American Journal of Analytical Chemistry
Vol.4 No.3(2013), Article ID:29053,7 pages DOI:10.4236/ajac.2013.43019
Stripping Voltammetric Determination of Uranium in Water Samples Using Hg-Thin Film Modified Multiwall Carbon Nanotube Incorporated Carbon Paste Electrode
Analytical Chemistry Division, Bhabha Atomic Research Centre, Mumbai, India
Email: *aksatpati@gmail.com
Received February 5, 2013; revised March 7, 2013; accepted March 18, 2013
Keywords: Uranium; Carbon Paste Electrode; Chloranilic Acid (CAA); MWCNT; Voltammetry; Impedance
ABSTRACT
The variables affecting determination of ultra trace levels of uranium (VI) in aqueous samples by differential pulse cathodic stripping voltammetry were examined in detail using Hg-thin film modified carbon paste and multiwall carbon nanotube (MWCNT) incorporated carbon paste electrode. Carbon paste electrode prepared in the laboratory was modified with Hg thin film and used as the working electrode. MWCNT was incorporated into the carbon paste for enhancement in sensitivity of the measurements. Electrochemical response for the uranium (VI) reduction peak was found to be well resolved on the thin mercury film modified carbon paste surface and also with the MWCNT modification. Characteristics of the adsorption preconcentration process were investigated using electrochemical impedance measurements. Electrochemical signals were observed to be enhanced on MWCNT modification of the carbon paste.
1. Introduction
Uranium is one of the important actinide in view of its use as a fuel in nuclear industry. Besides its utility as nuclear fuel, geochemical pathways of the element are extremely important due to its long term toxic implications. Compared to the other elements like Cu, Zn, Pb and Cd these aspects were studied more rigorously. Therefore determination of uranium is of immense importance in relation to the nuclear industry, environment [1-3] and also from a geochemical view point. Many methods for determination of uranium at various levels were reported but electrochemical methods provide simpler and cost effective analysis without any prior separation [4,5]. In addition, electrochemical methods using voltammetry and adsorptive pre-concentration have superior limit of detection compared to other methods. The levels of uranium in natural water samples are in 3 µg∙L−1 range and determination of uranium in this range demands special methodologies. Adsorptive pre-concentration of uranium at the microelectrode surface followed by cathodic stripping under the differential pulse mode (differential pulse cathodic stripping voltammetry, DPCSV) is an excellent analytical technique for the detection of uranium in µg∙L−1 concentration range. Chloranilic acid (CAA) has been used as the complexing agent by some workers and shown to have better activity in terms of the formation of surface-active complexes with uranium [6-11]. Many complexing agents, other than chloranilic acid has been used which includes, oxin [12], cupferron [13] propyl gallate [14], catechol [15], potassium hydrogen phthalate [16], aluminon [17], 8-hydroxy-quinoline [18], pyromellitic acid [19], arsenazo (III) [20] and humic acid [21]. In the entire above cases mercury drop as the working electrode was used. Use of mercury thin films reduces the use of toxic metallic mercury compared to the mercury drop. Modified electrodes by a number of ways of modifications of the solid-state electrodes have been investigated and a few among them are, propyl gallate [22], N-phenyl-cinnamohydroxamic acid [23], carbamoyl phosphonic acid [24], self-assembled monolayers of thiols [25,26], nafion [27], calixarenes [28,29], carbon nanotubes [30], and modified graphite electrodes [31]. Recently a relatively simpler method has been proposed based on 6-O-palmitoyl-l-ascorbic acid-modified graphite electrodes [32]. Thus, the general trend is to minimize the use of mercury drop electrode and to have modified electrode for the highly sensitive determination of uranium for its natural presence in water samples. Present method has been proposed based on the carbon paste electrode prepared from graphitic powder, onto which thin mercury film was deposited to form the sensor. Parameters for the optimal sensitivity and the minimal usage of metallic mercury have been optimized. Conditions and the mechanism of the adsorption pre-concentration process were investigated. The thin film electrode provides better physical stability over the mercury drop electrode for onsite application. In the present work carbon paste electrode was modified with Hg-thin film, which was used for the adsorptive stripping voltammetric determination of Uranium in natural water and sea water samples.
