American Journal of Analytical Chemistry
Vol.4 No.4(2013), Article ID:30455,10 pages DOI:10.4236/ajac.2013.44025
Square-Wave Adsorptive Cathodic Stripping Voltammeteric Determination of Manganese (II) Using a Carbon Paste Electrode Modified with Montmorillonite Clay
1Department of Chemistry, Faculty of Science, Kafr El-Sheikh University, Kafr El-Sheikh, Egypt
2Department of Chemistry, Faculty of Science, King Abdul Aziz University, Jeddah, Saudi Arabia
3Department of Chemistry, Faculty of Science, Tanta University, Tanta, Egypt
Email: mmghoneim@usa.net
Copyright © 2013 Amr M. Beltagi et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received January 24, 2013; revised March 26, 2013; accepted April 20, 2013
Keywords: Manganese; Stripping Voltammetry; Carbon Paste Electrode; Montmorillonite Clay
ABSTRACT
Manganese is an essential micronutrient for all organisms; however at high concentrations it has a toxic effect. Manganese toxicity is a serious constraint to crop cultivation since it is taken-up by plants and can easily be passed into the food chain again causing symptoms of Parkinson’s disease. A fully validated square-wave adsorptive cathodic stripping voltammetry method has been developed for determination of Mn (II) as a complex with 2-(5’-bromo-2’-pyridylazo) 5-diethylaminophenol in aqueous solutions using a carbon paste electrode (CPE) modified with montmorillonite-Na clay. The results showed that the modified CPE (90% (w/w) graphite powder and 10% (w/w) montmorillonite-Na clay) exhibited excellent electrochemical activity towards the investigated Mn (II) complex in acetate buffer of pH = 5.0. Factors affecting the performance of the modified carbon paste electrode and the sensitivity of the described squarewave stripping voltammetry method, including the electrode composition, concentration of ligand, pulse parameters and preconcentration conditions were examined. A detection limit (S/N = 3) of 0.015 mg·L−1 (2.73 × 10−10 mol·L−1) Mn (II) was achieved when a preconcentration time of 240 s was applied. Insignificant interferences from various inorganic and organic species were estimated. The described square-wave adsorptive cathodic stripping voltammetry method coupled with the modified carbon paste electrode has been successfully applied to Mn (II) analysis in different water samples.
1. Introduction
Manganese is an essential micronutrient for all organisms [1] but at high concentrations it has toxic effect [2] contributing for example to the early development of Parkinson’s disease symptoms in susceptible people [3]. Manganese toxicity is also a serious constraint to crop cultivation since it is taken-up by plants and can easily be passed into the food chain again causing symptoms of Parkinson’s disease [4].
Various analytical techniques have been applied for the determination of trace manganese in biochemical and environmental samples, such as spectrophotometry [5-8], X-ray fluorescence [9], potentiometry flow injection [10], flame atomic absorption spectrometry [11-16], graphite furnace atomic absorption spectrometry [17-21] and inductively coupled plasma-optical emission spectrometry [22,23]. However, equipments of most of these techniques are relatively expensive and not accurately reliable for the determination of ultra trace concentrations of metal ions. Moreover, most of the applied analytical methods suffer from serious matrix interferences.
Stripping voltammetry has shown numerous advantages including speed of analysis, good selectivity, sensitivity and inexpensive for determination of various metal ions [24]. However, anodic stripping voltammetric determination of Mn (II) at the hanging mercury drop electrode [25,26] or mercury film electrode [27] suffers from the low solubility of manganese in mercury, the closeness of its reduction potential to that of hydrogen ion (–1.7 V vs. SCE) and the formation of inter-metallic compounds at the mercury electrode.
Cathodic stripping voltammetry technique was successfully used for the determination of manganese at the hanging mercury drop electrode [28-30], glassy carbon electrode [31-33] and carbon paste electrode [34,35]. However, the toxicity of mercury limits the usage of the mercury electrodes in the analytical practice and excludes them from the out-of-laboratory applications. Moreover, the sensitivity of glassy carbon and carbon paste electrodes is relatively poor in the determination of metal ions. In order to improve this, a fascinating and effective way is to modify it with a unique substance. Clay minerals have become attractive electrode modifier since the first example of the use of clay as modifier was reported [36]. Montmorillonite-Na clay has well-layered lattice structure, high chemical and mechanical stability, high cationic exchange capacity and strong adsorptive properties attributed to the expandability of its internal layers. Montmorillonite clay was used successfully as a modifier in carbon paste electrode for determination of each of Cu (II) [37], Au (III) [38,39], Hg (II) [40,41], Eu (III) [42] and Pb (II) [43]. It has been used also in this laboratory for determination of some pharmaceutical compounds [44,45] and for simultaneous determination of Cd (II), Pb (II), Cu (II) and Hg (II) [46].
This work aimed to describe sensitive, precise and fully validated square-wave adsorptive cathodic stripping voltammetry method for determination of Mn (II) complexed with 2-(5’-bromo-2’-pyridylazo) 5-diethylamino phenol in aqueous solutions using a carbon paste electrode (CPE) modified with montmorillonite-Na clay.
2. Experimental
2.1. Apparatus
A computerized Electrochemical Trace Analyzer Model 394-PAR (Princeton Applied Research, Oak Ridge, TN, USA) controlled via 270/250 PAR software was used for the voltammetric measurements. A micro-voltammetric cell consisting of a C-2 stand (BAS model MF-2063) with a carbon paste electrode body (BAS model MF-2010), an Ag/AgCl/3 M KCl reference electrode (BAS model MF- 2079) and a platinum wire counter electrode (BAS model MW-4130) was used. The body of the carbon paste electrode was a Teflon rod with end cavity of 3 mm diameter and 1 mm deep bored at one end for paste filling. Contact was made with a copper wire through the center of the Teflon rod. A magnetic stirrer (PAR-305) with a Tefloncoated magnet was used to provide the convective transport during the preconcentration step. The whole measurements were automated and controlled through the programming capacity of the apparatus.
A Shimadzu Flame Atomic Absorption Spectrometer (FAAS) Model AA-670 interfaced with a data processor was used for determination of the examined metal ion. A Mettler balance (Toledo-AB104, Greifensee, Switzerland) was used for weighing the solid materials. A pH-meter (Crison, Barcelona, Spain) was used for measuring the pH of solutions. A micopipetter (Eppendorf—Multipette® plus) was used for transferring the solutions throughout the present experimental work.
