.32 min for chlorphenamine maleate standard and 4.31 min for diclofenac standard.
Figures 2-4 show the selectivity of the developed method with specific migration times for chlorphenamine maleate and diclofenac standards. Also the good resolution of the peaks of both standards show that the method proposed is selective.
3.1.2. Linearity
The regression curve was linear within the concentration range of 450 - 750 mg/L (Figure 5). The equation of
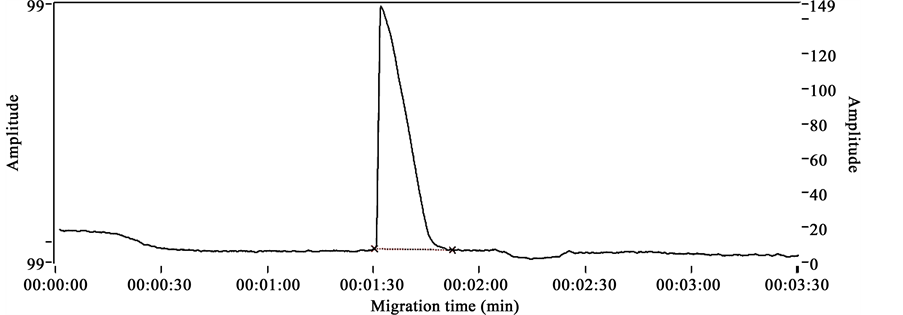
Figure 2. Chlorphenamine maleate standard’s electrophoregram.
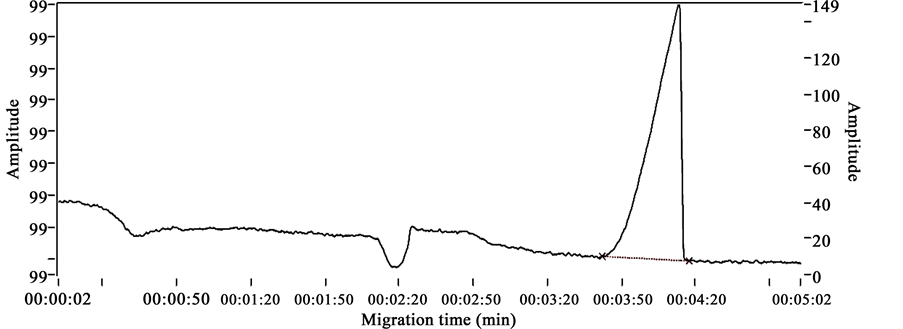
Figure 3. Diclofenac standard’s electrophoregram.
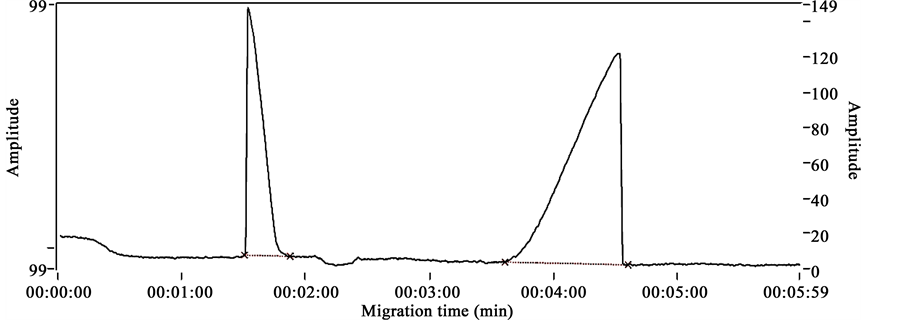
Figure 4. Mixture of chlorphenamine maleate and diclofenac standard’s electrophoregram.