2. Experimental Procedure
2.1. Instrumentation
All the experiments were carried out under potentiostatic control using Eco Chemie Potentiostat, AUTOLAB-100 with the VA663 stand. Electrochemical impedance measurements were carried out using the frequency response analyzer (FRA) module attached with Autolab-100 potentiostat. Impedance scan was measured at the frequency range of 10 kHz to 0.1 Hz with the amplitude of 10 mV. After obtaining the optimum frequency for maximum capacitive behavior at 50 Hz, impedance measurements were carried at a single frequency of 50 Hz at different applied potentials. All the potentials were applied and measured were with respect to the saturated calomel electrode, SCE. The electrolyte solution was purged for 300 s using high purity nitrogen gas before every electrochemical scan.
2.2. Reagents
Spectroscopic grade carbon powder obtained from EMerck, India limited was used as received for the preparation of the paste. MWCNT of 10 - 20 nm diameter and 10 - 30 µm length was used for the modification of the carbon paste. Silicon oil was obtained from SD fine chemicals, Mumbai, India and chloranilic acid (CAA) was obtained from Aldrich, used without further purification.
2.3. Preparation of the Carbon Paste Electrode
The spectroscopic grade carbon powder was heated at 400˚C for 4 h in inert atmosphere taking 0.5 g of the powder in a quartz boat. After heating, the powder was transferred to a beaker containing high purity paraffin wax dissolved in n-hexane. The mixture was kept in a fume hood maintained at negative pressure and the n-hexane was allowed to evaporate. After that the mixture was kept under hot air blower for complete removal of n-hexane. Heating removed the adsorbed gases from the surface of the carbon powder and upon paraffin impregnation the re-adsorption of gases was minimized. In this study ~1.5% (weight %) of paraffin wax was added to the carbon powder. After paraffin impregnation, required quantity of silicone oil was added as the binder (25% of the mass of modifier and 75% carbon powder) and grounded well to get homogeneous paste. The carbon paste was filled of a glass tube with 1.5 mm diameter from the bottom end. A platinum wire was used to connect the carbon paste electrode to the instrument.
3. Results and Discussions
Modification of the carbon paste electrode was carried out by depositing Hg film from 0.1 mM∙Hg2+ solution on bare carbon paste surface by applying −0.9 V potential for 300 s. The thin mercury film modified carbon paste electrode (CPE) was found to be physically stable in the experimental conditions and workability was comparable to the hanging mercury drop electrode (HMDE). Physical instability of HMDE restricts the stirring which is otherwise not a problem with the mercury thin film modified CPE. Mercury thin films on carbon substrates are stable in neutral to alkaline solution. At highly acidic solution (pH ~1) instability can be an issue. In the present case at pH 2.5, the electrode was stable up to 3 hrs. Electrochemical parameters for sensitive determination of uranium were optimized using the mercury thin film modified carbon paste electrode (MTFMCPE). The SEM micrograph of the thin mercury film modified carbon paste electrode was shown in Figure 1. High resolution image of mercury droplets on the carbon paste and also on CNT modified carbon paste surface was shown in Figures 2 and 3 respectively. Mercury droplets were uniformly distributed over the electrode surface. Droplets over the CNT modified surface were relatively smaller compared to those over bare carbon paste surface. The thin mercury film over the CPE surface was observed from the micrograph. Occasional mercury droplets were also observed on the surface.
3.1. Effect of Accumulation Potential on the Measurements
Selection of an appropriate accumulation potential for the optimum adsorption of the uranium-CAA complex is important in the present type of stripping voltammetric analysis as this could adversely affect the pre-concentration (marginal change in the accumulation potential). Additionally an easy accumulation is beneficial for the field application of the modified electrode. Effect of the accumulation potential was investigated by applying it from −0.8 V to 0.12 V (SCE) and the results were plotted in Figure 4. It was observed that the peak current was highest at around 0.05 to 0.08 V (SCE) of accumulation potential. An increased peak current was also observed at around −0.6 V and −0.7 V (SCE). Beyond (more positive) 0.12 V the peak current was reduced drastically possibly due to the oxidation of mercury films. Thus an accumulation potential of around 0.05 to 0.08 V was ideal for the optimum determination of uranium from the water samples. Interestingly it was observed that the open circuit
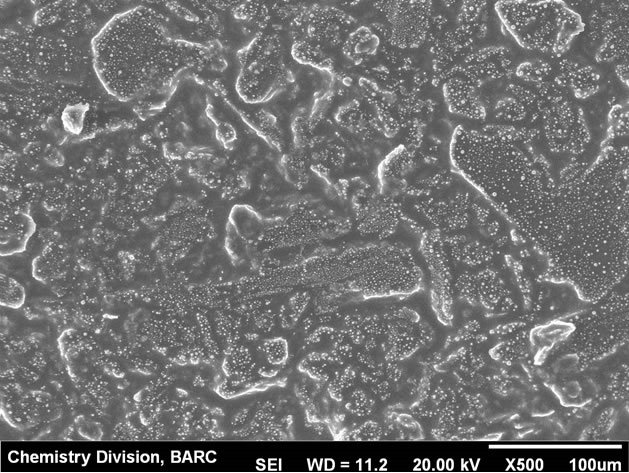
Figure 1. SEM micrograph of the MTFMCPE at lower resolution.