2.2. Reagents and Solutions
Britton-Robinson (B-R) universal buffer (pH 2.0 - 11.0), acetate buffer (pH 4.0 - 6.0), and phosphate buffer (pH 2.0 - 7.5) were prepared in de-ionized water and were tested as supporting electrolytes. A solution of 1 × 10−3 mol·L−1 2-(5’-bromo-2’-pyridylazo) 5-diethylaminophenol was prepared by dissolving an appropriate amount of the compound (Sigma) in spec-pure methanol. Desired standard solutions of K (I), Na (I), Mg (II), Ca (II), Al (III), Cu (II), Cd (II), Pb (II), Sb (III), Bi (III), Se (IV), Zn (II), Mn (II), Ni (II), Co (II) and Fe (III) were prepared by accurate dilution of their standard stock solutions (1000 mg·L−1 dissolved in aqueous 0.1 mol·L−1 HCl, supplied from Cica, Japan) by de-ionized water. Standard solutions of Cl−, 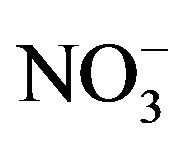 ,
, 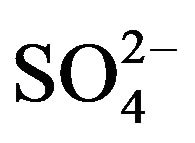 and
and 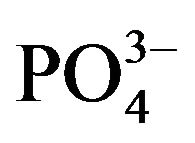 (each of 1000 mg·L−1) were prepared by dissolving appropriate amounts of KCl, KNO3, Na2SO4 and Na3PO4, respectively, in deionized water. Solution of 1.0% Triton X-100 was prepared in de-ionized water. All chemicals used were of analytical grade and were used without further purification.
(each of 1000 mg·L−1) were prepared by dissolving appropriate amounts of KCl, KNO3, Na2SO4 and Na3PO4, respectively, in deionized water. Solution of 1.0% Triton X-100 was prepared in de-ionized water. All chemicals used were of analytical grade and were used without further purification.
The de-ionized water used throughout the present work was obtained from a Purite-Still plus Deionizer connected to a Hamilton-Aqua Matic bidistillation water system (Hamilton Laboratory Glass LTD, Kent, UK).
2.3. Preparation of the Modified Carbon Paste Electrode
4.5 g of graphite powder (1 - 2 mm, Aldrich, Milwaukee, WI, USA) and 0.5 g of the montmorillonite-Na clay (Fine powder <5 mm, ECC America Inc., Southern Clay Products Subsidiary, Gonzales, Texas, USA) were mixed uniformly by milling in a small agate mortar, then 1.8 mL Nujol oil (Sigma, d = 0.84 g/mL) was added and milled again to give homogenous carbon paste modified with 5% - 20% (w/w) montmorillonite-Na clay (MMTNa). An amount of the prepared modified carbon paste was pressed into the end cavity (3 mm in diameter, 1 mm in depth) of the electrode body. Surface of the modified carbon paste working electrode was manually smoothed by polishing on clean paper before use.
2.4. General Analytical Procedure
A 10-mL volume of 0.1 mol·L−1 acetate buffer (pH = 5.0) was transferred to the micro-electrolysis cell and the smoothed modified CPE was then immersed in the supporting electrolyte and several sweeps were applied until obtaining a low background current. After that, aliquot of the Mn (II) standard solution was introduced into the electrolysis cell and a selected preconcentration potential was then applied to the modified CPE for a selected preconcentration time, while the solution was stirred at 400 rpm. At the end of the precocentration time, the stirring was stopped and a 5 s rest period was allowed for the solution to become quiescent. The voltammogram was then recorded by scanning the potential towards less positive direction using the square-wave potential waveform.
For regeneration of the electrode surface after recording each voltammogram, the modified CPE was transferred to a blank electrolyte solution in the voltammetric cell and series of cyclic scans were continued (between 1.2 to –0.2 V versus Ag/AgCl/3 M KCl) until a voltammogram corresponding to the residual current (lower background current) was obtained. The electrode was then ready for use in the next measurement.
2.5. Analyzed Environmental Water Samples
Various water samples were subjected to analysis by means of the described square-wave cathodic stripping voltammetry method utilizing the modified CPE. These include groundwater, bottled natural water (available in the Egyptian market) and coastal seawater samples (collected from the sea coast at Alexandria City, Egypt). The sea water sample was taken a few meters from the coast where the water was 3 - 4 m deep. Then, the seawater sample was UV-digested (3 hs) after acidification with HCl to pH 1.0 with a 1 KW high-pressure mercury-vapor lamp to avoid possible interferences caused by natural organic compounds and to breakdown organic-metal complexes.
3. Results and Discussion
3.1. Voltammetric Response of Mn (II) and 5-Br-PADAP Solutions at a Bare CPE
Square wave adsorptive cathodic stripping (SW-AdCS) voltammogram of 10 mg·L−1 Mn (II) in acetate buffer (0.1 mol·L−1) of pH = 5.0 at a bare CPE exhibited no any voltammetric peak over the studied potential range, even following preconcentration by adsorptive accumulation for 60 s (Figure 1, curve a). Similarly, the square-wave cathodic stripping voltammogram of 50 mmol·L−1 5-BrPADAP free ligand in acetate buffer of pH = 5 at the bare CPE showed no any voltammetric peak (Figure 1, curve b). These behaviors indicated that either Mn (II) nor 5-Br-PADAP species was electro-inactive at the bare CPE. On the other side, voltammogram of a solution containing 10 mg·L−1 Mn (II) and 50 mmol·L−1 5-BrPADAP exhibited a small cathodic peak at +0.76 V (vs. Ag/AgCl/3 M KCl) in acetate buffer of pH = 5.0, follow
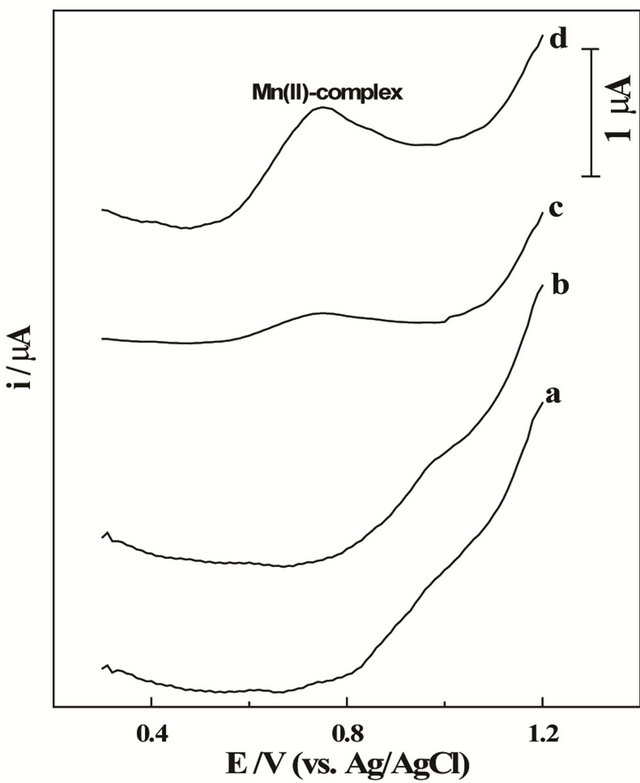
Figure 1. SW voltammograms of (a) 10 mg·L−1 Mn (II), (b) 50 mmol·L−1 5-Br-PADAP in acetate buffer (0.1 mol·L−1) of pH = 5.0 following preconcentration for 60 s, then (c) solutions of (a) + (b) under open circuit conditions and (d) following preconcentration by adsorptive accumulation at +1.2 V for 60 s at the bare CPE; f = 100 Hz, DEi = 10 mV and a = 25 mV.
ing preconcentration at open circuit conditions (Figure 1, curve c). This behavior suggests the formation of electro-active Mn (II)—5-Br-PADAP complex species in solution. However, following preconcentration at the bare CPE by adsorptive accumulation at +1.2 V for 60 s the cathodic peak of Mn (II)—5-Br-PADAP complex was better enhanced (Figure 1, curve d) indicating the interfacial adsorption nature of the formed Mn (II)—complex at the bare CPE.