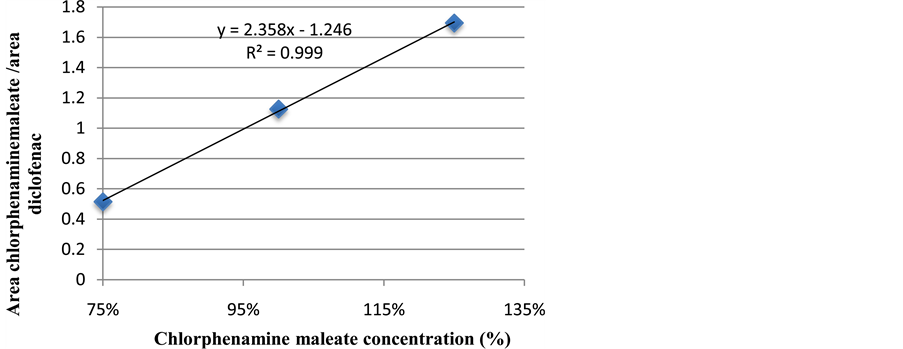
Figure 5. Chlorphenamine maleate regression line.
the regression was y = 2.3587x − 1.2468 with a correlation coefficient (R2) of 0.9996. Figure 5 shows a linear correlation between chlorphenamine maleate amount injected and the detector response.
These values are in accordance with ICH specifications [8] which consider a good linearity if R2 > 0.99.
3.1.3. Trueness, Intra-Day and Inter-Day Precision Studies
For trueness, recoveries were between 99.55% and 99.84%. For intra-day and inter-day precision the RSDs between 1.9% and 4.8% were lower than 5%. These values below 5% indicate a good precision confirmed by the accuracy profile (Figure 6) which clearly shows the good quantitative method performance throughout the validation field [10] . They confirm the absence of variation within and between days with this method.
3.1.4. Accuracy Profile
Considering the degrees of freedom equal to 6, an α risk of 5%, a Student t value of 3.7074, a tolerance on the accuracy was calculated. This was used to calculate higher and lower tolerance limits. Also, the upper and lower limits of trueness are 120% and 80% respectively. All these data summarized in Table 1, allowed to establish the accuracy profile (Figure 6).
3.1.5. Limit of Detection (LOD) and Limit of Quantification (LOQ)
The limits of detection (LOD) and quantification (LOQ) were respectively 5 mg/l and 15 mg/l and indicate the suitability of the proposed method for the quality control of drugs.
3.2. Chlorphenamine Maleate Content in Samples Analyzed
The validated method was applied to the control of the quality of 3 samples of chlorphenamine maleate-based drugs (Coded 170 to 172) provided by the Senegalese National Medicines Control Laboratory.
In order to confirm the results obtained with this newly developed method by CEB, the samples were also assayed by an official spectrophotometric UV-VISIBLE method described in the US Pharmacopoeia [3] . These confirmations were made at Senegalese National Medicines Control Laboratory (LNCM) and showed similar results for both methods with comparable precision (FISCHER F-test). Then, on the basis of the evidence, CEB appeared as accurate as spectrophotometric UV-VISIBLE for chlorphenamine maleate analysis. The assays and FISCHER F-test results are summarized in Table 2.

Table 1. Accuracy profile data.

Figure 6. Accuracy profile graphic.
Table 2. Chlorphenamine maleate assay results by CEB and UV-VISIBLE.
The application of this validated method for assessing the quality of drug samples containing chlorphenamine maleate showed that all samples contained the active ingredient declared with correct amount between 90% and 110% (Table 2). These results are comparable to those obtained in other studies [11] - [14] .
Comparing these results with those of a reference method gave similar results [3] . However this new proposed method combines the separating power of electrophoresis and the detection sensitivity in the ultraviolet. It therefore has an additional advantage of the molecular absorption spectrophotometry in the ultraviolet. The risk of possible excipients interference of impurities or degradation products that may be encountered in a direct determination by UV-VISIBLE spectrophotometry are better controlled or eliminated with capillary electrophoresis.
Similarly, the CEB has the advantage of greater sensitivity since the optical path corresponds to the internal diameter of the capillary that is 50 μm while the optical path in UV-VISIBLE spectrophotometry direct assay is generally 1 cm.
Sample volumes injected below the microliter show the economic nature of this method.
Also the CEB by its weight and its design is more portable and handy as a conventional UV-VISIBLE spectrophotometer. Indeed, the source of UV light traditionally used was replaced by a new type of light emitting diode (LED) having a wavelength of 255 nm. The advantage of LED is to provide not only better performance but also to be very small compared to traditional light sources.
4. Conclusion
The validated new method proposed is accurate, precise and selective for controlling the quality of medicines containing chlorphenamine maleate. It could be an alternative method and of major interest for pharmaceutical laboratories, medicines control laboratories, public and university structures because of its simplicity, ecological character, cheapness and short analysis time (approximately 5 min).
