Open Journal of Modern Neurosurgery
Vol.3 No.4(2013), Article ID:38002,22 pages DOI:10.4236/ojmn.2013.34016
New Developments in Drug Therapy and Research of Cerebral Vasospasm
1Department of Neurosurgery, Sana Klinikum Offenbach, Akademisches Lehrkrankenhaus der Universität, Frankfurt, Germany
2Department of Neurosurgery, Klinikum Traunstein, Akademisches Lehrkrankenhaus der Universität, München, Germany
Email: *neurosurgery@t-online.de
Copyright © 2013 Eleftherios Archavlis et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received August 27, 2013; revised September 27, 2013; accepted October 4, 2013
Keywords: Cerebral Vasospasm; Drug Therapy; Subarachnoid Hemorrhage; Delayed Cerebral Ischemia; New Developments; Treatment
ABSTRACT
In this manuscript a comprehensive coverage of recent developments in the drug therapy of vasospasm while providing the background information that neuroscientists need to understand its rationale. The range of new agents available for treatment of cerebral vasospasm is expanding rapidly along with rapid advances in pharmacology and physiology that are uncovering the mechanisms of this disease. Although there are many publications for treatment of cerebral vasospasm, most are focusing on different aspects of vasospasm treatment and many have limited value due to insufficient quality. Moreover, the complexity of this, in many cases deleterious condition, is enormous and the information needed to understand drug effects is accordingly often not readily available in a single source. A number of pharmacological and medical therapies are currently in use or being investigated in an attempt to reverse cerebral vasospasm, but only a few have proven to be useful. Current research efforts promise the eventual production of new medical therapies. At last, recommendations for the use of different treatment stages based on currently available clinical data are provided.
1. Definitions and Characteristics of Cerebral Vasospasm
Cerebral vasospasm is a condition of morphologic narrowing of cerebral arteries that is most commonly seen following aneurysmal subarachnoid haemorrhage (SAH) [1,2], but may also follow other intracranial hemorrhages, like intraventricular haemorrhage from arteriovenous malformations (AVM) [3], SAH of unknown etiology [4], traumatic brain injury with or without SAH [5], brain surgery and may be associated with hypothalamic injury, lumbar puncture, infection and preeclampsia [6]. The risk of mortality and morbidity after aneurysmal subarachnoid hemorrhage is one of the most discussed issues among neuroscientists. Although, considerable advances in diagnosis and surgical and/or interventional treatment have contributed to decrease the case fatality rate after SAH [7-9], cerebral vasospasm (CVS), remains a partially unsolved and not fully understood issue [10]. A recently proposed definition of symptomatic vasospasm based on treatment of cerebral vasospasm after SAH in Japan include: 1) the presence of neurological worsening; 2) no other identifiable cause of neurological worsening; and 3) confirmation of vasospasm by medical examinations [11]. The presence of vasospasm has been correlated with a threefold increase in mortality during the first 2 weeks after SAH [12].
The majority of research was focused on the assumption, CVS to be solely responsible for delayed cerebral ischemia (DCI). Today, it is well accepted that not all patients with CVS develop DCI. Inversely, DCI can occur in absence of CVS [13,14]. Recent review of the literature indicates that CVS is not the only cause of DCI and that the entire picture of DCI is multifactorial [10]. Mechanisms postulated more recently in the pathogenesis of DCI and consecutive poor outcome after SAH include early brain injury [15], cortical spreading depolarization [16,17], inflammation [18-21] and microthrombosis [7,15], as well as CVS [22]. The presence of vasospasm at the time of initial presentation of a ruptured aneurysm poses special considerations for management. The narrowing of critical blood vessels reduces blood flow, compromising perfusion to the point of ischemia and infarction. Classically, CVS has been associated with DCI occurring 4 - 10 days after ictus, which ultimately leads to cerebral infarction associated with morbidity and mortality.
The amount of subarachnoid blood correlates with the degree of vasospasm [23,24]. Angiographic evidence of vasospasm is apparent in 70% of patients with SAH [1]. However, angiographic vasospasm does not always correlate with clinical symptomatic vasospasm. Symptomatic vasospasm is diagnosed when a decreased level of consciousness and/or focal deficit occurs after SAH in the presence of angiographic vasospasm and the absence from other factors reducing cerebral perfusion. Clinical manifestation of vasospasm is present in one third of patients [14]. However, comparison of studies and validation of treatment impact is hampered due to the remaining inconsistency in terminology and definition, mainly mixing radiographic evidence of CVS with clinical features of DCI. There is an on-going debate about this issue, however, beside functional outcome, cerebral infarction on imaging studies seem to be the most appropriate measure of DCI [14,25].
Although being the subject of substantial research interest, effective and/or causative prophylaxis and treatment are not available. Therefore, CVS might be regarded as the remaining—potentially treatable—cause of morbidity and mortality in contemporary neurocritical care medicine.
2. Pathophysiology of Cerebral Vasospasm
Several mechanisms are identified causing acute as well as prolonged vascular spasm and are outlined below (Figure 1). Endothelin-1 (ET-1) is a major vasoconstric-
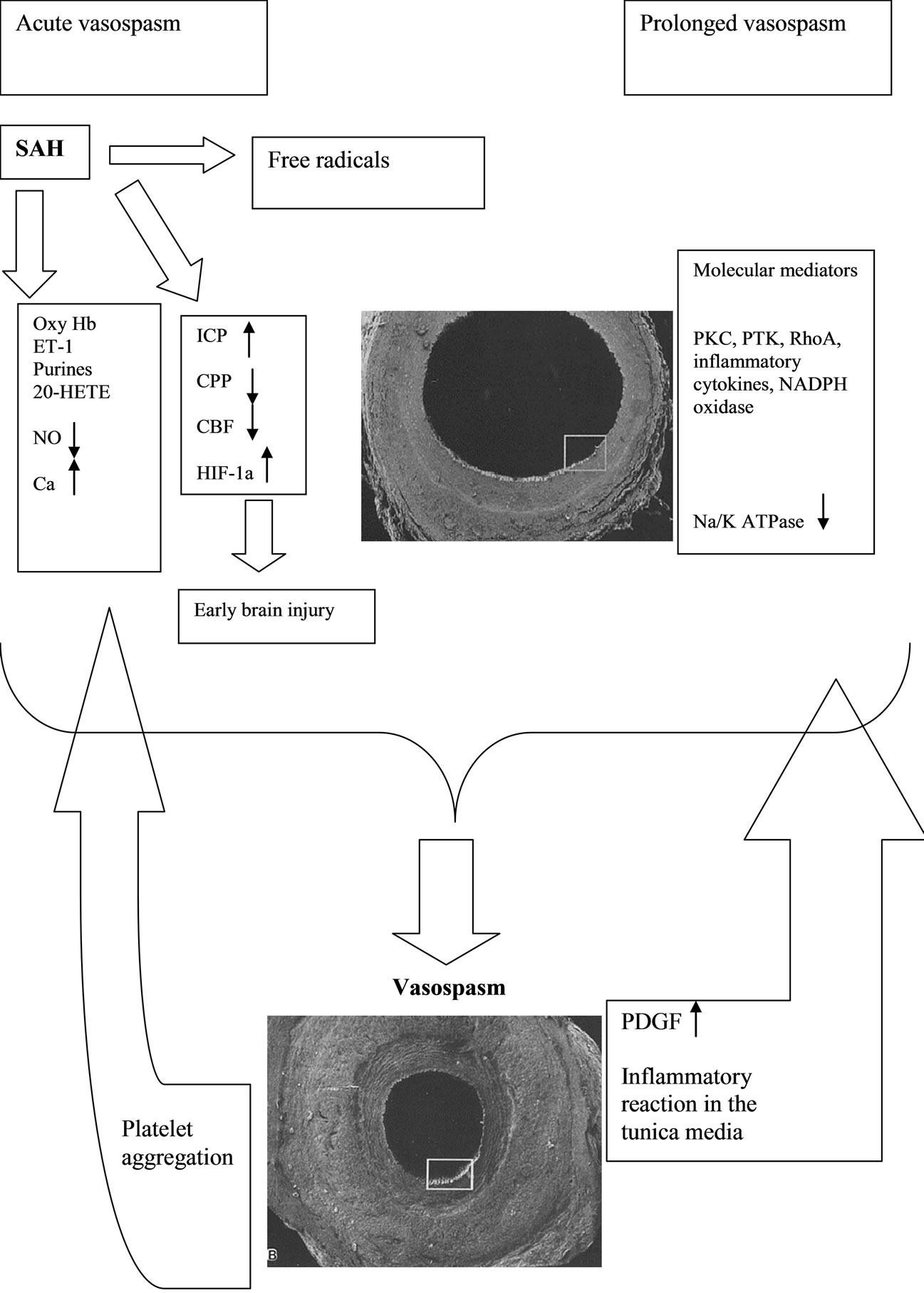
Figure 1. Humoral and molecular mechanisms of cerebral vasospasm in patients with SAH.
tor that induces cerebral vasospasm following SAH. The impact on ET-1-induced arterial narrowing, however, has not been determined. In addition, the concentrations of ET-1 that induce ischemia in experiments are by magnitudes higher than those measured in SAH patients [26, 27]. It may suggest that ET-1 is potentiated by other factors playing a consistent pathophysiological role in the development of vasospasm [27]. However, another plausible explanation is that not only the level of ET-1 but also an increase in the vascular density of ET and 5-hydroxytryptamine 1B (5HT1B) receptors underlie vascular narrowing after SAH [28]. The levels of ET-1 nearly equivalent to physiological levels are sufficient to induce submaximal contraction of cerebral arteries excised from rats after SAH. It is also known that ischemia upregulates endothelin receptors in the brain [29].
In the acute phase, cerebral vascular spasm occurs through a calcium-dependent mechanism. In response to hemoglobin exposure, the intracellular level of calcium increases and drives myosin light chain kinase (MLCK) to phosphorylate the myosin light chains, which produces transient contraction. However, the mechanism of the prolonged phase of vasospasm is largely calcium-independent. Near resting levels of calcium were detected during protracted vasospasm. This phenomenon can be caused by calcium sensitization mediated by several kinases acting in concert with ET-1 released by endothelial cells, astrocytes, and leukocytes in response to hemoglobin and acute cerebral ischemia [31]. ET-1 in general produces lasting receptor stimulation. Hirata et al. [32] first demonstrated that ET-1 can occupy its receptors for up to 48 h. The major involvement of Rho A kinase in the process of prolonged contraction has been also postulated [33]. ET-1 activates Rho A, which in turn activates Rho kinase. The activated Rho kinase enhances myosin light chain (MLC) phosphorylation through the inhibition of myosin phosphatase targeting subunit (MYPT1) of the myosin light chain phosphatase (MLCP) [34]. The increased phosphorylation of MLC is the key event which results in prolonged contraction without elevation in intracellular calcium [35]. Kikkawa et al. demonstrated the mechanism which potentiates the Ca2+ sensitization after SAH. The increased expression of protein kinase C (PKC), Rho-associated protein kinase (ROCK), PKC-potentiated phosphatase inhibitor of 17 kDa (CPI-17) and Myosin phosphatase target subunit 1 (MYPT1) as well the upregulation of ET(A) receptor expression are suggested to underlie the enhanced and prolonged Ca(2+) sensitization induced by ET-1 [36]. While vascular smooth muscle contraction normally involves phosphorylation of the MLC at Ser19, diohosphorylation at Ser19 and Thr18 has been observed in a canine model of cerebral vasospasm [37]. Indeed, diphosphorylation has been observed in association with several instances of pathological hypercotractility [38]. Diphosphorylation has been shown to reduce the rates of myosin dephosphorylation and relaxation, which could explain the prolonged contraction in response to certain physiological contractile stimuli under pathological conditions of cerebral vasospasm, intimal hyparplasia and hypertension [39]. The following diagram based on several relevant review articles of the literature, includes the signal transduction pathways to protein kinase in Ca2+- induced contractions and Ca2+ sensitization of contraction and relaxation [40-42] (Figure 2).
The reactive oxygen species (ROS) produced from enzymatic sources and formed during hemoglobin autoxidation constitute a major etiologic factor underlying the development of cerebral vasospasm after SAH (Figure 3).
Concordantly, antioxidants have been shown to attenuate arterial narrowing in experimental SAH produced by autologous blood injection [43,44]. There have been several mechanisms proposed to explain how ROS may contribute to cerebral vasospasm after SAH. ROS oxidize bilirubin to bilirubin oxidation products (BOXes) [42]. BOXes inhibit eNOS and, due to the reduced availability of NO, impair vasodilation mechanism. Nitric oxide is a major vasodilator produced principally by endothelial cells and acts through the stimulation of guanyl cyclase that produces cyclic guanosine monophosphate (cGMP). The relaxing effect of these compounds involves the activation of cGMP-dependent protein kinases (cGKI) with subsequent dephosphorylation of myosin light chains [45]. Therefore, the endothelial dysfunction and, more so, injury resulting in the reduction of NO level may contribute to vascular constriction after SAH. In addition, reactive oxygen species, together with thrombin and clotting cascade components, stimulate the production of 20-hydroxy-eicosatetraenoic acid, a vasoconstrictor metabolite of arachidonic acid which blocks calcium-activated potassium channels, thereby leading to a decrease in cerebral blood flow after SAH [46]. One of the major sources of free radicals after subarachnoid bleeding is NADPH oxidaseproducing superoxide. The superoxide radical can combine with nitric oxide to form peroxynitrite, which targets nitric oxide synthase (NOS) [47]. Hyperbaric oxygen (2.8 ATA for 2 h) has been shown to inhibit NADPH oxidase activity/expression and reduce the level of lipid peroxidation products in the cerebral tissues after SAH [48]. The pharmacological inhibitors of NADPH oxidase, diphenyleneiodonium and apocynin, potently reduced cerebral vasospasm in the rat model of SAH [49,50].