Figure 2. SEM micrograph of the MTFMCPE at high resolution.

Figure 3. SEM micrograph of the MWCNT modified MTFMCPE at high resolution.
potential of the MTFMCPE in the supporting electrolyte media was 0.07 V. Thus it is possible to pre-concentrate the uranium-CAA complex even under open circuit conditions which is an important observation for field application of this method. In all the present investigations pre-concentration was carried out at 0.08 V.
3.2. Impedance Measurements
Electrochemical impedance measurements were carried out to have an explanation about the adsorption process of uranium-CAA complex on the MTMCPE surface. From the frequency scan at open circuit potential, Nyquist plot was obtained and shown in Figure 5. Bare CPE was observed to show very high charge transfer resistance at open circuit potential. On deposition of mercury thin film on the CPE surface the charge transfer resistance was observed to reduce by almost two orders of magnitude (cf. Inset of Figure 5). Contribution due to the diffusion was observed at the lower frequency region.
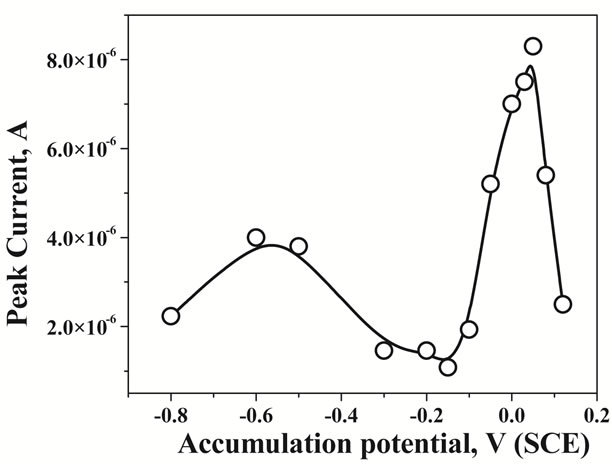
Figure 4. Effect of the variation of the accumulation potentials from −0.8 V to 0.12 V for 2 µg∙L−1 of U in 0.5 M KCl at pH 2.5.
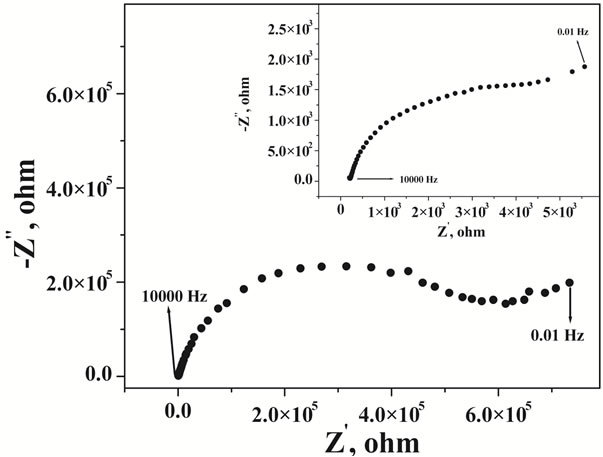
Figure 5. Nyquist plot of the bare carbon paste electrode, Inset: Nyquist plot of the mercury thin film modified carbon paste electrode at pH 2.5 with 1 × 10−5 M CAA and 10 µg∙L−1 of U.