Addition of 0.01% Triton X-100 to solution of the examined Mn (II) complex leads to strong suppression of the SW-AdCS voltammetric peak current of the investigated Mn (II)-complex. This behavior indicated that the Mn (II)-complex was adsorbed at the surface of the bare CPE. As reported in the literature, the molar ratio of Mn (II) to 5-Br-PADAP ligand in the complex was found to be 1:2 [47,48], therefore, the form of the present complex is Mn (II)-(5-Br-PADAP)2.
3.2. Voltammetric Response of Mn (II)— Complex at the Modified CPE
Voltammograms of 10 mg·L−1 Mn (II) in the presence of 50 mmol·L−1 5-Br-PADAP in 0.1 mol·L−1 acetate buffer (pH = 5.0) at the CPE modified with MMT-Na clay under various experimental conditions (Figure 2) demonstrated that the modified CPE remarkably improved the SW-AdCS voltammetric peak current magnitude of the investigated Mn (II) complex in comparison to that recorded at the bare CPE (Figure 2, curve a). This behavior
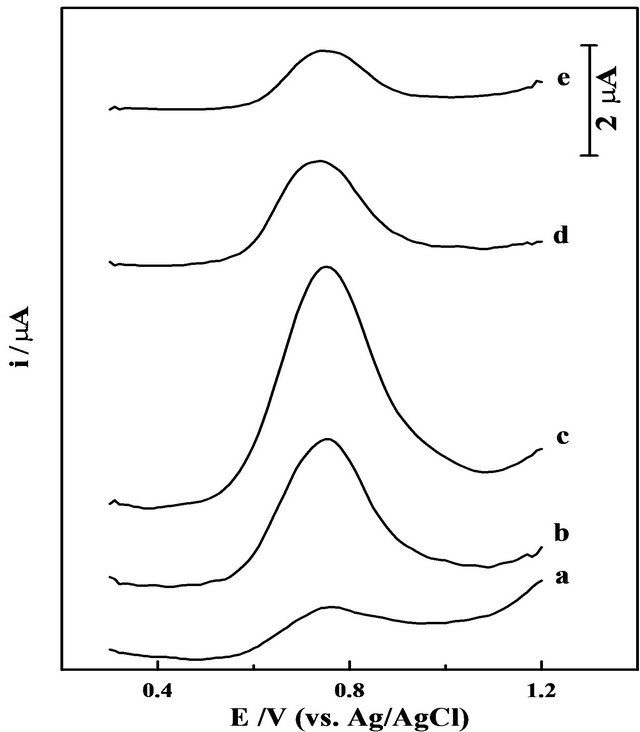
Figure 2. SW voltammograms of 10 mg·L−1 Mn (II) in presence of 50 mmol·L−1 5-Br-PADAP in acetate buffer (0.1 mol·L−1) of pH = 5.0 following preconcentration by adsorptive accumulation for 60 s at +1.2 V, at a bare CPE (a), then at the CPE modified with 5% (b), 10% (c), 15% (d) and 20% (w/w) MMT-Na clay (e). Other parameters are as those given in Figure 1.
indicated that modification of the CPE with MMT-Na clay significantly enhances the stripping peak current magnitude of Mn (II) complex under the experimental conditions which may be attributed to the superior adsorptive ability of MMT-Na clay. Following preconcentration of Mn (II)—complex by adsorptive accumulation at the modified CPE surface, it stripped out from the electrode surface by scanning the potential towards less positive direction resulting in a cathodic voltammetric peak (Figure 2). The overall electrode process may be expressed in two steps as:
1) Preconcentration (by adsorptive accumulation) step:
{Mn2+ − (5-Br-PADAP)2}(solution) ¾® {Mn2+ − (5-Br-PADAP)2}(surface)
(Formed immediately in solution)
2) Cathodic stripping step:
{Mn2+ − (5-Br-PADAP)2}(surface) + 2e– ¾® (Mn0) + 2 (5-Br-PADAP)
As shown in Figure 2 (curves b-e), the ratio (w/w) of MMT-Na clay in the graphite paste remarkably influences the square-wave cathodic stripping voltammetry peak current magnitude of the Mn (II) complex compared to that at the bare CPE (Figure 2, curve a). The stripping peak current magnitude firstly increased upon the increase of the MMT-Na clay ratio up to 10% (w/w) in the carbon paste (Figure 2, curve c) and then decreased at higher clay ratios (Figure 2, curves d and e).
Such enhancement of stripping peak current magnitude is expected due to the superior adsorptive ability of MMTNa clay. However, the conductivity of the modified CPE dropped with increasing the ratio of the non-conducting MMT-Na clay (curves d and e); hindering the electron transfer process and increasing the background current. Therefore, a CPE modified with 10% (w/w) of MMT-Na clay was used over the rest of the present analytical studies.
For optimization of a square-wave adsorptive cathodic stripping voltammetry (SW-AdCSV) method for sensitive determination of Mn (II) as a complex with 5-BrPADAP utilizing the modified CPE, the following were carried out.
3.3. Effect of Supporting Electrolyte and Its pH
SW-AdCS voltammograms of 10 mg·L−1 Mn (II) in the presence of 50 mmol·L−1 5-Br-PADAP were recorded in Britton-Robinson (pH 2.0 - 11.0), acetate (pH 4.0 - 6.0) and phosphate (pH 2.0 - 7.5) buffers following preconcentration of the Mn (II) complex at the modified CPE by adsorptive accumulation at +1.2 V (vs. Ag/AgCl/3 M KCl) for 60 s. The results showed that, better enhanced SWAdCSV peak current was achieved in the acetate buffer of pH values 4.0 - 5.0 (Figure 3) in comparison to that obtained in the other two media.
The dramatic decrease in the SW-AdCS voltammetric peak current magnitude at higher pH values (Figure 3) may be attributed to the precipitation of Mn (II) as Mn(OH)2
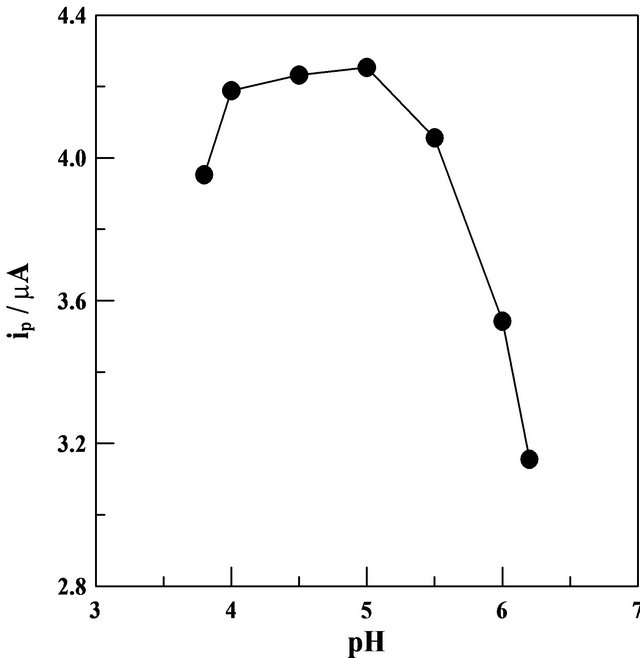
Figure 3. SW-AdCS voltammetry peak currents (ip) as a function of pH of acetate buffer (0.1 mol·L−1) for 10 mg·L−1 Mn (II) in presence of 50 mmol·L−1 5-Br-PADAP following preconcentration by adsorptive accumulation for 60 s at the CPE modified with 10% (w/w) MMT-Na clay. other parameters are as those given in Figure 1.
in alkaline media. In addition, the influence of ionic strength of the acetate buffer (0.05 - 0.20 mol·L−1) was also studied while keeping the pH value at 5.0. Better enhanced peak current was achieved in 0.10 mol·L−1 acetate buffer of pH = 5.0, therefore, it was chosen as a supporting electrolyte in the rest of the present analytical study.