The reduced bioavailability of NO can be also caused by the negative regulation of NOS by PKC activated after SAH [51]. Furthermore, the depletion of nitric oxide occurs due to scavenging by hemoglobin (sink effect) and the damage of nitric oxide-secreting cells in the vas-
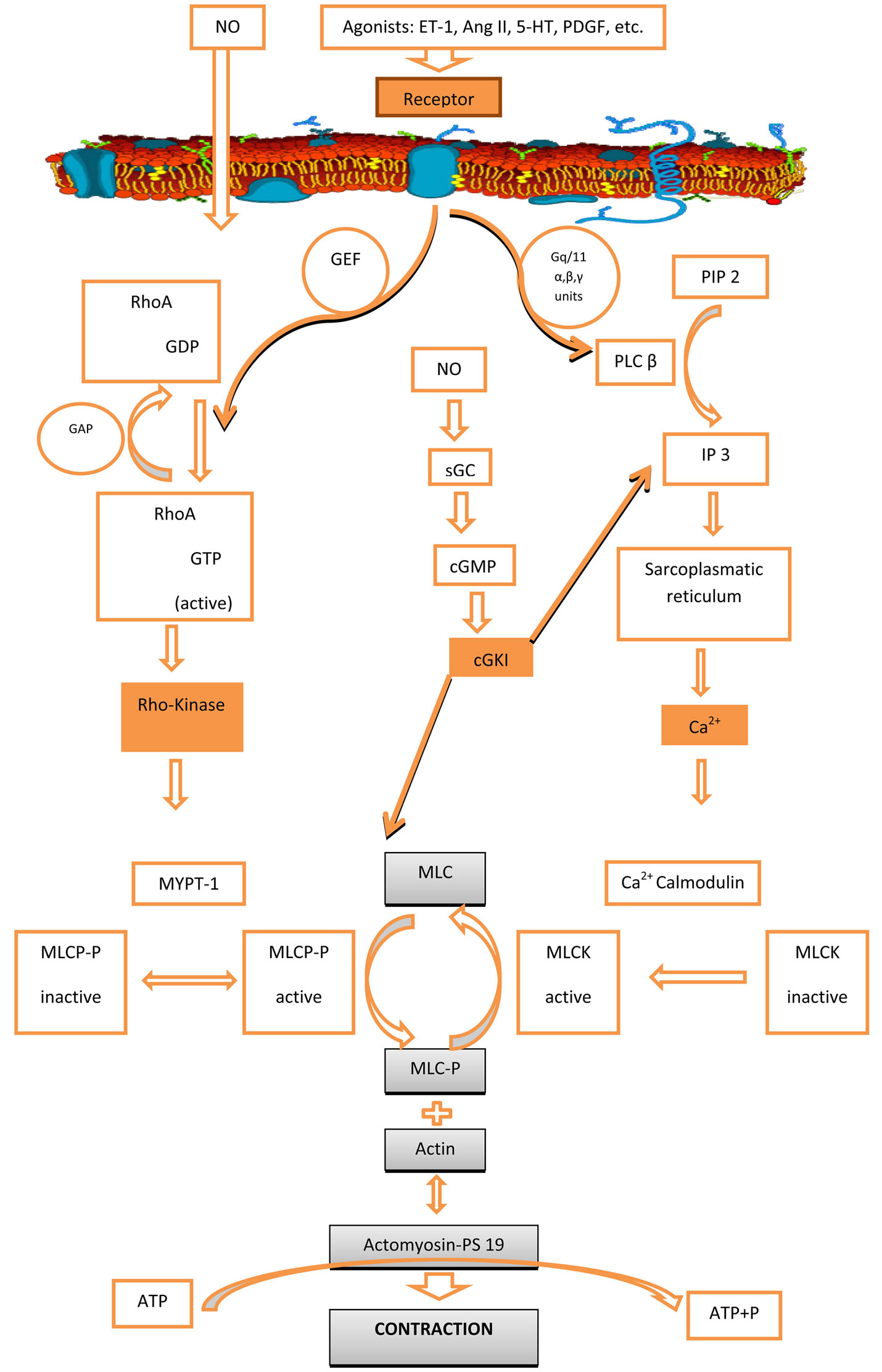
Figure 2. Signal transduction pathways leading to smooth muscle contraction. Abbreviations: cGKI: cGMP-dependent protein kinases, cGMP: cyclic guanosine monophosphate, ET-1: endothelin-1, 5-HT: 5-hydroxytryptamine (serotonin), GAP: GTPase activating proteins, GEF: guanine nucleotide exchange factors, Gq/11:members of the Gq/111 subfamily of heterotrimeric G proteins with α, β, γ subunits, IP3: inositol 1,4,5-triphosphate, MLCK: myosin light chain kinase, MLCP: myosin light chain phosphatase, MYPT-1: myosin phosphatase target subunit 1, PIP2: phosphatidylinositol 4,5-bisphosphatase, PLCβ: phospholipase Cβ isoform, PS19: phosphoserine at position 19 of LC20.
cular adventitia.
SAH triggers a cascade of molecular events leading to endothelial apoptosis [50]. The expression of active caspase-3 and the presence of DNA strand breaks have been detected in endothelial cells of spastic cerebral arteries after experimental SAH [50]. Electron microscopic in-
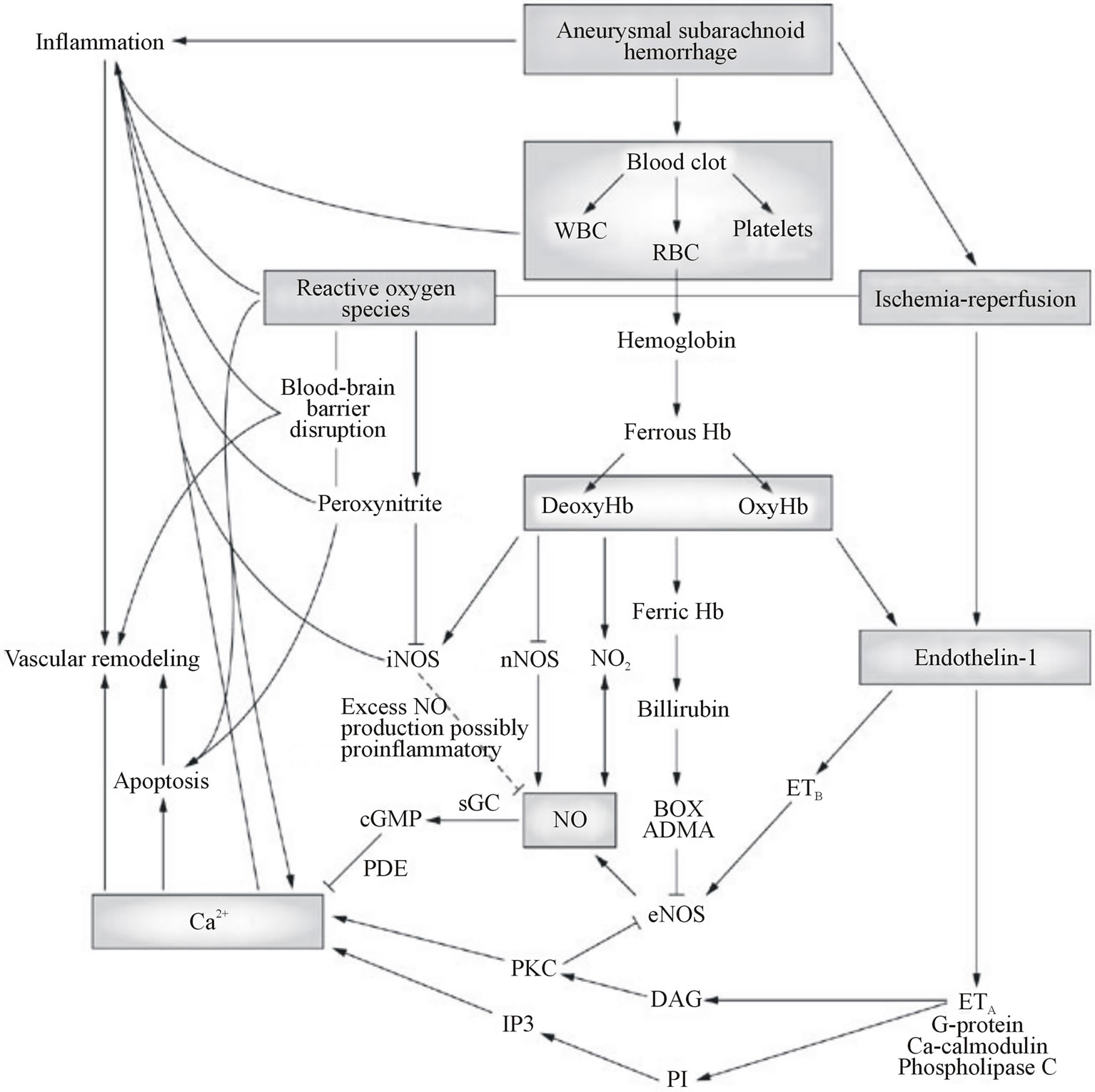
Figure 3: Extracellular processes leading to vasospasm by production of reactive oxygen species (ROS) and elevation of intracellular calcium level in smooth muscle cells. Abbreviations: ADMA, asymmetric dimethylarginine; Box, bilirubin oxidation products; DAG, diacylglycerol; eNOS, endothelial nitric oxide synthase; ETA, endothelin receptor, type A; ETB, endothelin receptor, type B; Hb, hemoglobin; ET-1, Endothelin 1; iNOS, inducible nitric oxide synthase; IP3, inositol 1,4,5-triphosphate; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; NO2, nitrogen dioxide; PDE, phosphodiesterase; PI, phosphatidylinositol; PKC, protein kinase C; RBC, red blood cells; sGC, soluble guanylyl cyclase; WBC, white blood cells.
vestigations revealed apoptotic changes in endothelial cells of cerebral arteries collected from a patient who died after suffering severe CVS caused by aneurysm rupture [51]. The loss of endothelial cells producing nitric oxide can affect the fragile balance between vasoconstrictors and vasodilators acting on the vascular wall. Studies have shown that the endothelial injury after SAH is associated with inflammatory mediators. Tumor necrosis factor α (TNF-α) and interleukin-1β (IL-1β) have been shown to exert a pro-apoptotic effect on cultured cerebral microvascular endothelial cells [52]. In the experimental settings, caspase inhibitors have been shown to attenuate CVS after SAH and to reduce levels of inflammatory mediators including IL-1β [53]. Other studies further corroborated these results by showing that treatment with a broad caspase inhibitor decreased TNF- α expression in arterial wall and attenuated CVS [50].
When the layer of endothelial cells is damaged, the underlying vascular smooth muscle cells are exposed to intraluminally acting spasmogens like ET-1. In addition, ET-1 released by invading mononuclear leukocytes may exert a potent vasoconstrictive effect abluminally.
Brain inflammation plays an important role in the development of cerebral vasospasm after SAH [54-56]. The extravasated blood induces local inflammatory reaction in the closest vicinity of cerebral arteries. In this setting, cerebral vasospasm develops in response to spasmogens such as ET-1 released from invading leukocytes [57] or thromboxane A2 and serotonin, both released from platelets [58,59]. It has been demonstrated that SAH increases the tissue expression of several inflammatory mediators, including intercellular adhesion molecule 1 (ICAM-1) and TNFα, in proximity to extravasated blood [60]. E-selectin , a soluble cell adhesion molecule plays a principal role in the development of cerebral vasospasm. Other proteins participating in cell adhesion that have been implicated in the mechanisms of CVS include ICAM-1, CD18, and L-selectin, all found elevated after SAH [61,62].
It has been postulated that the severity of the acute brain injury will have a profound impact on delayed vasospasm and long-term outcomes after subarachnoid hemorrhage. Several MAPK—including ERK1/2, JNK, and p38, with known roles in the development of vasospasm—were found activated in both brain parenchyma and cerebral vascular tissues acutely after SAH. One study found activated JNK in the cerebral vessels both on days 1 and 7 after SAH [40]. These findings may indicate that JNK activation is required but alone is not sufficient for the development of arterial spasm, which usually does not occur on day 1. Treatment with JNK inhibitor SP600125 reduced angiographic and morphological vasospasm after SAH [40].
Except for minor leaks, SAH has a profound ischemic impact on the brain [63-65]. It has been clinically demonstrated that SAH decreases brain tissue oxygen pressure and pH [66]. The acute cerebral ischemia after SAH has a protective aspect as it can reduce bleeding into the subarachnoid space. However, if the ischemia is prolonged beyond several minutes, it can trigger an injurious cascade of molecular events orchestrated by hypoxia inducible factor-1 (HIF-1) [67]. HIF-1 consists of regulatory α-subunit and constitutively expressed β-subunit (aka aryl hydrocarbon receptor nuclear translocator) [68]. The excessive activation of HIF-1 results in the overexpression of its target genes such as vascular endothelial growth factor (VEGF), responsible for increased BBB permeability, or BCL2/adenovirus E1B 19-kDa proteininteracting protein 3 (BNIP3) and Nip3-like protein X, both mediating apoptosis [69]. Thus, the hypoxic brain injury at the onset of SAH can induce apoptosis of endothelial cells in large arteries by the activation of HIF-1α and BNIP3 [70]. In addition, the pro-apoptotic p53 protein, known to be stabilized by HIF-1α, has been found upregulated after SAH [71]. Since p53 was found elevated in the vasospastic basilar arteries, it has been suggested that p53 may play an important role in the etiology of vasospasm with relation to SAH [63]. The results of other studies may also lend support to the hypothesis that HIF-1α plays an important role in CVS mechanisms. Rats treated with the HIF-1α inhibitor, 2-methoxyestradiol (2ME2), showed reduced expression of VEGF, BNIP3, and proliferating cell nuclear antigen in the basilar arteries. This change was associated with attenuation of vasospasm in the basilar artery after SAH [72]. The same group showed that cerebral vasospasm can also be reduced through pharmacological inhibition of p53 [73].