Acknowledgements
The authors thank the Swiss organization against counterfeit and substandard medicines (Pharmelp) for the low- cost CEB machine installed in Cheikh Anta DIOP University and the Senegalese National Medicine Control Laboratory’s staff for their technical assistance.
Cite this paper
Serigne Omar Sarr,Eddy Kevin Senan Ouendo,Thierno Mouhamed Wane,El Hadji Assane Diop,Diop Amadou,Serigne Momar Ndiaye,Rokhaya Gueye,Aminata Sarr,Ousmane Niass,Djibril Fall,Bara Ndiaye,Serge Rudaz,Yérim Mbagnick Diop,Khadidiatou Thiam, (2016) Validation of a Capillary Electrophoresis Method for Analyzing Chlorphenamine Maleate-Based Drugs Using the Accuracy Profile Approach. American Journal of Analytical Chemistry,07,452-459. doi: 10.4236/ajac.2016.75042
References
- 1. Bate, R., Tren, R., Hess, K., Mooney, L. and Porter, K. (2009) Pilot Study Comparing Technologies to Test for Substandard Drugs in Field Settings. African Journal of Pharmacy and Pharmacology, 3, 165-170.
- 2. WCPT (2011) Background Document on Counterfeit Medicines in Asia, Taipei. World Confederation for Physical Therapy, 1-26.
http://www.wcpt.org/node/44404 - 3. US Pharmacopeai (2013) USP 36-NF 31.
www.uspnf.com - 4. ICH. (2005) Validation of Analytical Procedures: Text and Methodology. International Conference on Harmonization (ICH), Q2(R1), Geneva, 17 p.
- 5. Amarouche, S. (2012) Characterization of a Measurement Method: Key Step in the Validation Process. Techniques de l’Ingénieur, 1040, 1-14.
- 6. Feinberg, M. (2010) Interpretation of the Accuracy Profile. Le Cahier des techniques de l’INRA, Special Volume, 45-60.
- 7. Hubert, P., Nguyen-Huu, J.J., Boulanger, B., Chapuzet, E., Cohen, N., Compagnon, P.A., Dewé, W., Feinberg, M., Laurentie, M., Mercier, N., Muzard, G. and Valat, L. (2003) Validation of Quantitative Analytical Procedures Harmonisation of Approaches. STP Pharma Pratiques, 13, 101-138.
- 8. ICH (1997) Validation of Analytical Procedures: Text and Methodology. International Conference of Harmonization, 62, 27463-27467.
- 9. Suntornsuk, L. (2001) Separation of Cold Medicine Ingredients by Capillary Electrophoresis. Electrophoresis, 22, 139-143.
http://dx.doi.org/10.1002/1522-2683(200101)22:1<139::AID-ELPS139>3.0.CO;2-D - 10. ORA LABS (2014) Validation and Verification Guidance for Human Drug. Analytical Method, Appendix 1, 17-19.
- 11. Diop, Y.M., Sarr, S.O., Diop, A., Ndiaye, B., Dadele, V.I.N., Fall, D. and Smine, A.K. (2008) Quality Control of Acetylsalicylic Acid and Paracetamol Used in Senegal. Therapie, 63, 403-404.
http://dx.doi.org/10.2515/therapie:2008055 - 12. Diop, A., Sarr, S.O., Diop, Y.M., Niang, A.A. and Ndiaye, B. (2008) Quality Control of Cotrimoxazole Used in Senegal. Therapie, 63, 405-408.
http://dx.doi.org/10.2515/therapie:2008056 - 13. Diop, Y.M., Sarr, S.O., Diop, A., Diedhiou, A., Dieye, A., Ndiaye, B., Gaye, O. and Smine, A. (2009) Quality Control of Antimalarias at Five Sentinel Sites for Malaria Epidemiological Surveillance. Annales des falsifications, de l’expertise chimique et toxicologique, 970, 55-62.
- 14. Yang, B., Mo, J. and Yang, X. (1999) Separation and Determination of the Ingredients of Cold Medicines by Capillary Electrophoresis with Amperometric Detection. Se Pu, 17, 477-479.
NOTES

*Corresponding author.