3.4. Effect of Concentration of 5-Br-PADAP as a Ligand
SW-AdCS voltammograms of 100 mg·L−1 Mn (II) in the presence of increased amount of 5-Br-PADAP (10 to 50 mmol·L−1) were recorded in acetate buffer of pH = 5.0 following preconcentration of the Mn (II) complex by adsorptive accumulation at the modified CPE at +1.2 V for 60 s. The voltammograms showed that the peak current (ip) magnitude of Mn (II) complex increased with concentration of 5-Br-PADAP up to ~ 20 mmol·L−1 and then leveled off. Therefore, 20 mmol·L−1 of the tested ligand was used in the rest of analytical study since it is sufficient enough for the formation of complex with Mn (II) ions in concentrations up to 100 mg·L−1. Reaction kinetics of Mn (II) with 5-Br-PADAP were identified from its voltammograms recorded after different times of mixing the reactants. The SW-AdCS voltammetric peak current (ip) magnitude of the examined Mn (II) complex was practically constant with the reaction time, indicating the immediate formation of Mn (II)—(5-Br-PADAP)2 complex at the time of mixing the reactants in the electrochemical cell. Therefore, heating of the reactants solution was not required in the present work.
3.5. Square-Wave Pulse Parameters
In order to obtain a well developed and better enhanced SW-AdCS voltammetric peak for 10 mg·L−1 Mn (II) in the presence of 20 mmol·L−1 5-Br-PADAP using the acetate buffer (0.1 mol·L−1) of pH = 5.0 as a supporting electrolyte following preconcentration at the modified CPE by adsorptive accumulation at +1.2 V for 60 s, the square-wave pulse parameters (frequency f, scan increment DEi and pulse-amplitude a) were optimized. The peak current magnitude of the examined Mn (II)—(5- Br-PADAP)2 complex was increased linearly with frequency within the range 20 - 120 Hz; its corresponding regression equation was:
ip (mA) = 0.0425 f (Hz) – 0.019 (r = 0.997)
On increasing the scan increment within the range 2 - 10 mV, the peak current magnitude of the examined Mn (II) complex was also increased linearly; its corresponding regression equation was:
ip (mA) = 0.3434 DEi (mV) + 1.859 (r = 0.998)
Although the peak current (ip) magnitude was increased linearly with the pulse-amplitude within the range 20 - 60 mV, the best peak morphology with the lower baseline was obtained at pulse-amplitude of 25 mV. Accordingly, the optimal square-wave pulse-parameters used over the rest of the present analytical study were: frequency f = 120 Hz, scan increment DEi = 10 mV and pulse-amplitude a = 25 mV.
3.6. Preconcentration Conditions
Effect of varying the preconcentration (accumulation) potential Eacc from +1.3 to +1.0 V (vs. Ag/AgCl/3M KCl) on the peak current magnitude of the SW-AdCS voltammograms for 10 mg·L−1 Mn (II) in acetate buffer (0.1 mol·L−1) of pH = 5.0 in the presence of 20 mmol·L−1 5-Br-PADAP was evaluated following its preconcentration at the modified CPE by adsorptive accumulation for 60 s (Figure 4). The results showed that better enhanced peak current magnitudes were achieved over the potential range +1.2 to +1.1 V. Therefore, a preconcentration potential of +1.1 V (vs. Ag/AgCl/3 M KCl) was used throughout the present analytical study.
On the other hand, SW-AdCS voltammograms of 5 and 10 mg·L−1 Mn (II) in the presence of 20 mmol·L−1 5-Br-PADAP were recorded under the optimum operational conditions following increased preconcentration time from 0.0 to 300 s by adsorptive accumulation at +1.1 V (Figure 5). The peak current magnitude of the Mn (II) complex was increased linearly with preconcentration time over the examined time period. At a higher concentration of Mn (II) (10 mg·L−1), adsorptive saturation of the electrode surface (adsorption equilibrium) was
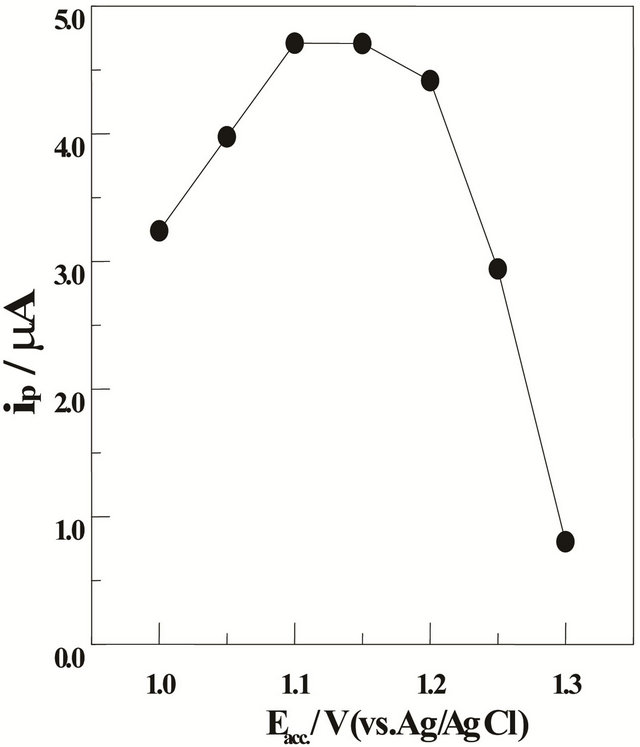
Figure 4. SW-AdCS voltammetric peak currents (ip) as a function of preconcentration potential (Eacc.) in acetate buffer (0.1 mol·L−1) of pH = 5.0 for 10 mg·L−1 Mn (II) in the presence of 20 mmol·L−1 5-Br-PADAP following preconcentration by adsorptive accumulation for 60 s at the CPE modified with 10% (w/w) MMT-Na; f = 120 Hz, DEi = 10 mV and a = 25 mV.
reached at 240 s and hence the peak current leveled off. Accordingly, the preconcentration time should be chosen according to the concentration level of Mn (II) in the examined solution.
3.7. Method Validation
3.7.1. Linearity Range and Limit of Detection
SW-AdCS voltammograms of various concentrations of Mn (II) in the presence of 20 mmol·L−1 5-Br-PADAP were recorded under the optimum operational conditions using the modified CPE (Figure 6). Rectilinear relations between the peak current magnitude (ip) and concentrations (C) of Mn (II) following preconcentration for various times (60, 120 and 240 s) at the modified CPE were obtained over the ranges shown in Table 1.