The aggravation of ischemic brain injury after SAH can occur due to cortical spreading depression (CSD) characterized by periodically generated waves of cortical depolarization accompanied by decreases in CBF. The presence of both events has been detected after SAH [73]. Their propagation in the cerebral cortex may exacerbate ischemic lesions formed as a result of arterial narrowing. The microarterial spasm and cell necrosis caused by CSD may further aggravate the neurological status of SAH patients [74]. Several factors inducing CSD have been identified in experimental studies including ET-1, oxyhemoglobin, and potassium ions [75]. In addition, one of the major triggers for CSD is a decrease in activity of Na+/K+-ATPase. It has been demonstrated that even its incomplete functional inactivation can cause SD-like anoxic depolarization in the hippocampus [76]. Interestingly, SAH decreases Na+/K+-ATPase activity in the synaptosomal membranes, collected 2 h after the induction of subarachnoid hemorrhage [77]. In an attempt to preserve the enzymatic activity of Na+/K+-ATPase in experimental SAH, Yufu et al. [77] used HBO at 2 ATA for 1 h, started at 30 min after hemorrhage induction. HBO significantly ameliorated a decrease in Na+/K+- ATPase activity, which allowed authors to suggest that HBO may be considered as a beneficial treatment for subarachnoid hemorrhage.
The treatment options which fall into the following categories are presented in detail.
3. Protection of CNS from Ischemic Injury
3.1. Calcium Channel Blockers
Calcium channel blockers also known as calcium antagonists block the “slow channel” of calcium influx which reduces the contraction of smooth and cardiac muscle, but does not affect skeletal muscle. It is thus theorized that the abnormal contraction of vascular smooth muscle that may contribute to vasospasm may be mitigated by the administration of calcium channel blockers. Oral nimodipine, a dihydropyridine calcium channel blocker, is the only drug to reduce the incidence of DCI and the risk of poor outcome in an evidence based manner (class I, level of evidence A) [7,10]. Nicardipine is another dihydropyridine calcium antagonist that displays regional selectivity on vascular smooth muscle. However, both showed a reduction of DCI without clear effect on CVS. Calcium channel blockers are more beneficial in neuroprotection than in preventing vasospasm. Results of the British aneurysm nimodipine trial demonstrated a reduction from 33% to 22% in the occurrence of cerebral infarction in patients treated with nimodipine, and a 40% reduction in poor outcomes [78, 79]. In a metaanalysis of nimodipine treatment after SAH, Barker and Ogilvy [80] demonstrated that the prophylactic use of this drug is effective in increasing the odds of a good outcome after SAH. This efficacy was both statistically and clinically significant. In addition, in a retrospective analysis comparing cost of treatment with increase in life expectancy it was shown that nimodipine is extremely cost-effective and is associated with minimal adverse effects [81]. Their beneficial impact derives from the fact that the red blood cell deformability and thus blood rheology is improved, calcium entry in ischemic cells and thus cerebral infarction is prevented [82]. Other possible effects may be an anti-platelet aggregation [83] and dilatation of collateral leptomeningeal arteries [84]. Therefore, a marked shift to new concepts for treatment and/or prophylaxis can be observed, rather addressing DCI than focusing on CVS.
3.2. NMDA Receptor Antagonists
N-methyl-D-aspartic acid (NMDA) antagonists address cortical spreading depolarization. NMDA is an amino acid derivative which acts as a specific agonist at the NMDA receptor mimicking the action of glutamate, the neurotransmitter which normally acts at that receptor. Unlike glutamate, NMDA only binds to and regulates the NMDA receptor and has no effect on other glutamate receptors (such as those for AMPA and kainate). The role of spreading depression, spreading depolarization and spreading ischemia is an emerging research field in the context of DCI [16,17]. One major factor contributing to neuronal death in penumbra is thought to be spreading depression (SD) waves, which are accompanied by depolarization of both astrocytes and neurons, collapsed ionic gradients and impaired ATP generation. Recently, seizure and brain ischemia [16] were reported to induce Cyclooxygenase-2 (COX-2) mRNA or protein. COX-2 is a key enzyme in conversion of arachidonic acid to prostaglandins and thromboxanes. The induction is reduced by NMDA receptor antagonists that block SD waves. Currently there is a multicenter diagnostic phase III single-arm study underway to determine the correlation of spreading depolarization and DCI: depolarisations in ischemia after subarachnoid hemorrhage 1 (DISCHARGE-1) [16]. In theory, other potential approaches for the suppression of cortical spreading depression include correction of cerebral glucose and hyperoxia [17].
3.3. Free Radical Scavengers
Free radical induced lipid peroxidation has been identified as a potentially important contributor to both the arterial narrowing of vasospasm and the final cascade of ischemic cell death. The 21-aminosteroid tirilazad mesylate was developed as a potent inhibitor of lipid peroxidation. Indeed, in experimental models of SAH and focal cerebral ischemia tirilazad has been shown to ameliorate vasospasm and improve cerebral blood flow as well as reduce the size of cerebral infarction. In addition, preliminary studies with this drug have shown it to be safe and unassociated with side effects such as hypotension, mental status changes or glucocorticoid toxicities. Tirilazad mesylate is a nonglucocorticoid 21-aminosteroid that exerts its anti-lipid peroxidation action through cooperative mechanisms: a radical scavenging action (i.e., chemocal antioxidant effect) and a physicochemical interaction with the cell membrane that serves to decrease membrane fluidity (i.e., membrane stabilization). Several multicenter, randomized, double-blind, vehicle-controled clinical trials have been organized throughout the world to test the efficacy of tirilazad mesylate in patients with aneurysmal SAH—including a North American study. The primary hypothesis to be tested was whether treatment with tirilazad reduced symptomatic vasospasm and improved overall outcome 3 months after SAH. Kassell et al. showed in this trial a reduction in symptomatic vasospasm in the group that received 6 mg/kg per day of tirilazad. However, the difference was not statistically significant and the benefits were predominantly shown in men rather than than in women. The lack of statistical significance and the less benefit in women may be explained by increased clearance of tirilazad in middleaged women (as compared to men) as well as use of phenytoin (administration of phenytoin or Dilantin with tirilazad increases clearance to approximately 50% in healthy male volunteers). Thus, additional phase III studies should be done to evaluate the efficacy of higher doses of tirilazad in the treatment of SAH. As a further support of the potential beneficial role of tirilazad, it has been shown to be of benefit in other forms of brain injury (which may involve lipid peroxidation) including traumatic brain injury. Results from a rabbit subarachnoid haemorrhage model have indicated that free radical reactions mediated by oxyHb may play an important role in the pathogenesis of cerebral vasospasm through the expression of Rho-kinase and this experimental technique looks promising for future application [86].
4. Direct Pharmacological Arterial Dilatation
4.1. ETA Antagonists (Clazosentan) and ETA/B Antagonists
Endothelin-1 has been the primary suspect in the pathogenesis of CVS [1]. The interplay between ET and NO is critical to the maintenance of cerebral vascular dilation. Endothelin-1 is an extremely potent vasoconstrictor, and investigators have demonstrated a close correlation between cerebral vasospasm and increasing levels of ET-1 in CSF and plasma. Human cerebral arteries express two types of ET-1 recep tors, ETA and ETB [87] Activation of the smooth-muscle ETA receptor induces vasoconstriction, whereas a release of NO resulting in vasodilation is mediated by the ETB receptor located on the endothelium. Therefore, the use of Clazosentan (Actelion Pharmaceuticals, Allschwil, Switzerland), a selective endothelin-1A receptor antagonist has been in the focus of research. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage 1 (CONSCIOUS-1), a randomized placebo-controlled phase IIa study, demonstrated a convincing and significant reduction of angiographic CVS in a dose-dependent manner. However, the reduction in the number of DCI was insignificant. A trend toward better clinical outcome could only be found after modifications implemented in the posthoc analysis [1,10,88,89]. Two phase III trials (CONSCIOUS-2 and CONSCIOUS-3) have been consecutively carried out in patients treated with aneurysm clipping and coiling, respectively. In the CONSCIOUS-2 trial, patients were randomly assigned to Clazosentan (5 mg/h) or placebo for up to 14 days [88,89]. The results showed that Clazosentan had no significant effect on mortality or CVS-related morbidity 6 weeks after SAH (primary endpoint). Clazosentan was only associated with an insignificant 17% relative risk reduction in the primary endpoint, in terms of a reduction in use of rescue therapy for angiographic CVS. There was no significant effect on poor functional outcome at week 12, defined as extended Glasgow outcome scale (GOS) at least four (secondary endpoint). As of adverse events, lung complications, anemia and hypotension were described [88]. The authors pointed out that the lack of improvement might have occurred because processes other than CVS contribute to DCI and consecutive poor outcome, which are not improved by prevention of CVS. Although the results of the CONSCIOUS-3 trial are not published yet, the results of CONSCIOUS-1 and CONSCIOUS-2 definitively called the importance of CVS in DCI and outcome into question and underlined the need for a paradigm shift.
4.2. Intraarterial Agents (Papaverine, Nimodipine)
The effects of intraarterial drug injections are considered as short-lived and either repeated several times, requiring multiple arterial catheterizations, or performed in combination with angioplasty, which in the hand of experienced interventional neuroradiologists, may have more profound effects. Papaverine is a nonspecific phosphodiesterase (PDE) inhibitor that has also been applied topically during surgery. When given intra-arterially, it effectively dilates the spastic arteries. However, the effect is short-lived and vasospasm usually recurs. Angiographic resolution of diameter reduction and angiographically assessed capillary blood flow together differed not significantly between papaverine and nimodipine treated groups. Papaverine infusion dilated all angiographic demonstrable vessels while nimodipine infusion was ineffective in 16% of the patients [90]. Capillary flow on preand post-infusion angiographies was not different between these two groups. The clinical implications remain to be established. A multimodal approach, perhaps combining different agents for intraarterial infusion in SAH patients, needs to be evaluated.
Endovascular treatment with the phosphodiesterase III inhibitor milrinone was studied in several non-randomized trials and demonstrated an efficacy to reverse cerebral vasospasm [91,92]. Phosphodiesterase III inhibitors combine vasodilating and inotropic properties, resulting from the increase in cAMP in the cytosol of vascular smooth muscle cells and cardiomyocytes. Among these agents, milrinone is widely used to treat patients with acute heart failure. The combination of potent vasodilatation with reinforcement of inotropy is also very attractive to treat aSAH. Milrinone should be compared randomly and blindly to another vasodilator such as nimodipidine in large trials to evaluate which drug is the best for chemical angioplasty in aSAH patients.
4.3. Intrathecal Thrombolytics
A consistently demonstrated risk factor for the prediction of CVS is the amount of subarachnoid blood, visualized in computed tomography [7,10,93]. Therefore, there is a strong rationale for interventions aimed at the clearance of blood. A very recent meta-analysis of five randomized controlled trials, enrolling 465 patients showed significant reductions in the development of poor outcome, DCI and CVS without any increment in haemorrhagic or infectious complications [93]. Either tissue plasminogen activator or urokinase was applied. However, due to the wide variation of the methodology, the results should be interpreted with caution. A routine use is currently not recommended [10,93].
4.4. Intracisternal Agents (NPRIs)
Local pharmacological therapies, in terms of direct intrathecal application, are promising. However, there is a risk of infection and the need of a surgical access. As systemic administration of vasoactive drugs has been associated with significant side effects and insufficient efficacy, intrathecal administration of nicardipine prolonged-release implants (NPRI) has been developed. Intraoperative implantation of nicardipine pro-longed-release implants, so-called pellets, into the basal cistern in close contact to the proximal cerebral vasculature has been reported to reduce the occurrence of CVS [94,95]. These nicardipine-loaded polymers release the drug over 14 days at a high and constant concentration. In a randomized, double-blind phase IIa study, the reduction in occurrence of CVS and cerebral infarction, as well as improvement in clinical outcome has been shown [94]. The disadvantage is that patients treated with endovascular coiling cannot profit from this therapy. The recently reported feasibility of intraventricular use of nicardipine prolonged-release implants might be a possibility to overcome this problem [96]. Further studies using nicardipine prolonged-release implants are warranted.
4.5. Sympatholytics
The cerebral vasculature, in particular the pial vessels, is densely supplied with noradrenergic sympathetic nerve fibers mainly originating in the superior cervical ganglion, accompanying the carotid artery, and projecting into the ipsilateral hemisphere. Intracerebral vessels constrict in response to cervical sympathetic stimulation and dilate when these fibers are interrupted. Activation of the sympathetic system leading to an elevation in circulating catecholamines has been well documented in patients after SAH [97]. This activation has been implicated in the development of cerebral vasospasm, yet there is no unequivocal linking evidence. Percutaneous blockade of the sympathetic chain at the superior cervical ganglion interrupts the innervation of adrenergic cerebral nerve fibers and can exclude any potential detrimental effect sympathetic hyperactivity may have in the occurrence and evolution of vasospasm. Cervical sympathectomy may contribute to improve cerebral perfusion when used as an adjunct to standard procedures or when standard procedures are not applicable or contraindicated [97]. A larger-scale trial that includes a control group comparison is not yet undertaken to firmly establish the value of the cervical block.
4.6. Inhibition of Inflammatory Reactions
The pathogenesis of cerebral vasospasm is closely associated with inflammation and immune response in arterial walls. In a n experimental research study, the peroxisome proliferator-activated receptor (PPAR) gamma agonist Rosiglitazone suppressed the SAH-induced inflammatory responses in basilar arteries by inhibiting the Toll-like receptor (TLR)4 signalling. Furthermore, rosiglitazone could attenuate cerebral vasospasm following SAH. Therefore, the authors suggested that PPAR-gamma agonists may be potential therapeutic agents for cerebral vasospasm [98]. Many animal studies claim reduced vasospasm in subarachnoid hemorrhage with the use of anti-inflammatory agents, radical scavengers, lipid peroxidation inhibitors, iron chelators, NSAIDs, glucocorticoids, and serine protease inhibitors, but these effects are not yet confirmed in human trials, where symptomatic vasospasm and outcome are the major endpoints [99]. Despite recent work on penetrating vessel constriction, there is a paucity of studies on inflammatory markers in the microcirculation [99].
4.7. Nitric Oxide Donors
The alteration in nitric oxide production is an undoubtedly important mechanism in the pathophysiology of CVS and DCI. The reduced availability of endovascular and perivascular nitric oxide has been shown to contribute to development of CVS in terms of deficient vessel relaxation [7,10,100]. In theory, there is a strong rationale for treatment with nitric oxide donors, and experimental studies showed promising results. However, the short half-life of nitric oxide demands its therapeutic substitution via nitric oxide donors. Classic nitric oxide donors such as sodium nitroprusside or nitroglycerin are challenging in the clinical setting due to their undesirable side effects, most notably their induction of hypotension [7,10,100]. Different modes of application have been tested, transdermal, i.v., intra-arterial, inhalational and intrathecal, with some promising results [7,10,100]. Currently, there is no evidence suggesting routine use of nitric oxide donors. If routine use would be required, sodium nitrite (NaNO2) could be a possible candidate. NaNO2 has been shown to prevent and more recently to reverse CVS, both in a primate SAH model [101]. Furthermore, the safety and feasibility of long-term i.v. NaNO2 infusion has been reported in healthy volunteers [102]. The latter result calls for a clinical trial in near future.