The Nyquist plot was characterized as a suppressed semicircle could be due to the uneven nature of the surface. Nature of the Nyquist plot was not altered on mercury film deposition. In order to obtain the charge transfer characteristics with respect to the applied potential, impedance measurements were carried out at different potentials at a fixed frequency of 50 Hz. Corresponding results of potential dependency of the real part of impedance was shown in Figure 6 for CPE and in the inset of Figure 6 for MTFMCPE. General pattern of the real impedance with respect to the applied potential was observed to be similar for the bare CPE and the MTFMCPE. A clear maximum was observed at around 0.19 V (SCE) in both the cases and in either side of 0.19 V real impedance decreased sharply. This maximum corresponds to the potential of zero charge (PZC) of the electrochemical interface. At a more positive potential to 0.19 V the mercury thin film could be oxidized. A minimum in the real impedance measurements was observed at around 0.08 V indicated the possibility of maximum adsorption in that potential region. As the maximum adsorption potential region is at negative direction to the PZC, the adsorptive species would be adsorbing through the positively polarized group of the uranium-CAA complex. Thus a lower pH of 2.5 should be ideal as the ligand would be protonated at a lower pH and made the adsorption process more feasible. The real impedance value increased at a more negative potential to 0 V thus, choosing an accumulation potential more negative to 0 V would be detrimental to the uranium-CAA adsorption process. Since the adaption of the CAA as the complexing agent for the determination of uranium in adsorptive stripping voltammetry an accumulation potential of around 0.05 V has been used [6-11]. Present investigation from the impedance measurements and the PZC explained the importance and the feasibility of the accumulation potential in the range of 0 to 0.1 V. An adsorption potential of around 0.08 V optimized in the present case should be ideal for selective and sensitive determination of uranium through the uranium-CAA complex adsorption pre-concentration process using MTFMCPE. Nyquist plot of CNT modified carbon paste electrode was shown in Figure 7, with the results of mercury thin film deposited CNT modified presented at the inset. Resistance was observed to be reduced drastically on thin film deposition over the CNT modified carbon paste electrodes. Resistance of CNT modified carbon paste electrode without mercury film was similar to that of bare carbon paste electrode.
3.3. Voltammetric Response
The well-defined reduction peak at around −0.068 V was obtained in KCl solution using the optimized electrochemical and chemical parameters was due to the reduction of U-CAA complex (at the U-centre) (cf. Figure 8). With increase in the standard U concentration the voltammetric peak height increased proportionately. Effects of commonly occurring interfering agents like, Pb, Cu, Cd, Zn, Cr, V, Mo and Fe were studied. The method was found
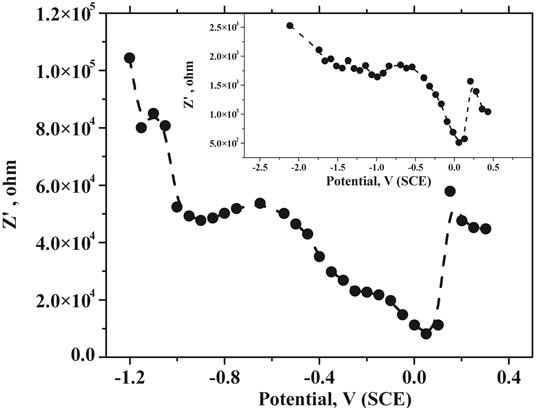
Figure 6. Plot of the change in the real impedance with the change in the applied potential V vs SCE at pH 2.5 with 1 × 10−5 M CAA and 10 µg∙L−1 of U.
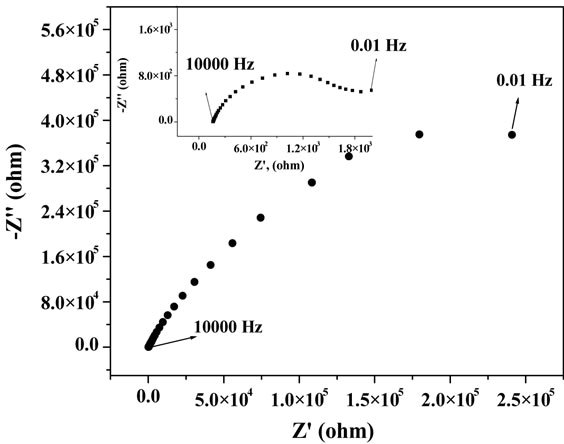
Figure 7. Nyquist plot of the CNT modified carbon paste electrode. Inset: Nyquist plot of the mercury thin film deposited CNT modified carbon paste electrode at pH 2.5 with 1 × 10−5 M CAA and 10 µg∙L−1 of U.