Limits of detection (LOD) of Mn (II) as 5-Br-PADAP complex for three linearity ranges were estimated as 3 S/N (signal-to-noise) [49]. The achieved LOD following preconcentration of the Mn (II) complex onto the modified CPE for 240 s under the optimum operational conditions was 0.015 mg·L−1 (2.73 × 10−10 mol·L−1) (Table 1). The results shown in Table 1 confirmed the sensitivity and reliability of the developed electrode {90% (w/w) CP +10% (w/w) MMT-Na clay} coupled with the optimized SW-AdCSV method for ultra-trace determination of Mn (II) as a complex with 5Br-PADAP over most of the reported electrochemical methods [25-35].
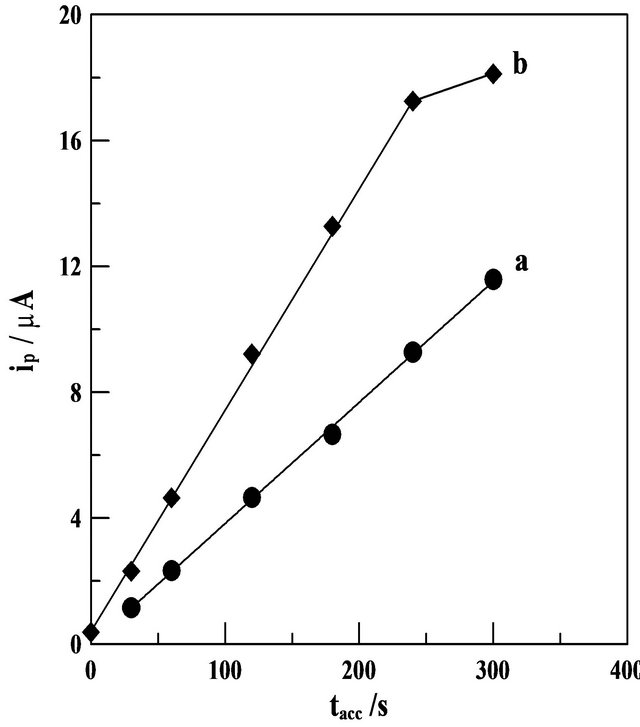
Figure 5. SW-AdCS voltammetric peak currents (ip) as a function of the preconcentration time (tacc) in acetate buffer (0.1 mol·L−1) of pH = 5.0 for a solution containing; (a) 5 and (b) 10 mg·L−1 Mn (II) in presence of 20 mM 5-Br-PADAP following preconcentration at the CPE modified with 10% (w/w) MMT-Na clay by adsorptive accumulation at +1.1 V. other parameters are as those given in Figure 4.
3.7.2. Accuracy and Precision
Accuracy and precision of the described SW-AdCSV method for determination of Mn (II) complex utilizing the modified CPE were estimated as recovery (% R) and standard deviation (% SD) by analyzing three reference standard concentration levels of Mn (II) for three times (Table 2) under the optimized operational conditions. Mean percentage recoveries and standard deviations obtained by applying the calibration curve and standard addition methods (Table 2) indicated the accuracy and precision of the described SW-AdCSV method coupled with the modified CPE for determination of Mn (II) as 5- Br-PADAP complex.
Moreover, the results obtained by the described stripping voltammetry method, applying the calibration curve, were statistically compared with those obtained by flame atomic absorption spectrometry (FAAS). The results showed that the described voltammetric method utilizing the modified CPE is much more sensitive than the FAAS method. Since the calculated value of F-statistic does not exceed the theoretical one (Table 2), there was no significant difference between the described SW-AdCSV and FAAS methods with respect to reproducibility [48]. Also, no significant differences were noticed between the two methods regarding accuracy and precision as revealed by t-values [50] (Table 2).
3.7.3. Interferences
Interferences from some foreign cations (K+, Na+, Mg2+,
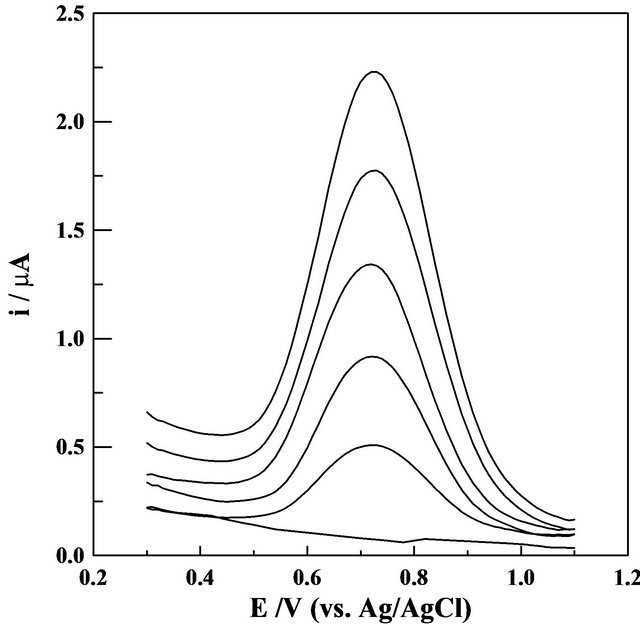
Figure 6. SW-AdCS voltammograms for successive additions of Mn (II), recorded in acetate buffer (0.1 mol·L−1) of pH = 5.0 containing 20 mmol·L−1 Br-PADAP following preconcentration by adsorptive accumulation at +1.1 V for 240 s at the modified CPE with 10% (w/w) MMT-Na clay (Each addition affected a 0.2 mg·L−1 Mn (II)). Dotted line represents the blank solution. other parameters are as those given in Figure 4.
Table 1. Characteristics of the calibration curves for the determination of Mn (II) as 5-Br-PADAP complex following preconcentration by adsorptive accumulation at the modified CPE with 10% (w/w) MMT-Na, Eacc. = +1.1 V.
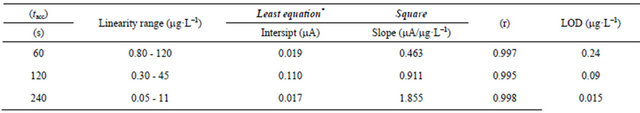 *Average of three determinations.
*Average of three determinations.
Table 2. Accuracy and precision of the described SW-AdCS voltammetry method as recovery (%R) and standard deviation (%SD) for determination of Mn (II) as 5-Br-PADAP complex following preconcentration by adsorptive accumulation at the modified CPE with 10% (w/w) MMT-Na clay.
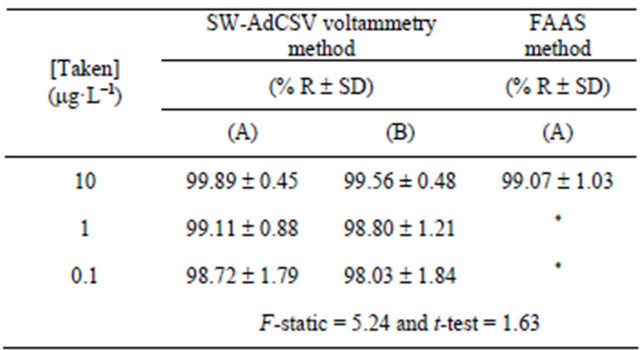
(A) Calibration curve method and (B) Standard addition method. *Under limit of detection. Theoretical F-statistic = 6.39 and t-test = 2.3 at 95% confidence limit for n1 = n2 = 5.