4.8. Inhibitors of Rho-Associated Kinase (ROCK)
The identification of Rho-associated kinase (ROCK) as a mediator of inflammation and oxidative stress provides insight into the development of new therapies. There are two isoforms of Rho-kinase, ROCK1 and ROCK2, and they have different functions with ROCK1 for circulating inflammatory cells and ROCK2 for vascular smooth muscle cells. Accumulating evidence suggests that ROCK inhibitors are useful for the treatmment of several cardiovascular diseases in humans [103]. The activity and the expression of ROCKs are enhanced at the inflammatory/arteriosclerotic coronary lesions. ROCK inhibitors significantly reduce cyclophilin A secretion from vascular smooth muscle cells in animals. Blocking the malignant cycle that augments oxygen ions and peroxides through cyclophilin A secretion may be partially involved in the beneficial effect of ROCK inhibitors [104]. Fasudil, the best known ROCK inhibitor has been reported to reduce CVS [105], however, without any effect on clinical outcome [10]. Recent animal studies suggest that Hydroxyfasudil, an active metabolite of Fasudil, is a selective ROCK inhibitor and contributes to the potency of fasudil as a therapeutic agent against cerebral vasospasm and hyperviscosity [106]. Velat et al. reviwed the RCTs and meta-analyses in the literature regarding the treatment and prevention of cerebral vasospasm following aneurysmal SAH and concluded that as far as the effectiveness of fasudil is conserned, further multicenter RCTs need to be performed [107]. Zhao et al. conducted a randomized, open trial to compare the efficacy and safety of vasudil compared with nimodipine. The results of this study suggested that fasudil was equally safe and eequally or more effective than nimodipine for the prevention of cerebral vasospasm and subsuquent ischemic injury in patients undergoing surgery for SAH [108]. Nakamura et al. showed that the selective intra-arterial infusion of fasudil was signifficantly effective for the treatment of delayed vasospasm and more beneficial than the nonselective treatment [109]. A very recent meta-analysis, analyzing eight eligible studies with a total of 843 patients, found a significant reduction in the occurrence of CVS and cerebral infarction as well as an improvement in clinical outcome as assessed by GOS [110]. However, the results should be interpreted with caution, as the impact of combined medications in those trials was not considered, and the number of trials and samples were quite limited. Further-more, there is a considerable risk of bias. More potent and selective ROCK inhibitors, e.g. Y-27632, have emerged more recently [103], which may prove more useful therapeutically as ROCK may also be involved in the central mechanisms of sympathetic nerve activity [104].
5. Indirect Arterial Dilatation through Hyperdynamic Therapy
At present, the mainstay of medical treatment of cerebral vasospasm, alongside calcium channel blockers, is hyperdynamic or triple-H therapy, referring to hypervolemia, hypertension, and hemodilution. The rationale behind this treatment is that maintenance of high circulating blood volume, increased perfusion pressures, and decreased blood viscosity will enhance CBF in the setting of vasoconstriction. Triple H therapy has been widely used in the management of CVS. On the basis of the current knowledge, there is insufficient evidence for the effectiveness of prophylactic triple H. In particular hypervolemia and hemodilution might not produce a net benefit and there is evidence of harm using aggressive hydration [7,111]. There is an accumulating body of evidence against the utility of hemodilution in management of CVS [112,113]. Induced hypertension is strongly recommended in case of suspected DCI, as anecdotal reports linked induced hypertension with neurologic improvement, however, the evidence is moderate [112]. Another concern is the optimal hemoglobin concentration in patients with high risk for CVS and/or DCI. Higher hemoglobin has been associated with better outcome, whereas packed red blood cell transfusions with worse outcome [113]. Naidech et al. [114] performed a prospective pilot trial of higher goal hemoglobin. The preliminary results showed higher goal hemoglobin in patients with SAH to be well tolerated and feasible. A phase II study is currently recruiting patients (ClinicalTrials.gov identi-fier NCT00968227). Mechanical hemodynamic support may be an additional tool to improve cerebral hypoperfusion. The use of NeuroFlo (CoAxia Inc., Maple Grove, Minnesota, USA) has been proposed recently [115]. By inflation of two balloons in the aorta, 35% - 50% of the blood flow is redirected to the brain. Although the feasibility and technical safety has been reported, the impact on outcome is unknown.
6. Drugs with Rheologic Properties
The improvement of rheologic properties of intravascular blood to enhance perfusion of ischemic zones is a generally accepted concept. Mannitol, low molecular weight dextran, albumin and perfluorocarbons are some of the drugs included to achieve volume expansion. Mannitol has been widely accepted by treating physicians in helping control elevated intracranial pressure (ICP), and control of ICP has been shown to improve outcome. Mannitol is the most commonly utilized agent for this indication. It is widely available, effective, and has a low side-effect profile. Large doses for long term periods have been shown to be safe, but its usefulness is limited by elevation of serum osmolarity and potential alteration in renal function, and its efficacy seems to diminish with repeated doses. Rebound intracranial hypertension has been reported after discontinuation of large doses. The maintainance of the circulation volume might have a therapeutic effect in order prevent cerebral vasospasm. Mori et al. have focused on maintaining the circulation volume in order to prevent cerebral vasospasm. But to maintain the central venous pressure, huge infusion volume was required, and hyponatremia was frequently observed due to natriuresis and osmotic diuresis. Excessive natriuresis and diuresis cannot be managed through sodium and water replacement, since sodium replacement induces further natriuresis and diuresis (desalination), and water replacement induces hyponatremia. They therefore administered fludrocortisone and hydrocortisone to inhibit excessive natriuresis and diuresis. The efficacy of sodium reabsorption therapy was extremely high to maintain the circulation volume [116].
7. Novel Agents
7.1. Magnesium Sulphate
As magnesium (Mg2+) is the fourth most common cation in the body and the second most common intracellular cation after potassium, it has a fundamental role as a co-factor in numerous enzymatic reactions involving energy metabolism and nucleic acid synthesis. It is also involved in several processes including hormone receptor binding, gating of calcium channels, transmebrane ion flux and regulation of adenylate cyclase; muscle contraction; neuronal activity; control of vasomotor tone; cardiac excitability and neurotransmitter release. In many of its actions has been likened to a physiological calcium antagonist [117]. Magnesium sulphate has been considered to reduce the incidence of CVS and DCI, as shown in animal studies [7,10,118]. Phase I studies have established the feasibility of continuous intravenous (i.v.) MgSO4 infusion, and a couple of randomized phase II studies showed potential benefits [7,10,118,119]. Nevertheless, their interpretation and comparison had its limitation due to small sample size, different primary outcome measures and different dosage regimens [118]. In the first phase III trial, the intra-venous magnesium sulfate for aneurysmal subarachnoid hemorrhage (IMASH) study, patients were randomly assigned to MgSO4 infusion titrated to a serum Mg2+ concentration twice the baseline or to a placebo for 10 - 14 days [119]. The primary outcome analysis showed no significant difference in the number of patients with favorable outcome at 6 months, defined as extended GOS five to eight. Secondary outcome analyses—based on modified Rankin scale, Barthel index, short form 36 and clinical CVS—did not show any significant differences either. The most recent meta-analysis with six eligible studies and a total of 875 patients could not lend support to a beneficial effect [118]. Thus, current evidence does not support the routine use of continuous i.v. MgSO4 infusion [112,118]. The results from another phase III study, the magnesium in aneurysmal subarachnoid hemorrhage II (MASH-II) study are expected soon. In this study, MgSO4 64 mmol/day or placebo is continuously administered until day 20 after hemorrhage [120]. The latter study might shed more light on the efficacy of i.v. MgSO4 infusion. As of possible side effects, hypo-tension, hypocalcaemia and bradycardia have been discussed [7,10,118].
An interesting approach in the future might be the direct intrathecal administration of MgSO4, as one possible explanation for the lack of action could be the low cerebrospinal fluid penetration of peripherally infused MgSO4 [119]. Animal studies using intracisternal MgSO4 therapy showed promising results [121].
7.2. Statins
The use of statins in the acute phase after SAH has been proposed, as statins might ameliorate glutamate-mediated excitotoxicity, attenuate production of reactive oxygen species, upregulate endothelial nitric oxide synthase, and diminish inflammatory reaction by modulation of the cytokine response. The possibility that statins could act via prevention of geranylgeralnylation of the small GTPase RhoA (which is required for RhoA to activate ROCK) should be considered. Two small, randomized phase II trials treated by simvastatin and pravastatin, respectively, could show a reduction of CVS and occurrence of DCI, though subsequent studies could not reproduce the results [7,10]. A very recent meta-analysis including six randomized controlled trials reported a significant reduction in DCI with statins, whereas the effect on CVS was more difficult to determine, due to differences in definitions used among those studies. Analyzing only the four high-quality trials, treatment with statins reduced DCI as well as mortality [122]. As of side effects, asymptomatic elevation in liver function has been reported, though these side effects were not significantly different compared with the placebo group. On the basis of the current evidence, immediate statins therapy appears to be well tolerated, with some beneficial evidence [112,122]. There are no results of a phase III trial available yet. One phase III trial is currently under-way: The simvastatin in aneurysmal subarachnoid hemorrhage (STASH) trial assesses the effects of simvastatin given at a 40 mg dose for 21 days versus placebo (http://www.stashtrial.com) [7,10].
7.3. Sildenafil Citrate
Nitric oxide (NO) is a primary endogenous vasodilator that affects smooth muscle relaxation. NO regulates the relaxation of vascular smooth muscle cells through activation of soluble guanylate cyclase. The cyclase converts guanosine triphosphate to cyclic guanosine monophosphate (cGMP) in the cells of cerebral arteries. Accumulation of cGMP relaxes the smooth muscles and its reduction leads to vasoconstriction. On the contrary, selective Phosphodiesterase (PDE) inhibitors prevent the hydrolysis of cGMP and thereby mediate vasodilatationThus, the amount of cGMP in the vascular smooth muscle cells is influenced by both NO and PDE. Currently, 11 families of PDEs have been identified in humans. PDE1 and PDE5 are present in vascular smooth muscle cells. Brain tissue has PDE1, PDE2, PDE4, and PDE10 [123]. Sildenafil has good selectivity for PDE5, but the selectivity is not exclusive as it has some action on PDE1, PDE2, and PDE4. The selectivity ratio for PDE5 to PDE1, PDE2, and PDE4 is 1:80:19,000:2057, respectively [124]. Sildanefil was originally used for angina and later for erectile dysfunction. It has been used in pulmonary hypertension, Raynaud’s phenomenon and vertebrobasilar insufficiency [123]. The mechanism of action of sildenafil may be more complex. Rats administered sildenafil in the lateral ventricles had remarkable tachycardia without significant change in basal arterial pressure [124]. Sildenafil elicited an increase in sympathetic nerve activity that is not baroreflex mediated, suggesting that this drug is able to elicit an autonomic imbalance of central origin. It has been shown that inhibitors of PDE2, PDE4, PDE5, PDE9, and PDE10 improve a wide range of cognitive processes, including information processing, attention, learning, memory, executive functioning, and response inhibition, in various behavioral models within different species. It is argued that it is unlikely that blood flow is the mechanism underlying these procognitive effects. It is felt that long-term potentiation appears to be a better substrate for the cognition-enhancing properties of PDE inhibitors [123]. Nitrates, which increase cGMP by increasing NO, have been used in cerebral vasospasm by intrathecal and intraventricular routes [123]. The approach is invasive with accompanying complications and infection. Administering oral PDE inhibitor in the form of sildenafil is a novel approach. It is easier to administer with less side effects. Sildenafil citrate is documented to be useful to treat cerebral vasospasm in animal studies [124]. The initial results of oral sildenafil in human subjects showed that it can be useful in severe refractory vasospasm. But, this study lacks outcome measures and there is no control group. Larger studies should be carried out to find out the efficacy of sildenafil in refractory vasospasm.
8. Established Methods and Other Treatments
8.1. Lumbar Drainage
Another approach to address the clotting of the subarachnoid blood is the early application of a lumbar drainage. Preliminary studies showed a markedly diminished incidence of CVS and improvement in clinical outcome [7,125]. One hypothesis claims that the likelihood of angiographic vasospasm to occur is related to the amount of blood in the basal cisterns. According to this consideration, one prophylactic strategy is to remove as much of this blood as early as possible. If clipping of the aneurysm is performed this can be achieved intraoperatively by opening the terminal lamina and irrigating the blood from the basal cisterns. Albeit promising, studies addressing the efficacy of this measure show inconclusive results [126]. This approach is not feasible if the aneurysm is secured using an endovascular approach. Excess removal of cerebral spinal fluid (CSF) via an external ventricular drain fails to prevent vasospasm and may lead to a higher incidence of posthemorrhagic shunt dependency [127]. Supposedly this is because after aneurysmal SAH, the blood settles and clots in the basal cisterns and therefore only CSF, being more lightweight, is removed via the ventricular drain. Application of a lumbar drain has been proposed as an alternative approach to address clotting of the blood in the basal cisterns. In two retrospective studies in patients after aneurysmal SAH, the safety of this approach was shown [7,125]. One of these studies addressed the radiologic and clinical outcome after surgical clipping, while the other addressed the outcome after endovascular coiling. Both studies led to a markedly diminished incidence of angiographic vasospasm and improvement in clinical outcome measured by the Glasgow Outcome Scale (GOS). Therefore two prospective randomized studies addressing the efficacy of this approach are currently in progress: lumbar drainage in subarachnoid hemorrhage (LUMAS) (ClinicalTrials.gov identifier NCT00842049) and early lumbar cerebrospinal fluid drainage in aneurysmal subarachnoid hemorrhage (EARLYDRAIN) [125].