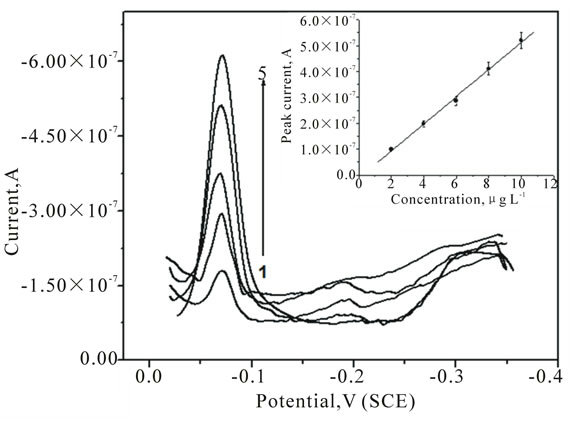
Figure 8. Adsorptive stripping voltammetric plots of U in 0.5 M KCl solution at pH 2.5 using MTFMCPE with the addition of 2, 4, 6, 8 and 10 µg∙L−1 of U standard solution.
to be free from interference due to the presence of Cu, Pb, Cd, Zn etc. at 104 fold higher concentrations than uranium. However, U stripping peak was found to be affected with the presence of Cr, V, Mo and Fe present at more than 500 times the concentration of uranium in KCl solution. Electrochemical response on MTFMCPE was found to be better compared to HMDE surface in terms of the shape of the peak [11]. The baseline was more stable on MTFMCPE compared to HMDE. Linear calibration plot was shown at the inset of Figure 8, observed to be linear. Three sigma detection limit of 0.52 μg∙L−1 was obtained with open circuit pre-concentration for 10 min. The modified carbon paste electrode was better option for the direct and also online determination of uranium in water samples due to their better stability compared to the HMDE. Additionally on MTFMCPE surface the UCAA complex adsorbed without application of any potential, this has an additional advantage on onsite determination.
Carbon paste electrode was surface modified using MWCNT by drop casting MWCNT-methanol suspension onto the carbon paste electrode surface. This mercury film was deposited by electrodeposition process onto the MWCNT modified carbon paste electrode surface. Electrodeposition was performed at −0.6 V deposition potential in 0.1 mM Hg2+ solution for 300 s. A less negative deposition potential was more appropriate in a MWCNT modified surface compared to the bare carbon paste surface due to the enhanced possibility of hydrogen evolution on MWCNT. This electrode was used as the working electrode for the adsorptive pre-concentration of uranium-CAA complex. For this electrode, the open circuit potential was not suitable for the adsorption pre-concentration thus a deposition potential of 0.05 V was applied for the pre-concentration on the electrode surface. Corresponding votammogram with increasing concentration of uranium was shown in Figure 9. Stripping peak was observed at −0.11 V, slightly negatively shifted compared to the previous case where MWCNT was not used. The negative shifting of the stripping is indicative of the stronger interaction of uranium-CAA complex on the MWCNT-mercury composite thin film surface. Stripping peak current was increasing proportionately with the addition of uranium standard concentration in the solution. The peak current was observed to be around two orders of magnitude higher on MWCNT modification. However, the stripping peaks were broadened on MWCNT modifications. The calibration plot was shown at the inset of Figure 9. Data points were fitted well with the linear correlation and the three sigma detection limit was obtained as 0.45 µg∙L−1. Linear dynamic range for the measurements has not improved on mercury thin films over mercury drop electrodes even with CNT modification. However, on real sample analysis accumulation time can be varied to obtain optimized signal.