Table 3. Interferences of some inorganic species in the simultaneous determination of 10 mg·L−1 Mn(II) as 5-BrPADAP complex by the described SW-AdCSV method utilizing the developed 10% (w/w) MMT-Na modified CPE.
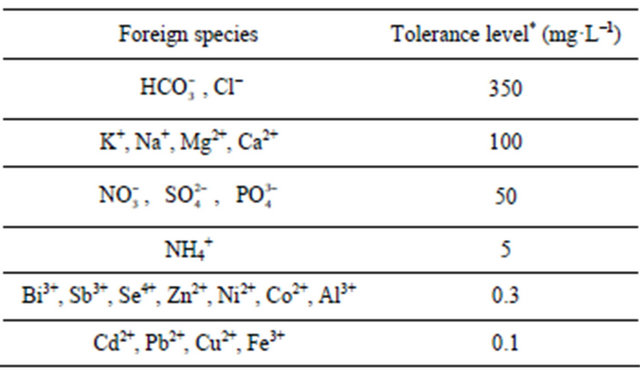
*For 6% error.
Ca2+, Cd2+, Pb2+, Cu2+, Bi3+, Sb3+, Se4+, Zn2+, Ni2+, Co2+, Fe3+, Al3+ and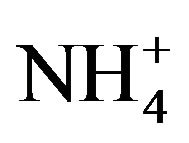 ) and inorganic anions (
) and inorganic anions (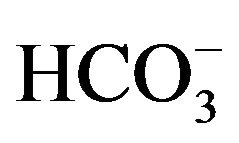 , Cl-,
, Cl-, 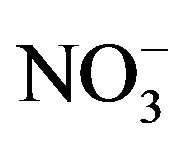 ,
, 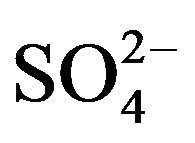 and
and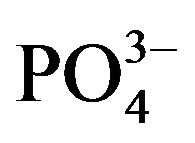 ) to the assay of 10 mg·L−1 Mn (II) as 5-Br-PADAP complex were evaluated. As shown in Table 3, the investigated foreign species, below tolerance level, have insignificant effect on the peak current magnitudes of 10 mg·L−1 Mn (II) complex since the relative errors are below 6%. Interference from Triton X-100
) to the assay of 10 mg·L−1 Mn (II) as 5-Br-PADAP complex were evaluated. As shown in Table 3, the investigated foreign species, below tolerance level, have insignificant effect on the peak current magnitudes of 10 mg·L−1 Mn (II) complex since the relative errors are below 6%. Interference from Triton X-100
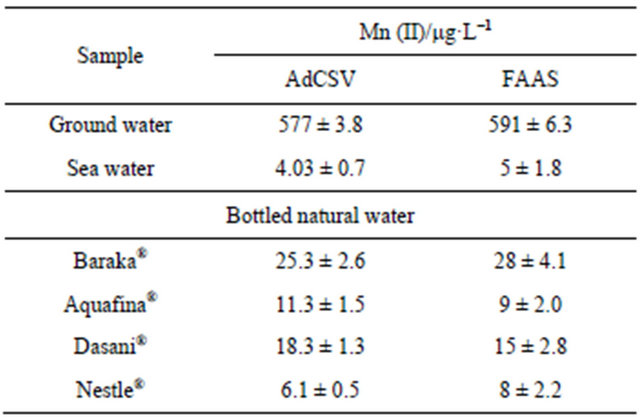
Table 4. Determination of Mn(II) as 5-Br-PADAP complex in ground, bottled and sea water samples by the described SW-AdCS voltammetry method and flame atomic absorption spectrometry (FAAS).
as non-ionic surfactant to the assay of the investigated metal ion was insignificant up to 0.001%. Higher concentrations of the surfactant cause deformation and great suppression of the voltammetric peak current magnitude of Mn (II) complex. Influence of surfactants, if present in the analyzed water samples, can be completely eliminated by thorough mineralization of water sample prior to the analysis.
3.8. Application
The described SW-AdCSV method coupled with the modified CPE was successfully utilized for determination of Mn (II) in ground water, bottled natural water and sea water samples. The same water samples were also analyzed by FAAS. Comparison of the results obtained by the described SW-AdCSV method with those obtained by FAAS (Table 4) revealed the capability of the modified CPE for determination of Mn (II) at trace and ultratrace concentrations.
4. Conclusion
A simple, precise and accurate fully validated squarewave adsorptive cathodic stripping voltammetry method has been developed for determination of Mn (II) as a complex with 2-(5’-bromo-2’-pyridylazo) 5-diethylaminophenol in aqueous solutions using a carbon paste electrode modified with 10% (w/w) montmorillonite-Na clay. The developed voltammetric method coupled with the modified carbon paste electrode has been successfully applied for analysis of Mn (II) in different water samples without interferences from various cations and anions. The modified carbon paste electrode is much more sensitive than most of the previously applied electrodes for determination of manganese in various matrices due to the superior adsorption property of montmorillonite-Na clay as a modifier.
REFERENCES
- B. H. J. Rehm and G. Reed, “Microbial Degradations,” VCH, Weinheim, 1986.