8.2. Direct Mechanical Vasodilatation (Angioplasty)
Anecdotal reports described a potential benefit from endovascular therapy in treating CVS [7,128-130]. These include intraarterial papaverine, verapamil or nicardipine administration (among others) and balloon angioplasty. On the basis of the recent literature review, endovascular intervention is recommended in CVS refractory to medical management only, however, the level of evidence is low [7,128-130]. A controlled randomized study is near completion that compares invasive endovascular therapy with conventional therapy after proof of perfusion relevant CVS: invasive diagnostic and therapeutic management of cerebral vasospasm after aneurysmatic subarachnoid hemorrhage (IMCVS) (ClinicalTrials.gov identifier NCT01400360). As of novel treatment strategies, Albanese et al. [131] reported about a prolonged intraarterial verapamil infusion through an indwelling microcatheter. This strategy might overcome the common problem of the transient nature of endovascular therapies. Concerning balloon angioplasty, controversy regarding the best balloon (compliant versus noncompliant) has been reported. A recent study could not find any difference between compliant and noncompliant balloons for therapeutic angioplasty in preventing recurrence or need for repetitive angioplasty in patients with CVS [132]. The constant development and improvement in balloon technology might influence the success of balloon angioplasty.
8.3. Surgical and Endovascular Aneurysm Treatments
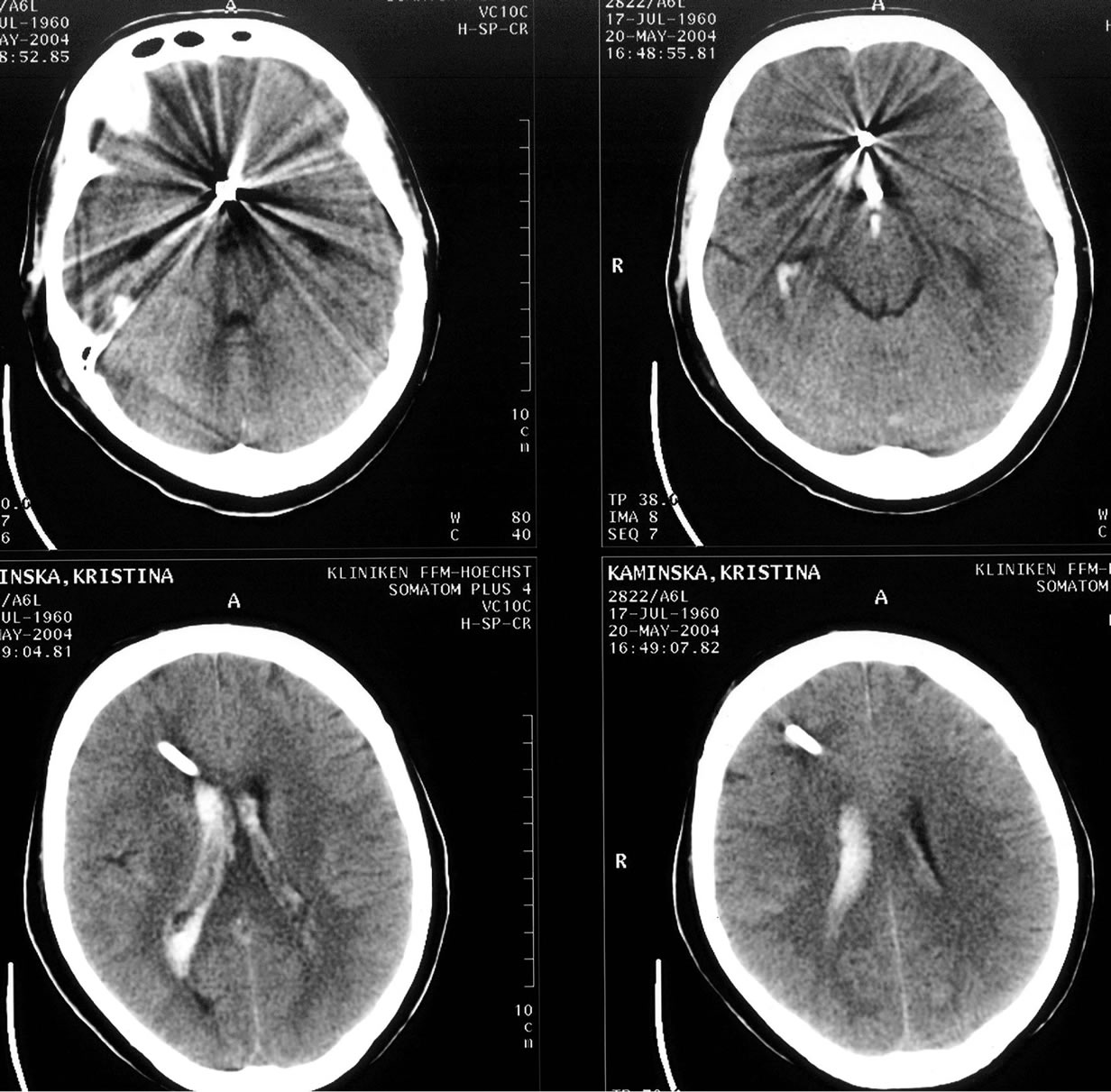
It is still controversial if the selection of treatment modality (clipping or coiling) affects cerebral vasospasm development following aSAH. Although several recent studies have suggested that the incidence of vasospasm after aSAH is lower in patients undergoing coiling as compared with clipping [133], other studies have had conflicting results, showing no influence on the incidence or duration of vasospasm and no significant difference in outcome at discharge or follow up [134,135]. The concept that coiling, as a less invasive form of treatment, can protect against vasospasm, has not a justified level of evidence. As a matter of fact, vasospasm does not only develop on the arterial territories from aneurysmatic compromised arteries, but also on neighbouring and even on far away located areas (Figure 4).
Particularly in cases with additional mass occupying hematomas, the initial surgical treatment not only reduces the increased intracranial pressure, but also in many cases prevents the development of vasospasm (Figure 5).
8.4. Kinetic Therapy
As mentioned before, there is a strong rationale for interventions aimed at the clearance of subarachnoid blood. A kinetic therapy using a head shaker or lateral rotational therapy in combination with local thrombolytic therapy has been described [7,136,137]. A recent analysis of three randomized studies, including a combination of thrombolytic and kinetic treatment, revealed a decrease in DCI, CVS and better clinical outcome in two studies [136,137]. No further progress in this therapy concept was reported recently.
8.5. Other Treatments
Other systemic drug therapies recently evaluated or currently in evaluation, include dantrolene [138], erythropoietin [10,139] and complementary Chinese herbs [140]. Although microthrombosis and increase of procoagulant factors have been discussed as additional factors contributing to DCI, preliminary clinical trials that target the coagulation system could not show a clear benefit. A systemic review in the Cochrane Database showed a trend toward better outcome. It has been discussed that hemorrhagic complications of these drugs might have counterbalanced the beneficial effects [7,10]. Therefore, more specific strategies are required. No novel treatment modalities addressing the coagulation system have been evaluated more recently. The inflammatory response with expression of cytokines, in particular interleukin 6 and leukocyte adhesion molecules, has been discussed to be involved in the pathogenesis of DCI and CVS [15,18,21]. Blocking or antagonizing cytokines and/or adhesion molecules might be promising treatment concepts, which need to be evaluated [19]. Signal transduction components mediating vasoconstrictor receptor
 (a)
(a)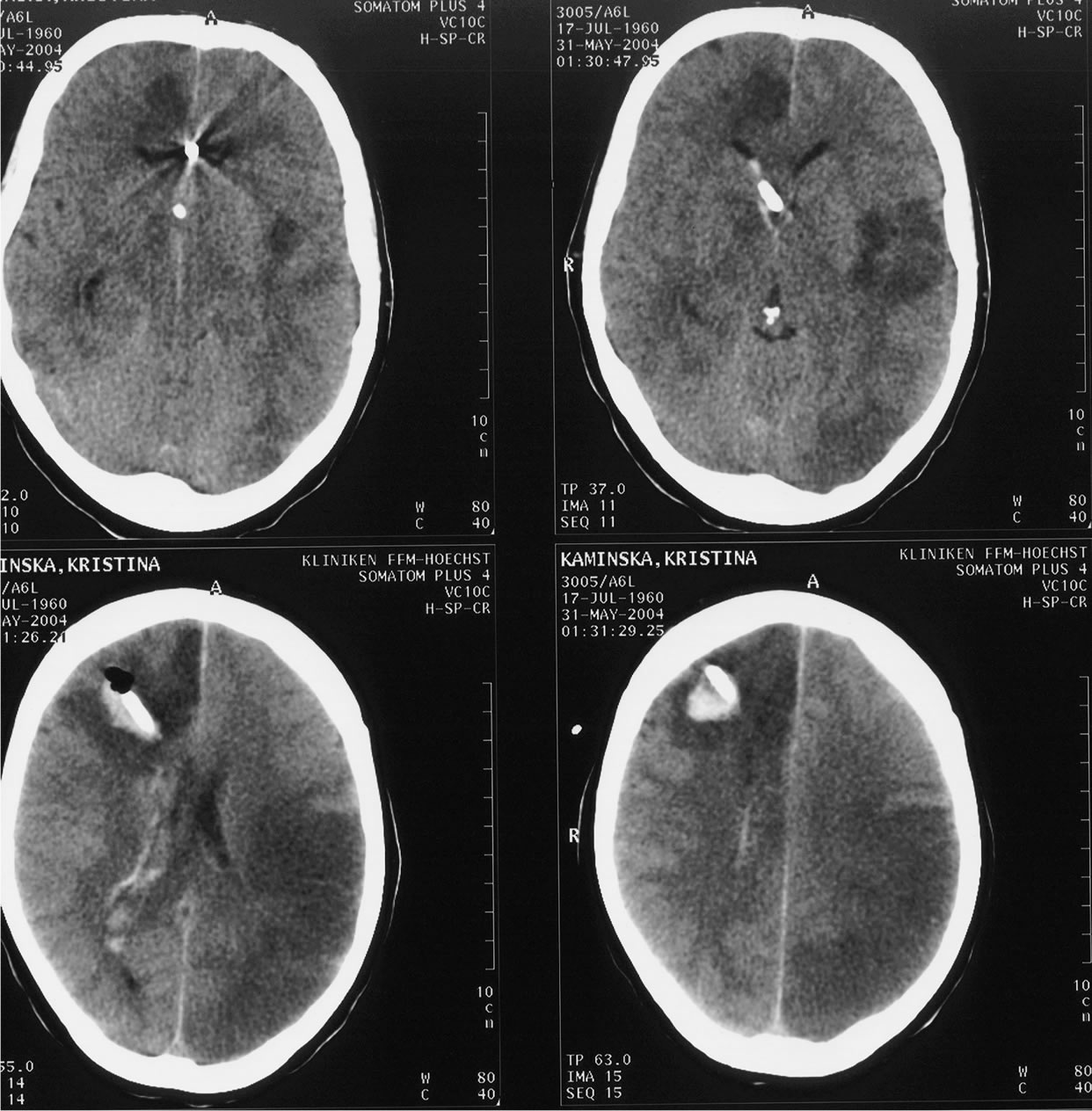 (b)
(b)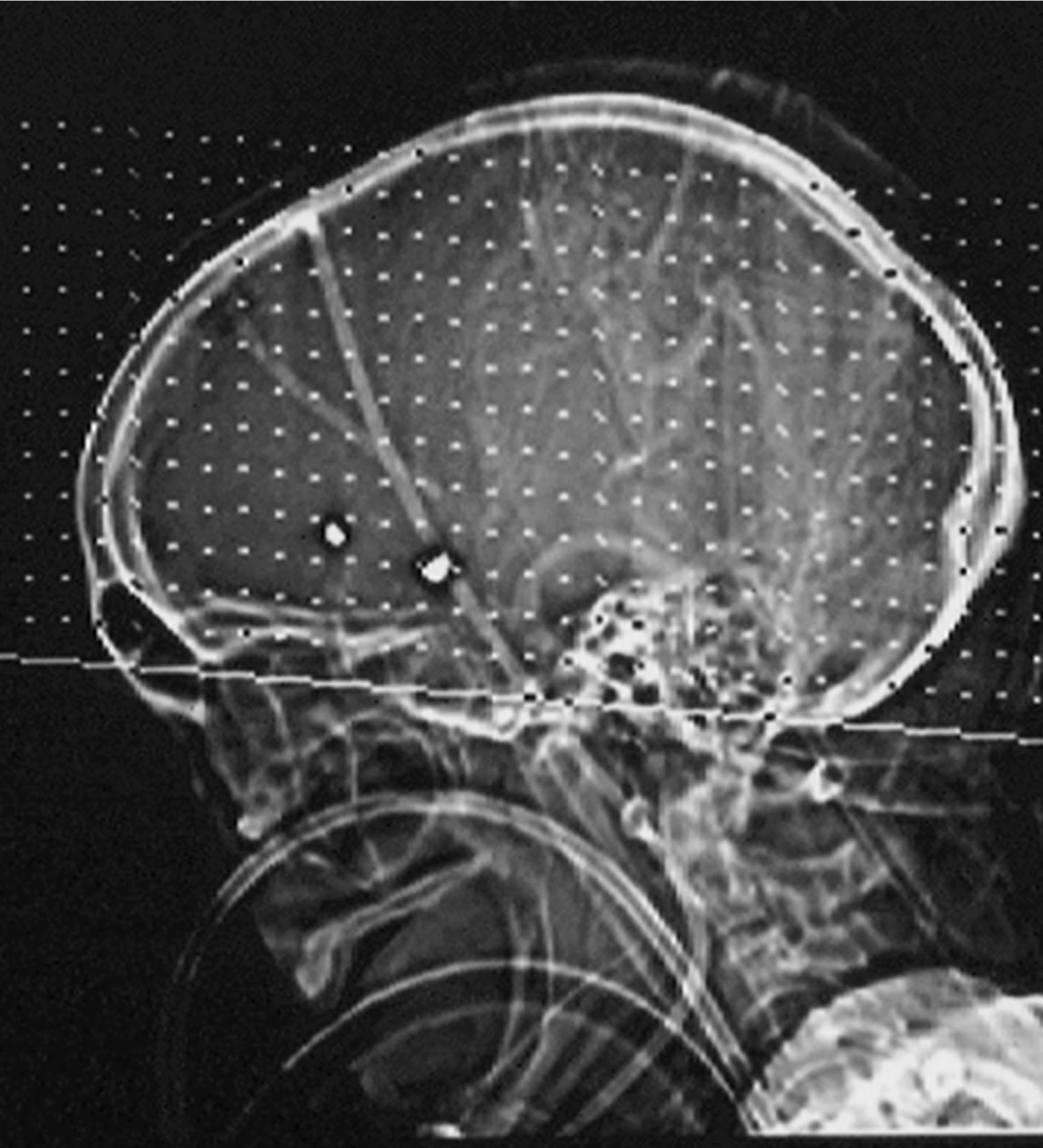 (c)
(c)
Figure 4. We present a 45 years old patient with two acute coiled aneurysms on the anterior communicating artery ACoA and pericallosal artery (a). Despite the intensive care management, the patient developed delayed neurological deficits accompanied by multiple cerebral infarctions following vasospasm (b). This case illustrates that vasospasm occurs even on non compromised arterial territories, free of aneurysm. In this case, a decompressive craniotomy was still necessary to reduce the increased intracranial pressure (c).
upregulation might be another pharmacological target [21].