3.4. Sample Analysis
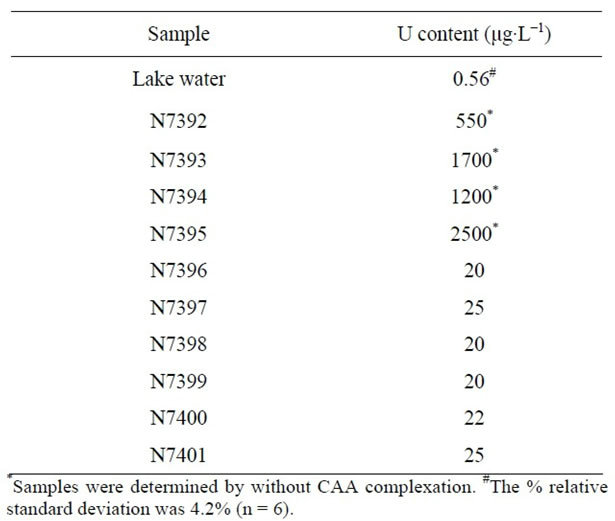
One lake water sample and some processed water samples were analysed using the MTFMCPE and the results were tabulated in Table 1. The contaminated water samples were coded from N7392-N7395 received from the Desalination Division of Bhabha Atomic Research Centre. A special recovery procedure was employed to recover the uranium from the contaminated water samples and the analysis result of the recovered water samples were coded as N7396-N7401. All the samples were analysed as received, no pre-treatment procedures were employed before analysis. Lake water sample was collected from Nainital Lake situated at the footstep of Himalayan range. This water is used for the drinking purpose and also used as the clean water source for the water quality
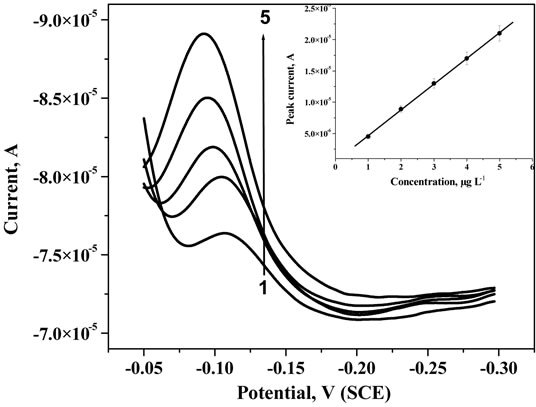
Figure 9. Adsorptive stripping voltammetric plots of U in 0.5 M KCl solution at pH 2.5 using MWCNT modified MTFMCPE with the addition of 1, 2, 3, 4 and 5 µg∙L−1 of U standard solution.
Table 1. Uranium content determined in lake water and contaminated water samples.
parameter.
4. Conclusion
A method was developed for the determination of uranium on mercury thin film modified carbon paste electrode using adsorptive cathodic stripping voltammetric method. Electrochemical impedance measurements were employed to investigate the adsorption preconcentration process. Three sigma detection limit of 0.52 μg∙L−1 was obtained with open circuit pre-concentration for 10 min using MTFMCPE. Three sigma detection limit was obtained as 0.45 µg∙L−1 with MWCNT modified MTFMCPE. The method was applied for the determination of uranium in sea lake water and contaminated water samples.
REFERENCES
- M. B. Gholivand, H. R. Nassab and H. Fazeli, “Cathodic Adsorptive Stripping Voltammetric Determination of Uranium(VI) Complexed with 2,6-Pyridinedicarboxylic Acid,” Talanta, Vol. 65, No. 1, 2005, pp. 62-66. doi:10.1016/j.talanta.2004.05.019
- T. P. Rao, Metilda and J. M. Gladis, “Preconcentration Techniques for Uranium(VI) and Thorium(IV) Prior to Analytical Determination—An Overview,” Talanta, Vol. 68, No. 4, 2006, pp. 1047-1064. doi:10.1016/j.talanta.2005.07.021
- E. Andreotti, A. Credaro, C. Dossi, A. Giuliani, D. Monticelli, M. Pellicciari and S. Sangiorgio, “Determination of Uranium Content in Water Using Cathodic Stripping Voltammetry and Gamma-Spectrometry,” Journal of Radioanalytical and Nuclear Chemistry, Vol. 277, No. 2, 2008, pp. 413-417. doi:10.1007/s10967-007-7018-1
- S. F. Marsh, M. R. Betts and J. E. Rein, “Determination of Submicromolar Amounts of Uranium (VI) by Complexometric Titration with Pyridine-2,6-Dicarboxylic Acid,” Analytica Chimica Acta, Vol. 119, No. 2, 1980, pp. 401- 404. doi:10.1016/S0003-2670(01)93644-X
- C. M. G. Van den Berg and Z. Q. Huang, “Determination of Uranium(VI) in Sea Water by Cathodic Stripping Voltammetry of Complexes with Catechol,” Analytica Chimica Acta, Vol. 164, 1984, pp. 209-222. doi:10.1016/S0003-2670(00)85632-9
- S. Sander and G. Henze, “Adsorption Voltammetric Techniques for the Determination as Complex Forming Reagent,” Fresenius’ Journal of Analytical Chemistry, Vol. 349, 1994, pp. 654-658. doi:10.1007/BF00323474