- G. M. Gadd and A. J. Griffiths, “Microorganisms and Heavy Metal Toxicity,” Microbial Ecology, Vol. 4, No. 4, 1978, pp. 303-317. doi:10.1007/BF02013274
- R. Witholt, R. H. Gwiazda and D. R. Smith, “The Neurobehavioral Effects of Subchronic Manganese Exposure in the Presence and Absence of Pre-Parkinsonism,” Neurotoxicology and Teratology, Vol. 22, No. 6, 2000, pp. 851- 861. doi:10.1016/S0892-0362(00)00108-2
- N. V. Hue, S. Vega and J. A. Silva, “Manganese Toxicity in a Hawaiian Oxisol Affected by Soil pH and Organic Amendments,” Soil Science Society of America Journal, Vol. 65, No. 1, 2001, pp. 153-160. doi:10.2136/sssaj2001.651153x
- N. Maniasso and E. A. G. Zagatto, “Flow-Injection Spectrophotometric Catalytic Determination of Manganese in Plants Exploiting the Aerial Oxidation of Diphenyl Carbazone,” Analytica Chimica Acta, Vol. 366, No. 1, 1998, pp. 87-92. doi:10.1016/S0003-2670(97)00682-X
- A. A. Abdel-Shafi, “Spectrophotometric Determination of Manganese by Using Redox Reaction of Tris (2,2’-bipyridine) Osmium(II) with Mn7+,” Analytical Sciences, Vol. 22, No. 6, 2006, pp. 825-828. doi:10.2116/analsci.22.825
- H. Sedaira, “Simultaneous Determination of Manganese and Zinc in Mixtures Using Firstand Second-Derivative Spectrophotometry,” Talanta, Vol. 51, No. 1, 2000, pp. 39-48. doi:10.1016/S0039-9140(99)00244-1
- Q. Wei, L. G. Yan, G. H. Chang and Q. Y. Ou, “Kinetic Spectrophotometric Determination of Trace Manganese (II) with Dahlia Violet in Nonionic Microemulsion Medium,” Talanta, Vol. 59, No. 2, 2003, pp. 253-259. doi:10.1016/S0039-9140(02)00498-8
- H. Bilinski, Z. Kwokal and M. Branica, “Formation of Some Manganese Minerals from Ferromanganese Factory Waste Disposed in the Krka River Estuary,” Water Research, Vol. 30, No. 3, 1996, pp. 495-500. doi:10.1016/0043-1354(95)00207-3
- H. Ohura, Y. Ishibashi, T. Imato and S. Yamasaki, “Potentiometric Flow Injection Determination of Manganese (II) by Using a Hexacyanoferrate (III)-Hexacyanoferrate (II) Potential Buffer,” Talanta, Vol. 60, No. 1, 2003, pp. 177- 184. doi:10.1016/S0039-9140(03)00120-6
- V. O. Doroschuk, S. O. Lelyushok, V. B. Ishchenko and S. A. Kulichenko, “Flame Atomic Absorption Determination of Manganese (II) in Natural Water after Cloud Point Extraction,” Talanta, Vol. 64, No. 4, 2004, pp. 853-856. doi:10.1016/j.talanta.2004.03.056
- R. G. O. Araujo, F. S. Dias, S. M. Macedo, W. N. L. dos Santos and S. L. C. Ferreira, “Method Development for the Determination of Manganese in Wheat Flour by Slurry Sampling Flame Atomic Absorption Spectrometry,” Food Chemistry, Vol. 101, No. 1, 2007, pp. 397-400. doi:10.1016/j.foodchem.2005.10.024
- V. A. Lemos, L. N. Santos and M. A. Bezerra, “Determination of Cobalt and Manganese in Food Seasonings by Flame Atomic Absorption Spectrometry after Preconcentration with 2-Hydroxyacetophenone-Functionalized Polyurethane Foam,” Journal of Food Composition and Analysis, Vol. 23, No. 3, 2010, pp. 277-281.
- Ç. A. Sahin, M. Efeçınar and N. Şatıroğlu, “Combination of Cloud Point Extraction and Flame Atomic Absorption Spectrometry for Preconcentration and Determination of Nickel and Manganese Ions in Water and Food Samples,” Journal of Hazardous Materials, Vol. 176, No. 1-3, 2010, pp. 672-677.
- A. P. dos Anjos, L. Cornejo-Ponce, S. Cadore and N. Baccan, “Determination of Manganese by Flame Atomic Absorption Spectrometry after Its Adsorption onto Naphthalene Modified with 1-(2-Pyridylazo)-2-naphthol (PAN),” Talanta, Vol. 71, No. 3, 2007, pp. 1252-1256. doi:10.1016/j.talanta.2006.06.026
- V. A. Lemos and G. T. David, “An On-Line Cloud Point Extraction System for Flame Atomic Absorption Spectrometric Determination of Trace Manganese in Food Samples,” Microchemical Journal, Vol. 94, No. 1, 2010, pp. 42-47. doi:10.1016/j.microc.2009.08.008
- P. R. M. Correia, E. de Oliveira and P. V. Oliveira, “Simultaneous Determination of Manganese and Selenium in Serum by Electrothermal Atomic Absorption Spectrometry,” Talanta, Vol. 57, No. 3, 2002, pp. 527-535. doi:10.1016/S0039-9140(02)00069-3
- P. G. Su and S. D. Huang, “Direct and Simultaneous Determination of Copper and Manganese in Seawater with a Multielement Graphite Furnace Atomic Absorption Spectrometer,” Spectrochimica Acta Part B: Atomic Spectroscopy, Vol. 53, No. 5, 1998, pp. 699-708.
- H. M. Liu, S. J. J. Tsai, F. C. Cheng and S. Y. Chung, “Determination of Trace Manganese in the Brain of Mice Subjected to Manganese Deposition by Graphite Furnace Atomic Absorption Spectrometry,” Analytica Chimica Acta, Vol. 405, No. 1-2, 2000, pp. 197-203.
- G. P. Brandão, R. C. de Campos, E. V. R. de Castro and H. C. de Jesus, “Determination of Manganese in Diesel, Gasoline and Naphtha by Graphite Furnace Atomic Absorption Spectrometry Using Microemulsion Medium for Sample Stabilization,” Spectrochimica Acta Part B: Atomic Spectroscopy, Vol. 63, No. 8, 2008, pp. 880-884. doi:10.1016/j.sab.2008.04.019
- M. Hsiang, Y. Sung and S. Huang, “Direct and Simultaneous Determination of Arsenic, Manganese, Cobalt and Nickel in Urine with a Multielement Graphite Furnace Atomic Absorption Spectrometer,” Talanta, Vol. 62, No. 4, 2004, pp. 791-799. doi:10.1016/j.talanta.2003.09.031
- K. Uysal, Y. Emre and E. Kose, “The Determination of Heavy Metal Accumulation Ratios in Muscle, Skin and Gills of Some Migratory Fish Species by Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES) in Beymelek Lagoon (Antalya/Turkey),” Microchemical Journal, Vol. 90, No. 1, 2008, pp. 67-70. doi:10.1016/j.microc.2008.03.005
- M. A. Bezerra, S. M. D. Maeda, E. P. Oliveira, M. D. B. de Carvalho and R. E. Santelli, “Internal Standardization for the Determination of Cadmium, Cobalt, Chromium and Manganese in Saline Produced Water from Petroleum Industry by Inductively Coupled Plasma Optical Emission Spectrometry after Cloud Point Extraction,” Spectrochimica Acta Part B: Atomic Spectroscopy, Vol. 62, No. 9, 2007, pp. 985-991. doi:10.1016/j.sab.2007.06.009
- J. Wang, “Stripping Analysis: Principles, Instrumentation and Application,” VCH, Deerfield Beach, 1985.
- M. M. Ghoneim, A. M. Hassanein, E. Hammam and A. M. Beltagi, “Simultaneous Determination of Cd, Pb, Cu, Sb, Bi, Se, Zn, Mn, Ni, Co and Fe in Water Samples by Differential Pulse Stripping Voltammetry at a Hanging Mercury Drop Electrode,” Fresenius’ Journal of Analytical Chemistry, Vol. 367, No. 4, 2000, pp. 378-383. doi:10.1007/s002160000410
- C. Locatelli and G. Torsi, “Voltammetric Trace Metal Determinations by Cathodic and Anodic Stripping Voltammetry in Environmental Matrices in the Presence of Mutual Interference,” Journal of Electroanalytical Chemistry, Vol. 509, No. 1, 2001, pp. 80-89.
- R. Piech, B. Baś and W. W. Kubiak, “The Cyclic Renewable Mercury Film Silver Based Electrode for Determination of Manganese (II) Traces Using Anodic Stripping Voltammetry,” Journal of Electroanalytical Chemistry, Vol. 621, No. 1, 2008, pp. 43-48.