 (a)
(a)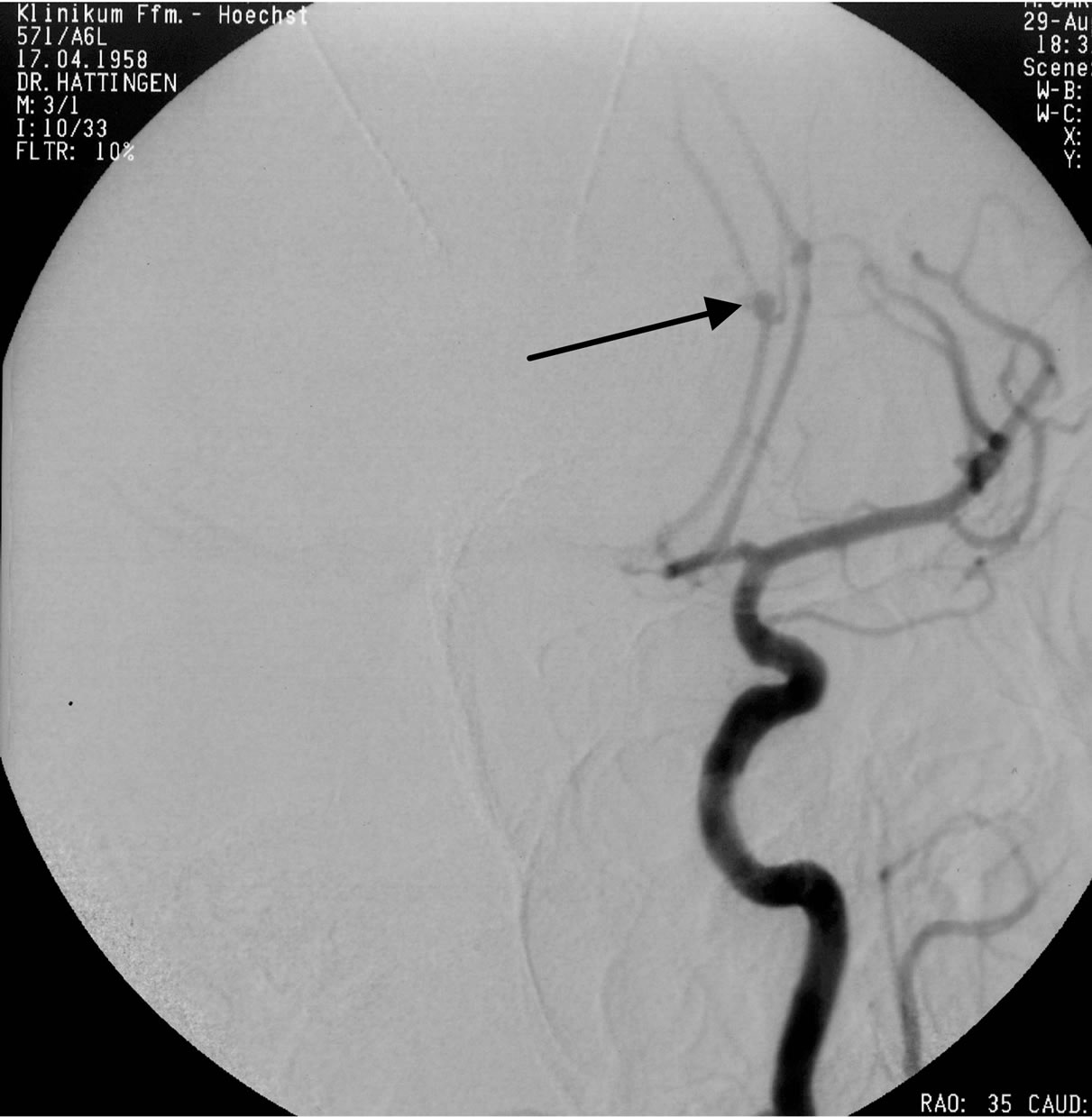 (b)
(b)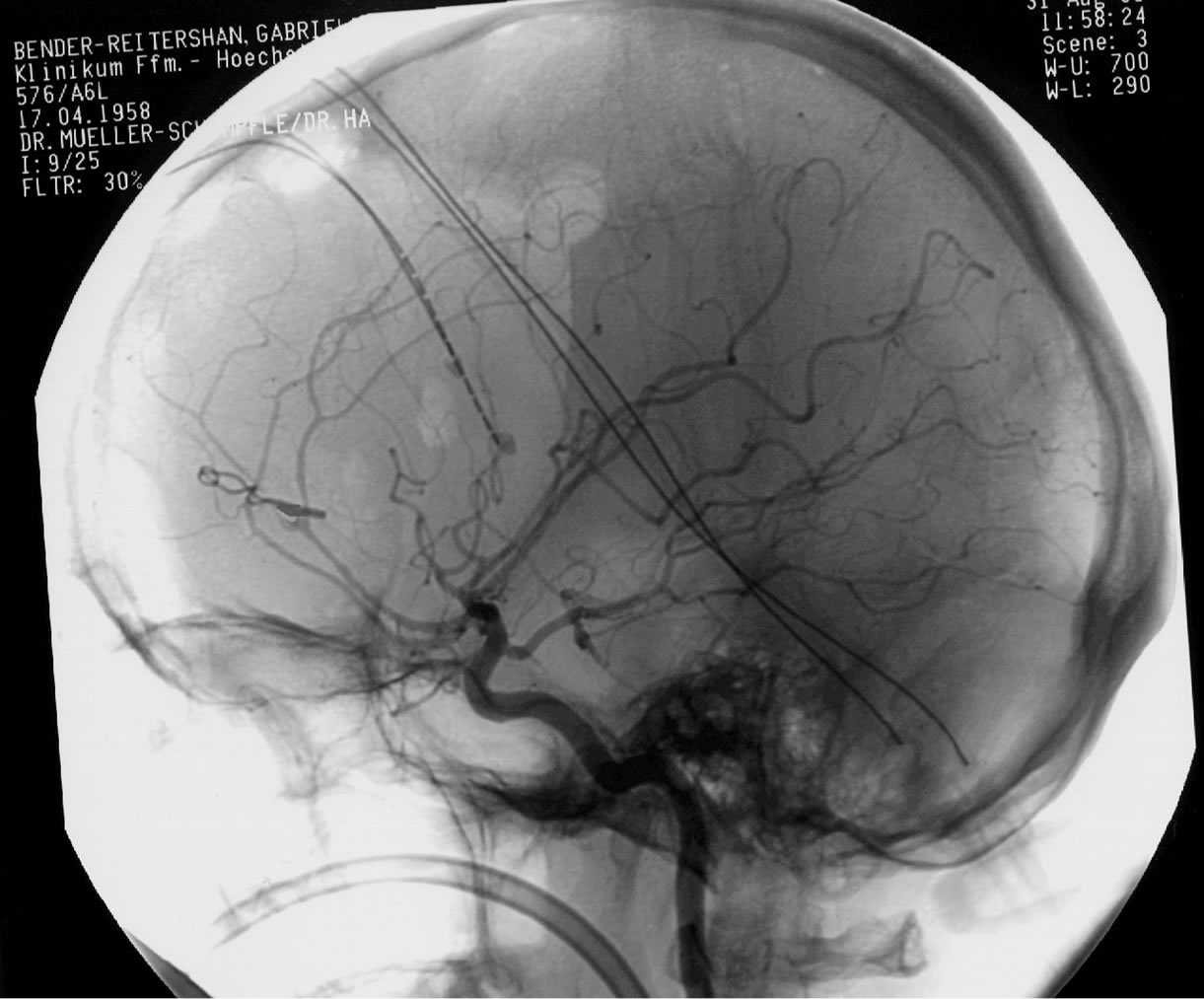 (c)
(c)
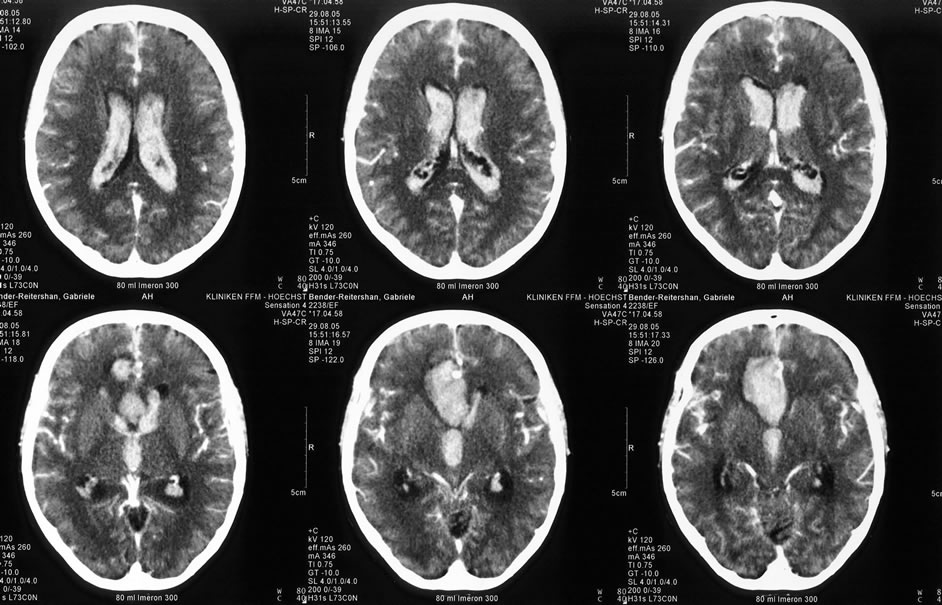
Figure 5. A 52 years old patient was admitted in the clinic with massive headaches and onfusion. The CT scan presented a massive intraventricular bleeding, a mass occupying interhemispheric hematoma and a diffuse SAH in a patient with a ruptured pericallosal aneurysm (a). The DSA (b) identified the aneurysm on the left pericallosal artery. The clipping of the aneurysm, the suction of the hematoma and the additional decompressive craniotomie allowed a satisfactory patient’s evolution. The angiographic pictures (c) confirm the complete aneurysm occlusion, without associated cerebral vasospasm.
9. Treatment of Non SAH Vasospasm
Cerebral vasospasm not associated with SAH is commonly described in the literature as reversible cerebral vasoconstriction syndrome (RCVS) [141]. It may be associated with the post partum period [142], many vasoactive and other drugs [143], sexual activity, and noumerous other conditions including pheochromocytoma and porphyria [141]. But frequently no cause can be identified [144]. The pathophysiology of RCVS remains unknown. There is evidence to suggest that many factors associated with vasospasm from aneurysmal SAH may underly in similar clinico-radiological mechanisms for RCVS [145]. The pathophysiology of RCVS and the disturbance in the control of cerebrovascular tone is likely to be multifactorial, dependent on vascular receptor activity and sensitivity. This may explain the strong association to sensory afferents from the first division of the trigeminal nerve, cervical root (C2), female sex hormones and autonomic nervous system especially when it is exposed to vasoactive drugs [146]. Patients tend to be middle-aged women but reports of all age groups are present in the literature. The primary clinical manifestation is a thunderclap headache which tends to recur daily and last for a period of around 2 weeks, often accopmpanied by nausea, vomoting, photophobia, confusion and blurred vision [145]. Most patients with RCVS recover without sequelae; however, relapse has been reported in a small proportion of patients [145]. The potential complications of RCVS include seizures, posterior reversible leukoencephalopathy, hemorrhagic events whithin the first week as localized convexity nonaneurysmal SAH, intracerebral hemorrhage and ischemic events during the second week including transient ischemic attacks and cerebral infarction [147]. Magnetic resonance imaging including angiography and venography and lumbar punctures are the studies of choise, whereas catheter angiography should not be implemented routinely [147]. Transcranial ultrasound should be used to follow the course of cerebral arerial vasospasm [148]. Patients with a flow velocity of the middle cerebral artery greater than 120 cm/s have a greater risk of ischemic complications than those without. Open-label trials showed calcium channel blockers, such as nimodipine may be an effective treatment in prevention of thunderclap headache attacks [149]. A commonly reported regimen consists of an initial intravenous administration of nimodipine at a rate of 1 - 2 mg/h, followed by an oral regimen (30 - 60 mg every 4 h) tapered over the course of several weeks. The optimal duration of treatment is unclear. In severe cases, intra-arterial therapy may be considered. The use of intra-arterial nimodipine, papaverine or the phosphodiesterase inhibitore milrinone has been reported with googd results, although experience is limited to case reports [150-152].