- S. Sander, W. Wagner and G. Henze, “Direct Determination of Uranium Traces by Adsorptive Stripping Voltammetry,” Analytica Chimica Acta, Vol. 305, No. 1-3, 1995, pp.154-158. doi:10.1016/0003-2670(94)00481-Z
- L. Lovotny, T. Navratil, S. Sander and P. Basova, “Electrocapillary Activity and Adsorptive Accumulation of U(VI)-Cupferron and U(VI)-Chloranilic Acid Complexes on Mercury Electrode,” Electroanalysis, Vol. 15, No. 21, 2003, pp. 1687-1692. doi:10.1002/elan.200302746
- S. Sander, “Simultaneous Adsorptive Stripping Voltammetric Determination of Molybdenum(VI), Uranium(VI), Vanadium(V), and Antimony(III),” Analytica Chimica Acta, Vol. 394, No. 1, 1999, pp. 81-89. doi:10.1016/S0003-2670(99)00218-4
- S. Sander, W. Wagner and G. Henze, “Possibilities for the Automated Determination of Trace Concentrations of Uranium in Water Samples by Adsorptive Stripping Voltammetry,” Analytica Chimica Acta, Vol. 349, No. 1-3, 1997, pp. 93-99. doi:10.1016/S0003-2670(96)00591-0
- A. K. Satpati, S. Sahoo, P. V. Ravindran and G. Venkateswaran, “Stripping Voltammetric Determination of Uranium Traces in Sea Water Samples: Effect of Surfactants on the Measurements,” Analytical Letters, Vol. 43, No. 4, 2010, pp. 1-14. doi:10.1080/00032710903406904
- J. Wang, R. Setiadji, L. Chen, J. Lu and S. Morten, “Automated System for Online Adsorptive Stripping Voltammetric Monitoring of Trace Levels of Uranium,” Electroanalysis, Vol. 4, No. 2, 1992, pp. 161-165. doi:10.1002/elan.1140040206
- J. Wang and R. Setiadji, “Selective Determination of Trace Uranium by Stripping Voltammetry Following Adsorptive Accumulation of the Uranium-Cupferron Complex,” Analytica Chimica Acta, Vol. 264, No. 2, 1992, pp. 205- 211. doi:10.1016/0003-2670(92)87007-8
- J. Wang, J. Lu and K. Olsen, “Adsorptive Stripping Voltammetry of Trace Uranium: Critical Comparison of Various Chelating Agents,” Analytica Chimica Acta, Vol. 292, No. 1-2, 1994, pp. 91-97. doi:10.1016/0003-2670(94)00067-0
- C. M. G. van den Berg and Z. Q. Huang, “Determination of Uranium(VI) in Sea Water by Cathodic Stripping Voltammetry of Complexes with Catechol,” Analytica Chimica Acta, Vol. 164, 1984, pp. 209-222. doi:10.1016/S0003-2670(00)85632-9
- O. A. Farghaly and M. A. Ghandour, “Cathodic Adsorptive Stripping Voltammetric Determination of Uranium with Potassium Hydrogen Phthalate,” Talanta, Vol. 49, No. 1, 1999, pp. 31-40. doi:10.1016/S0039-9140(98)00338-5
- K. W. Cha, C. H. Park and S. H. Park, “Simultaneous Determination of Trace Uranium(VI) and Zinc(II) by Adsorptive Cathodic Stripping Voltammetry with Aluminon Ligand,” Talanta, Vol. 52, No. 6, 2000, pp. 983-989. doi:10.1016/S0039-9140(00)00472-0
- F. Vernon, T. W. Kyffin and K. M. Nyo, “The Spectrophotometric Determination of Uranium with 8-Hydroxyquinoline,” Analytica Chimica Acta, Vol. 87, No. 2, 1976, pp. 491-493. doi:10.1016/S0003-2670(01)82280-7
- M. B. Gholivand and H. Rashidi Nassab, “Highly Selective Adsorptive Cathodic Stripping Voltammetric Determination of Uranium in the Presence of Pyromellitic Acid,” Electroanalysis, Vol. 17, No. 8, 2005, pp. 719-723. doi:10.1002/elan.200403145