- A. Romanus, H. Muller and D. Kirsch, “Application of Adsorptive Stripping Voltammetry (AdSV) for the Analysis of Trace-Metals in Brine 1. Batch Voltammetric Measurements,” Fresenius’ Journal of Analytical Chemistry, Vol. 340, No. 6, 1991, pp. 363-370. doi:10.1007/BF00321583
- L. Z. Wang, C. S. Ma, X. L. Zhang and J. G. Wang, “Determination of Trace Manganese by Adsorption Voltammetry,” Analytical Letters, Vol. 26, No. 8, 1993, pp. 1711- 1717. doi:10.1080/00032719308021491
- J. Wang and J. M. Lu, “Adsorptive Stripping Voltammetry of Trace Manganese in the Presence of 2-(5’-Bromo- 2’-pyridylazo)-5-diethylamino Phenol (5-Br-PADAP),” Talanta, Vol. 42, No. 3, 1995, pp. 331-335. doi:10.1016/0039-9140(95)01396-S
- N. A. El-Maali and D. A. El-Hady, “Square-Wave Adsorptive Stripping Voltammetry at Glassy Carbon Electrode for Selective Determination of Manganese. Application to Some Industrial Samples,” Analytica Chimica Acta, Vol. 370, No. 2, 1998, pp. 239-249. doi:10.1016/S0003-2670(98)00300-6
- J. W. Di and F. Zhang, “Voltammetry Determination of Trace Manganese with Pretreatment Glassy Carbon Electrode by Linear Sweep Voltammetry,” Talanta, Vol. 60, No. 1, 2003, pp. 31-36.
- J. S. Roitz and K. W. Bruland, “Determination of Dissolved Manganese (II) in Coastal and Estuarine Waters by Differential Pulse Cathodic Stripping Voltammetry,” Analytica Chimica Acta, Vol. 344, No. 3, 1997, pp. 175-180. doi:10.1016/S0003-2670(97)00041-X
- M. Rievaj, P. Tomcik, Z. Janosikova, D. Bustin and R. G. Compton, “Determination of Trace Mn (II) in Pharmaceutical Diet Supplements by Cathodic Stripping Voltammetry on Bare Carbon Paste Electrode,” Chemia Analityczna, Vol. 53, No. 1, 2008, pp. 153-161.
- E. M. Ghoneim, “Simultaneous Determination of Mn (II), Cu (II) and Fe (III) as 2-(5’-Bromo-2’-pyridylazo)-5-diethylaminophenol Complexes by Adsorptive Cathodic Stripping Voltammetry at a Carbon Paste Electrode,” Talanta, Vol. 82, No. 2, 2010, pp. 646-652. doi:10.1016/j.talanta.2010.05.025
- P. K. Ghosh and A. J. Bard, “Clay Modified Electrodes,” Journal of the American Chemical Society, Vol. 105, No. 17, 1983, pp. 5691-5693. doi:10.1021/ja00355a030
- P. Kula and Z. Navrátilová, “Voltammetric Copper (II) Determination with a Montmorillonite-Modified Carbon Paste Electrode,” Fresenius’ Journal of Analytical Chemistry, Vol. 354, No. 5-6, 1996, pp. 692-695.
- Z. Navrátilová and P. Kula, “Determination of Gold Using Clay Modified Carbon Paste Electrode,” Fresenius’ Journal of Analytical Chemistry, Vol. 367, No. 4, 2000, pp. 369- 372. doi:10.1007/s002160000401
- P. Kula and Z. Navrátilová, “Anion Exchange of Gold Chloro Complexes on Carbon Paste Electrode Modified with Montmorillonite for Determination of Gold in Pharmaceuticals,” Electroanalysis, Vol. 13, No. 8-9, 2001, pp. 795-798.
- P. Kula, Z. Navrátilová, P. Kulová and M. Kotoucek, “Sorption and Determination of Hg (II) on Clay Modified Carbon Paste Electrodes,” Analytica Chimica Acta, Vol. 385, No. 1, 1999, pp. 91-101. doi:10.1016/S0003-2670(98)00697-7
- W. S. Huang, C. H. Yang and S. H. Zhang, “Anodic Stripping Voltammetric Determination of Mercury by Use of a Sodium Montmorillonite-Modified Carbon-Paste Electrode,” Analytical and Bioanalytical Chemistry, Vol. 374, No. 6, 2002, pp. 998-1001. doi:10.1007/s00216-002-1438-0
- K. B. Ji and S. S. Hu, “Square Wave Voltammetric Determination of Trace Amounts of Europium (III) at Montmorillonite-Modified Carbon Paste Electrodes,” Collection of Czechoslovak Chemical Communications, Vol. 69, No. 8, 2004, pp. 1590-1599. doi:10.1135/cccc20041590
- D. Sun, C. D. Wan, G. Li and K. B. Wu, “Electrochemical Determination of Lead (II) Using a Montmorillonite Calcium-Modified Carbon Paste Electrode,” Microchimica Acta, Vol. 158, No. 3-4, 2007, pp. 255-260. doi:10.1007/s00604-006-0686-7
- A. M. Beltagi, “Utilization of a Montmorillonite-CaModified Carbon Paste Electrode for the Stripping Voltammetric Determination of Diflunisal in Its Pharmaceutical Formulations and Human Blood,” Journal of Applied Electrochemistry, Vol. 39, No. 12, 2009, pp. 2375-2384. doi:10.1007/s10800-009-9924-0
- H. S. El-Desoky and M. M. Ghoneim, “Stripping Voltammetric Determination of Silymarin in Formulations and Human Blood Utilizing Bare and Modified Carbon Paste Electrodes,” Talanta, Vol. 84, No. 1, 2011, pp. 223- 234. doi:10.1016/j.talanta.2011.01.027
- A. M. Beltagi, E. M. Ghoneim and M. M. Ghoneim, “Simultaneous Determination of Cadmium (II), Lead (II), Copper (II) and Mercury (II) by Square-Wave Anodic Stripping Voltammetry at a Montmorillonite-Calcium Modified Carbon Paste Electrode,” Internnational Journal of Environmental Analytical Chemistry, Vol. 91, No. 1, 2011, pp. 17-32. doi:10.1080/03067310902962577
- X. C Qiu, Y. S. Zhang and Y. Q. Zhu, “Rapid Spectrophotometric Determination of Water-Soluble Manganese in Soils Using 2-(5-Bromo-2-pyridylazo)-5-diethyl-amino-phenol,” Soil Science, Vol. 138, No. 6, 1984, pp. 432- 435. doi:10.1097/00010694-198412000-00007
- Z. T. Jiang, R. Li, J. B. Xi and B. Q. Yi, “Determination of Trace Amounts of Manganese by Beta-Cyclodextrin Polymer Solid Phase Spectrophotometry Using 2-(5- Bromo-2-pyridylazo)-5-diethylaminophenol,” Analytica Chimica Acta, Vol. 392, No. 2-3, 1999, pp. 247-253. doi:10.1016/S0003-2670(99)00175-0
- M. Swartz and I. S. Krull, “Analytical Method Development and Validation,” Marcel Dekker, New York, 1997.
- G. D. Christian, “Analytical Chemistry,” 5th Edition, Willey, Hoboken, 1994.