10. Recomendations and Conclusion
On the basis of the review of the most recent literature, there is increasing evidence that treatment of CVS alone does not prevent DCI and consecutive poor outcome. Despite the considerable number of studies, a convincing breakthrough has not been achieved. Oral nimodipine remains the only evidence-based pharmacological treatment. The most important recent findings are the negative results of the CONSCIOUS studies underlining the fact that prophylaxis and treatment of morphologic CVS alone might not be sufficient. A couple of newer treatment concepts are in evaluation. Further understanding and therapeutic approaches addressing mechanisms of endothelial dysfunction, microcirculation dysfunction, inflammation, platelet activation and microthrombi formation are warranted. Promising agents in trials are currently, nicardipine prolonged release implants placed intra-op in the cisterns, the selective endothelin IA receptor antagonist clazosentan and the routine use of statins (simvastatin 80 mg/d, pravastatin 40 mg/d). Considering the numerous treatments and the extensive literature about different agents and techniques that were
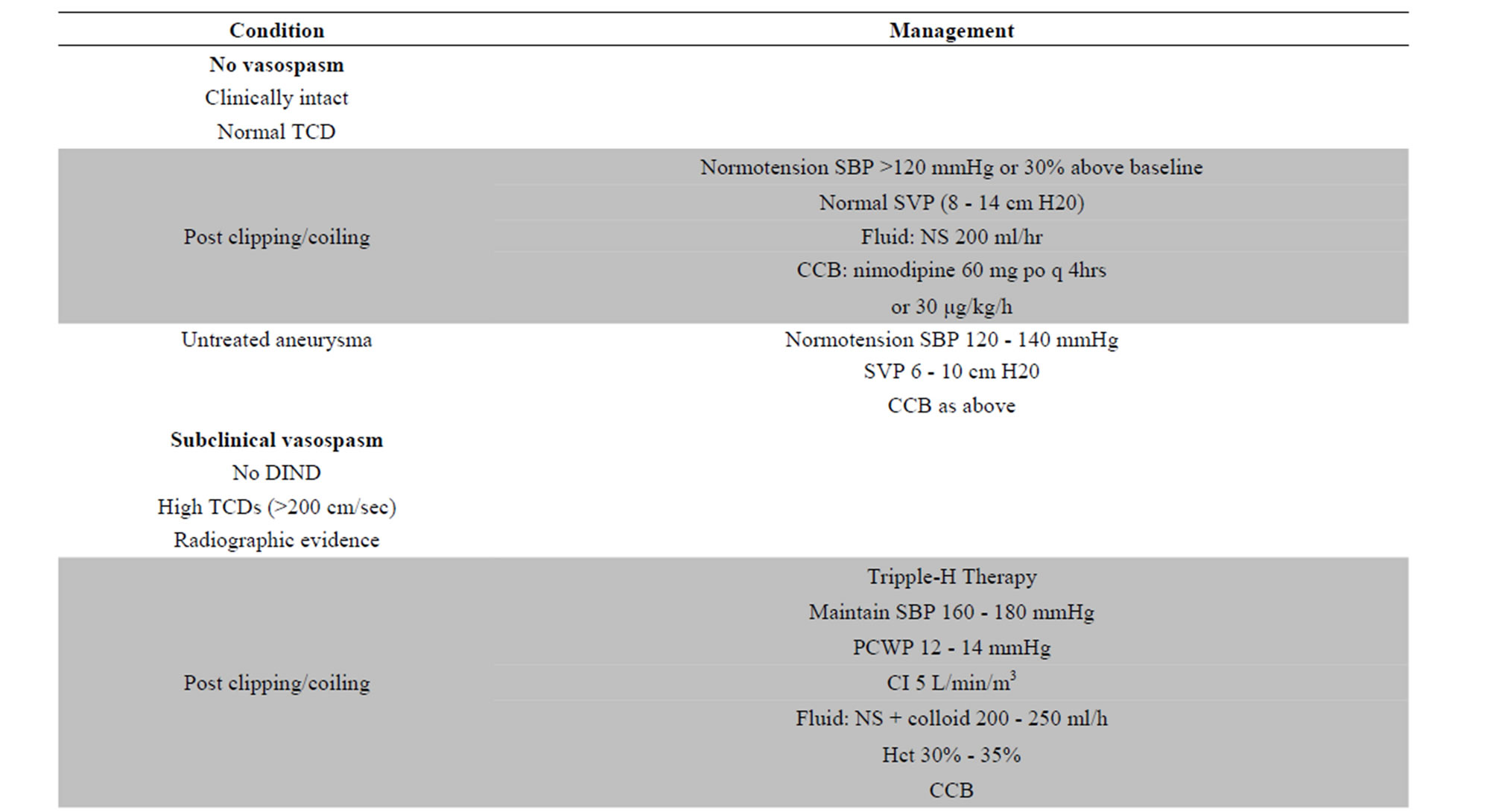
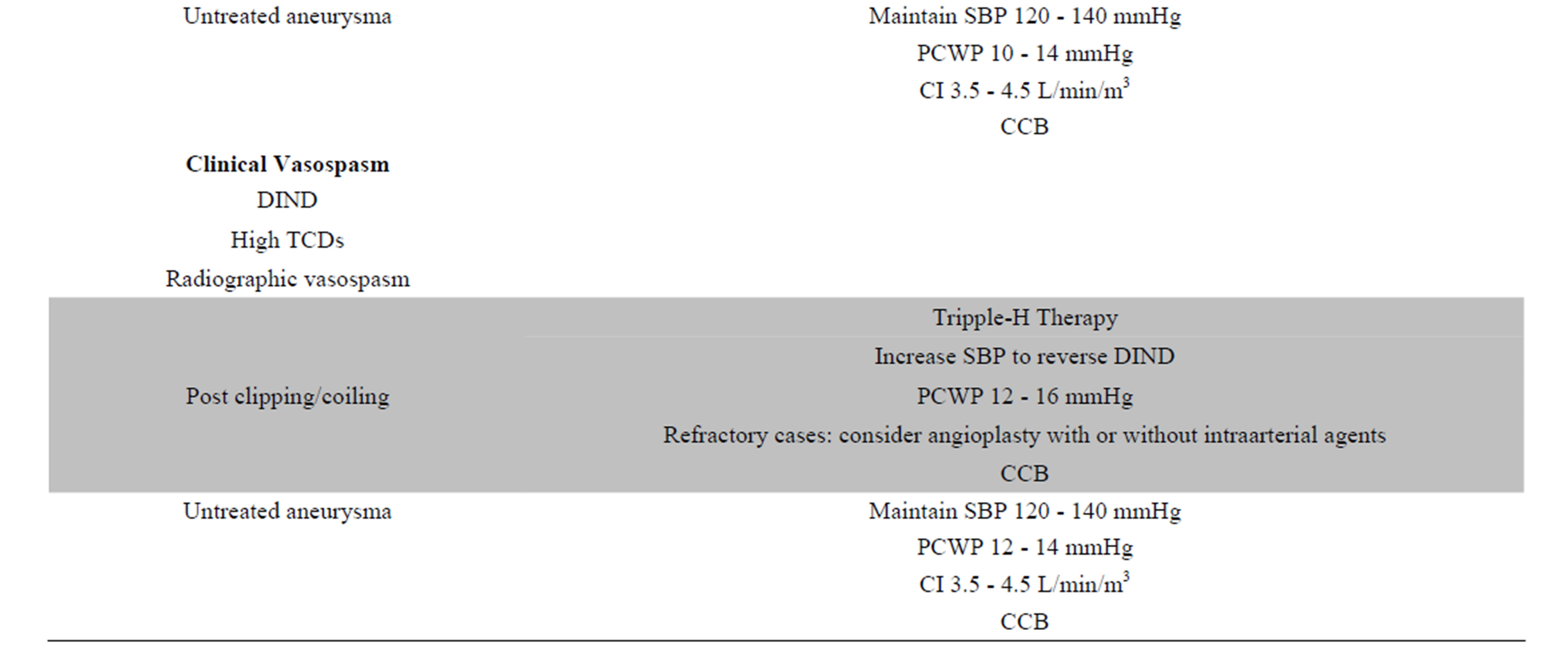
Table 1. Reference guide for the management of vasospasm in patients with SAH. Abbreviations: CCB: Calcium channel blockers, CI: cardiac index, DIND: delayed ischemic neurologic deficit, NS: normalsaline, PCWP: pulmonary capillary wedge pressure, SBP: systolic blood pressure, SVP: systemic venous pressure, TCD: trancranial Doppler.
studied until now in the literature we created the following reference guide about the recommended management of vasospasm after aneurysmatic SAH (Table 1).
REFERENCES
- N. F. Kassell, T. Sasaki, A. R. Colohan, et al., “Cerebral Vasospasm Following Aneurysmal Subarachnoid Hemorrhage,” Stroke, Vol. 16, No. 4, 1985, pp. 562-572. http://dx.doi.org/10.1161/01.STR.16.4.562
- N. F. Kassell, J. C. Torner, E. C. Haley Jr., et al., “The International Cooperative Study on the Timing of Aneurysm Surgery. Part 1: Overall Management Results,” Journal of Neurosurgery, Vol. 73, No. 1, 1990, pp. 18-36. http://dx.doi.org/10.3171/jns.1990.73.1.0018
- K. Maeda, H. Kurita, T. Nakamura, et al., “Occurrence of Severe Vasospasm Following Intraventricular Hemorrhage from an Arteriovenous Malformation. Report of Two Cases,” Journal of Neurosurgery, Vol. 87, No. 3, 1997, pp. 436-439. http://dx.doi.org/10.3171/jns.1997.87.3.0436
- M. N. Carvi y Nievas and E. Archavlis, “Atypical Causes of Nontraumatic Intracranial Subarachnoid Hemorrhage,” Clinical Neurology and Neurosurgery, Vol. 111, No. 4, 2009, pp. 354-358. http://dx.doi.org/10.1016/j.clineuro.2008.11.010
- N. A. Martin, C. Doberstein, C. Zane, M. J. Caron, K. Thomas and D. P. Becker, “Posttraumatic Cerebral Arterial Spasm: Transcranial Doppler Ultrasound, Cerebral Blood Flow, and Angiographic Findings,” Journal of Neurosurgery, Vol. 77, No. 4, 1992, pp. 575-583. http://dx.doi.org/10.3171/jns.1992.77.4.0575
- E. C. Raps, S. L. Galetta, M. Broderick and S. W. Atlas, “Delayed Peripartum Vasculopathy: Cerebral Eclampsia Revisited,” Annals of Neurology, Vol. 33, No. 2, 1993, pp. 222-225. http://dx.doi.org/10.1002/ana.410330215
- A. A. Rabinstein, G. Lanzino, E. F. Wijdicks, “Multidisciplinary Management and Emerging Therapeutic Strategies in Aneurysmal Subarachnoid Haemorrhage,” Lancet Neurology, Vol. 9, No. 5, 2010, pp. 504-519. http://dx.doi.org/10.1016/S1474-4422(10)70087-9
- O. Samuels, A. Webb, S. Culler, et al., “Impact of a Dedicated Neurocritical Care Team in Treating Patients with Aneurysmal Subarachnoid Hemorrhage,” Neurocritical Care, Vol. 14, No. 3, 2011, pp. 334-340. http://dx.doi.org/10.1007/s12028-011-9505-z
- E. Archavlis and M. N. Carvi y Nievas, “Acute Management of Poor Condition Subarachnoid Hemorrhage Patients,” Vascular Health and Risk Management, Vol. 3, No. 6, 2007, pp. 1075-1082.
- D. Castanares-Zapatero and P. Hantson, “Pharmacological Treatment of Delayed Cerebral Ischemia and Vasospasm in Subarachnoid Hemorrhage,” Annals of Intensive Care, Vol. 1, No. 1, 2011, p. 12. http://dx.doi.org/10.1186/2110-5820-1-12
- S. Shirao, H. Yoneda, H. Ishihara, K. Kajiwara and M. Suzuki, “Survey Study Members of Japan Neurosurgical Society. A Proposed Definition of Symptomatic Vasospasm Based on Treatment of Cerebral Vasospasm after Subarachnoid Hemorrhage in Japan: Consensus 2009, a Project of the 25 Spasm Symposium,” Surgical Neurology International, Vol. 2, 2011, p. 74.
- M. D. Vergouwen, “Participants in the International MultiDisciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Vasospasm Versus Delayed Cerebral Ischemia as an Outcome Event in Clinical Trials and Observational Studies,” Neurocritical Care, Vol. 15, No. 2, 2011, pp. 308-311. http://dx.doi.org/10.1007/s12028-011-9586-8
- M. M. Treggiari-Venzi, P. M. Suter and J. A. Romand, “Review of Medical Prevention of Vasospasm after Aneurysmal Subarachnoid Hemorrhage: A Problem of Neurointensive Care,” Neurosurgery, Vol. 48, No. 2, 2001, pp. 249-261. http://dx.doi.org/10.1227/00006123-200102000-00001
- M. D. Vergouwen, D. Ilodigwe and R. L. Macdonald, “Cerebral Infarction after Sub-Arachnoid Hemorrhage Contributes to Poor Outcome by Vasospasm-Dependent and Independent Effects,” Stroke, Vol. 42, No. 4, 2011, pp. 924-929. http://dx.doi.org/10.1161/STROKEAHA.110.597914
- F. A. Sehba, R. M. Pluta and J. H. Zhang, “Metamorphosis of Subarachnoid Hemorrhage Research: From Delayed Vasospasm to Early Brain Injury,” Molecular Neurobiology, Vol. 43, No. 1, 2011, pp. 27-40. http://dx.doi.org/10.1007/s12035-010-8155-z
- J. P. Dreier, “The Role of Spreading Depression, Spreading Depolarization and Spreading Ischemia in Neurological Disease,” Nature Medicine, Vol. 17, No. 4, 2011, pp. 439-447. http://dx.doi.org/10.1038/nm.2333
- L. Z. Leng, M. E. Fink and C. Iadecola, “Spreading Depolarization: A Possible New Culprit in the Delayed Cerebral Ischemia of Subarachnoid Hemorrhage,” Archives of Neurology, Vol. 68, No. 1, 2011, pp. 31-36. http://dx.doi.org/10.1001/archneurol.2010.226
- C. Muroi, S. Mink, M. Seule, et al., “Monitoring of the Inflammatory Response after Aneurysmal Subarachnoid Haemorrhage in the Clinical Setting: Review of Literature and Report of Preliminary Clinical Experience,” Acta Neurochirurgica Supplement, Vol. 110, Pt. 1, 2011, pp. 191- 196.