- A. Komersova, M. Bartos, K. Kalcher and K. Vytras, “Indirect Determination of Uranium Traces by Adsorptive Stripping Voltammetry with Arsenazo III,” Electroanalysis, Vol. 10, No. 6, 1998, pp. 442-445. doi:10.1002/(SICI)1521-4109(199805)10:6<442::AID-ELAN442>3.0.CO;2-R
- M. Mlakar, “Square-Wave Voltammetry of Uranyl-Humate Complex,” Analytica Chimica Acta, Vol. 276, No. 2, 1993, pp. 367-372. doi:10.1016/0003-2670(93)80406-B
- J. Wang, J. Li, D. D. Larson and K. Olsen, “Voltammetric Sensor for Uranium Based on the Propyl Gallate-Modified Carbon Paste Electrode,” Electroanalysis, Vol. 7, No. 3, 1995, pp. 247-250. doi:10.1002/elan.1140070308
- H. Alemu and B. S. Chandravanshi, “Electrochemical Behavior of N-Phenylcinnamohydroxamic Acid Incorporated into Carbon Paste Electrode and Adsorbed Metal Ions,” Electroanalysis, Vol. 10, No. 2, 1998, pp. 116-120. doi:10.1002/(SICI)1521-4109(199802)10:2<116::AID-ELAN116>3.0.CO;2-B
- W. Yantasee, Y. Lin, G. E. Fryxell and Z. Wang, “Carbon Paste Electrode Modified with Carbamoylphosphonic Acid Functionalized Mesoporous Silica: A New MercuryFree Sensor for Uranium Detection,” Electroanalysis, Vol. 16, No. 10, 2004, pp. 870-873. doi:10.1002/elan.200302868
- A. Becker, H. Tobias and D. Mandler, “Electrochemical Determination of Uranyl Ions Using a Self-Assembled Monolayer,” Analytic Chemistry, Vol. 81, No. 20, 2009, pp. 8627-8631. doi:10.1021/ac901092t
- S. Betelu, C. Vautrn-Ul, J. Ly and A. Chausse, “ScreenPrinted Electrografted Electrode for Trace Uranium Analysis,” Talanta, Vol. 80, No. 1, 2009, pp. 372-3769. doi:10.1016/j.talanta.2009.06.076
- P. Ugo, B. Ballarin, S. Daniele and G. A. Mazzocchin, “Electrochemical Behaviour and Preconcentration of Uranyl(VI) at Nafion-Coated Glassy Carbon Electrodes,” Journal of Electroanalytical Chemistry, Vol. 324, No. 1-2, 1992, pp. 145-159. doi:10.1016/0022-0728(92)80042-3
- A. Becker, H. Tobias, Z. Porat and D. Mandler, “Detection of Uranium(VI) in Aqueous Solution by a Calix arene Modified Electrode,” Journal of Electroanalytical Chemistry, Vol. 621, No. 2, 2008, pp. 214-221. doi:10.1016/j.jelechem.2007.11.009
- C. J. Evans and G. P. Nicholson, “A Self-Assembled Electrochemical Sensor for Uranium,” Sensors and Actuators B: Chemistry, Vol. 105, No. 2, 2005, pp. 204-207. doi:10.1016/j.snb.2004.06.001
- A. N. Golikand, M. Asgari, M. G. Maragheh and E. Lohrasbi, “Carbon Nanotube-Modified Glassy Carbon Electrode for Anodic Stripping Voltammetric Detection of Uranyle,” Journal of Applied Electrochemistry, Vol. 39, No. 1, 2009, pp. 65-70. doi:10.1007/s10800-008-9647-7
- R. K. Shervedani and S. A. Mozaffari, “Preparation and Electrochemical Characterization of a New Nanosensor Based on Self-Assembled Monolayer of Cysteamine Functionalized with Phosphate Groups,” Surface and Coatings Technology, Vol. 198, No. 1-3, 2005, pp. 123-128. doi:10.1016/j.surfcoat.2004.10.025
- P. A. Dimovasilis and M. I. Prodromidis, “An Electrochemical Sensor for Trace Uranium Determination Based on 6-O-Palmitoyl-l-Ascorbic Acid-Modified Graphite Electrodes,” Sensors and Actuators B, Vol. 156, No. 2, 2011, pp. 689-694. doi:10.1016/j.snb.2011.02.019
NOTES
*Corresponding author.