- T. Nakura, K. Osuka, T. Inukai, et al., “Soluble gp130 Regulatess Interleukin-6 in Cerebrospinal Fluid after Subarachnoid Haemorrhage,” Journal of Neurology, Neurosurgery, and Psychiatry, Vol. 82, No. 9, 2011, pp. 952- 954. http://dx.doi.org/10.1136/jnnp.2009.197244
- A. Sarrafzadeh, F. Schlenk, C. Gericke and P. Vajkoczy, “Relevance of Cerebral Interleukin-6 after Aneurysmal Subarachnoid Hemorrhage,” Neurocritical Care, Vol. 13, No. 3, 2010, pp. 339-346. http://dx.doi.org/10.1007/s12028-010-9432-4
- L. Edvinsson and G. K. Povlsen, “Late Cerebral Ischaemia after Subarachnoid Haemorrhage: Is Cerebrovascular Receptor Upregulation the Mechanism Behind?” Acta Physiologica, Vol. 203, No. 1, 2011, pp. 209-224. http://dx.doi.org/10.1111/j.1748-1716.2010.02227.x
- R. W. Crowley, R. Medel, A. S. Dumont, et al., “Angiographic Vasospasm Is Strongly Correlated with Cerebral Infarction after Subarachnoid Hemorrhage,” Stroke, Vol. 42, No. 4, 2011, pp. 919-923. http://dx.doi.org/10.1161/STROKEAHA.110.597005
- J. P. Kistler, R. M. Crowell, K. R. Davis, et al., “The Relation of Cerebral Vasospasm to the Extent and Location of Subarachnoid Blood Visualized by CT Scan: A Prospective Study,” Neurology, Vol. 33, No. 4, 1983, pp. 424-436. http://dx.doi.org/10.1212/WNL.33.4.424
- C. M. Fisher, J. P. Kistler and J. M. Davis, “Relation of Cerebral Vasospasm to Subarachnoid Hemorrhage Visualized by Computerized Tomographic Scanning,” Neurosurgery, Vol. 6, No. 1, 1980, pp. 1-9. http://dx.doi.org/10.1227/00006123-198001000-00001
- M. D. Vergouwen, N. Etminan, D. Ilodigwe and R. L. Macdonald, “Lower Incidence of Cerebral Infarction Correlates with Improved Functional Outcome after Aneurysmal Subarachnoid Hemorrhage,” Journal of Cerebral Blood Flow & Metabolism, Vol. 31, No. 7, 2011, pp. 1545- 1553. http://dx.doi.org/10.1038/jcbfm.2011.56
- J. Sharkey, S. P. Butcher and J. S. Kelly, “Endothelin-1 Induced Middle Cerebral Artery Occlusion: Pathological Consequences and Neuroprotective Effects of MK801,” Journal of the Autonomic Nervous System, Vol. 49, 1994, pp. S177-S185. http://dx.doi.org/10.1016/0165-1838(94)90109-0
- P. Gaetani, R. Baena, G. Grignani, G. Spanu, L. Pacchiarini and P. Paoletti, “Endothelin and Aneurysmal Subarachnoid Haemorrhage: A Study of Subarachnoid Cisternal Cerebrospinal Fluid,” Journal of Neurology, Neurosurgery, and Psychiatry, Vol. 57, No. 1, 1994, pp. 66- 72. http://dx.doi.org/10.1136/jnnp.57.1.66
- J. Hansen-Schwartz, S. Ansar and L. Edvinsson, “Cerebral Vasoconstriction after Subarachnoid Hemorrhage— Role of Changes in Vascular Receptor Phenotype,” Frontiers in Bioscience, Vol. 13, 2008, pp. 2160-2164.
- J. Hansen-Schwartz, N. L. Hoel, M. Zhou, C. B. Xu, N. A. Svendgaard and L. Edvinsson, “Subarachnoid Hemorrhage Enhances Endothelin Receptor Expression and Function in Rat Cerebral Arteries,” Neurosurgery, Vol. 52, No. 5, 2003, pp. 1188-1194. http://dx.doi.org/10.1227/01.NEU.0000058467.82442.64
- L. Edvinsson, “Cerebrovascular Endothelin Receptor Upregulation in Cerebral Ischemia,” Current Vascular Pharmacology, Vol. 7, No. 1, 2009, pp. 26-33. http://dx.doi.org/10.2174/157016109787354178
- R. D. Rothoerl and F. Ringel, “Molecular Mechanisms of Cerebral Vasospasm Following Aneurysmal SAH,” Neurological Research, Vol. 29, No. 7, 2007, pp. 636-642. http://dx.doi.org/10.1179/016164107X240224
- Y. Hirata, H. Yoshimi, S. Takaichi, M. Yanagisawa and T. Masaki, “Binding and Receptor Down-Regulation of a Novel Vasoconstrictor Endothelin in Cultured Rat Vascular Smooth Muscle Cells,” FEBS Letters, Vol. 239, No. 1, 1988, pp. 13-17. http://dx.doi.org/10.1016/0014-5793(88)80536-2
- C. Lan, D. Das, A. Wloskowicz and B. Vollrath, “Endothelin-1 Modulates Hemoglobin-Mediated Signaling in Cerebrovascular Smooth Muscle via RhoA/Rho Kinase and Protein Kinase C,” American Journal of PhysiologyHeart and Circulatory Physiology, Vol. 286, No. 1, 2004, pp. H165-H173. http://dx.doi.org/10.1152/ajpheart.00664.2003
- M. E. Grassie, L. D. Moffat, M. P. Walsh and J. A. MacDonald, “The Myosin Phosphatase Targeting Protein (MYPT) Family: A Regulated Mechanism for Achieving Substrate Specificity of the Catalytic Subunit of Protein Phosphatase Type 1δ,” Archives of Biochemistry and Biophysics, Vol. 510, No. 2, 2011, pp. 147-159. http://dx.doi.org/10.1016/j.abb.2011.01.018
- L. Miao, Y. Dai and J. Zhang, “Mechanism of RhoA/Rho Kinase Activation in Endothelin-1-Induced Contraction in Rabbit Basilar Artery,” American Journal of Physiology—Heart and Circulatory Physiology, Vol. 283, No. 3, 2002, pp. H983-H989.
- Y. Kikkawa, S. Matsuo, K. Kameda, M. Hirano, A. Nakamizo, T. Sasaki and K. Hirano, “Mechanisms Underlying Potentiation of Endothelin-1-Induced Myofilament Ca2+ Sensitization after Subarachnoid Hemorrhage,” Journal of Cerebral Blood Flow & Metabolism, Vol. 32, No. 2, 2012, pp. 341-352. http://dx.doi.org/10.1038/jcbfm.2011.132
- K. Obara, S. Nishizawa, M. Koide, K. Nozawa, A. Mitate, T. Ishikawa and K. Nakayama, “Interactive Role of Protein Kinase C-Delta with Rho-Kinase in the Development of Cerebral Vasospasm in a Canine Two-Hemorrhage Model,” Journal of Vascular Research, Vol. 42, No. 1, 2005, pp. 67-76. http://dx.doi.org/10.1159/000083093
- M. P. Walsh, “Vascular Smooth Muscle Myosin Light Chain Diphosphorylation: Mechanism, Function, and Pathological Implications,” IUBMB Life, Vol. 63, No. 11, 2011, pp. 987-1000. http://dx.doi.org/10.1002/iub.527
- C. Sutherland and M. P. Walsh, “Myosin Regulatory Light Chain Diphosphorylation Slows Relaxation of Arterial Smooth Muscle,” Journal of Biological Chemistry, Vol. 287, No. 29, 2012, pp. 24064-24076. http://dx.doi.org/10.1074/jbc.M112.371609
- B. G. Allen and M. P. Walsh, “The Biochemical Basis of the Regulation of Smooth-Muscle Contraction,” Trends in Biochemical Sciences, Vol. 19, No. 9, 1994, pp. 362-368. http://dx.doi.org/10.1016/0968-0004(94)90112-0
- A. P. Somlyo and A. V. Somlyo, “Ca2+ Sensitivity of Smooth Muscle and Nonmuscle Myosin II: Modulated by G Proteins, Kinases, and Myosin Phosphatase,” Physiological Reviews, Vol. 83, No. 4, 2003, pp. 1325-1358.
- F. Hofmann, “The Biology of Cyclic GMP-Dependent Protein Kinases,” Journal of Biological Chemistry, Vol. 280, No. 1, 2005, pp. 1-4.
- O. Guney, F. Erdi, H. Esen, A. Kiyici and Y. Kocaogullar, “N-Acetylcysteine Prevents Vasospasm after Subarachnoid Hemorrhage,” Surgical Neurology, Vol. 73, No. 1, 2010, pp. 42-49.
- A. Munakata, H. Ohkuma and N. Shimamura, “Effect of a Free Radical Scavenger, Edaravone, on Free Radical Reactions: Related Signal Transduction and Cerebral Vasospasm in the Rabbit Subarachnoid Hemorrhage Model,” Acta Neurochirurgica Supplement, Vol. 110. No. 2, 2011, pp. 17-22.
- K. Nakamura, Y. Koga, H. Sakai, K. Homma and M. Ikebe, “cGMP-Dependent Relaxation of Smooth Muscle Is Coupled with the Change in the Phosphorylation of Myosin Phosphatase,” Circulation Research, Vol. 101, No. 7, 2007, pp. 712-722. http://dx.doi.org/10.1161/CIRCRESAHA.107.153981
- F. Kehl, L. Cambj-Sapunar, K. G. Maier, N. Miyata, S. Kametani, H. Okamoto, et al., “20-HETE Contributes to the Acute Fall in Cerebral Blood Flow after Subarachnoid Hemorrhage in the Rat,” American Journal of Physiology—Heart and Circulatory Physiology, Vol. 282, No. 4, 2002, pp. H1556-H1565.
- P. Pacher, J. S. Beckman and L. Liaudet, “Nitric Oxide and Peroxynitrite in Health and Disease,” Physiological Reviews, Vol. 87, No. 1, 2007, pp. 315-424. http://dx.doi.org/10.1152/physrev.00029.2006
- D. E. Kim, Y. S. Suh, M. S. Lee, K. Y. Kim, J. H. Lee, H. S. Lee, et al., “Vascular NAD(P)H Oxidase Triggers Delayed Cerebral Vasospasm after Subarachnoid Hemorrhage in Rats,” Stroke, Vol. 33, No. 11, 2002, pp. 2687-2691. http://dx.doi.org/10.1161/01.STR.0000033071.99143.9E
- J. S. Zheng, R. Y. Zhan, S. S. Zheng, Y. Q. Zhou, Y. Tong and S. Wan, “Inhibition of NADPH Oxidase Attenuates Vasospasm after Experimental Subarachnoid Hemorrhage in Rats,” Stroke, Vol. 36, No. 5, 2005, pp. 1059- 1064.
- C. Zhou, M. Yamaguchi, G. Kusaka, C. Schonholz, A. Nanda and J. H. Zhang, “Caspase Inhibitors Prevent Endothelial Apoptosis and Cerebral Vasospasm in Dog Model of Experimental Subarachnoid Hemorrhage,” Journal of Cerebral Blood Flow & Metabolism, Vol. 24, No. 4, 2004, pp. 419-431. http://dx.doi.org/10.1097/00004647-200404000-00007
- A. Y. Zubkov, K. Ogihara, D. H. Bernanke, A. D. Parent and J. Zhang, “Apoptosis of Endothelial Cells in Vessels Affected by Cerebral Vasospasm,” Surgical Neurology, Vol. 53, No. 3, 2000, pp. 260-266. http://dx.doi.org/10.1016/S0090-3019(99)00187-1
- H. Kimura, I. Gules, T. Meguro and J. H. Zhang, “Cytotoxicity of Cytokines in Cerebral Microvascular Endothelial Cell,” Brain Research, Vol. 990, No. 1-2, 2003, pp. 148-156. http://dx.doi.org/10.1016/S0006-8993(03)03450-4
- K. Iseda, S. Ono, K. Onoda, M. Satoh, H. Manabe, M. Nishiguchi, et al., “Antivasospastic and Antiinflammatory Effects of Caspase Inhibitor in Experimental Subarachnoid Hemorrhage,” Journal of Neurosurgery, Vol. 107, No. 1, 2007, pp. 128-135. http://dx.doi.org/10.3171/JNS-07/07/0128
- R. M. Pluta, “Dysfunction of Nitric Oxide Synthases as a Cause and Therapeutic Target in Delayed Cerebral Vasospasm after SAH,” Acta Neurochirurgica Supplement, Vol. 104, 2008, pp. 139-147.
- A. S. Dumont, R. J. Dumont, M. M. Chow, et al., “Cerebral Vasospasm after Subarachnoid Hemorrhage: Putative Role of Inflammation,” Neurosurgery, Vol. 53, No. 1, 2003, pp. 123-133. http://dx.doi.org/10.1227/01.NEU.0000068863.37133.9E
- T. Sozen, R. Tsuchiyama, Y. Hasegawa, et al., “Role of Interleukin-1beta in Early Brain Injury after Subarachnoid Hemorrhage in Mice,” Stroke, Vol. 40, No. 7, 2009, pp. 2519-2525. http://dx.doi.org/10.1161/STROKEAHA.109.549592
- K. Fassbender, B. Hodapp, S. Rossol, et al., “Endothelin- 1 in Subarachnoid Hemorrhage: An Acute-Phase Reactant Produced by Cerebrospinal Fluid Leukocytes,” Stroke, Vol. 31, No. 12, 2000, pp. 2971-2975. http://dx.doi.org/10.1161/01.STR.31.12.2971
- H. Takeuchi, M. Tanabe, H. Okamoto and M. Yamazaki, “Effects of Thromboxane Synthetase Inhibitor (RS-5186) on Experimentally-Induced Cerebral Vasospasm,” Neurological Research, Vol. 21, No. 5, 1999, pp. 513-516.
- S. Satoh, Y. Suzuki, T. Harada, et al., “The Role of Platelets in the Development of Cerebral Vasospasm,” Brain Research Bulletin, Vol. 27, No. 5, 1991, pp. 663- 668. http://dx.doi.org/10.1016/0361-9230(91)90042-I
- G. F. Prunell, N. A. Svendgaard, K. Alkass and T. Mathiesen, “Inflammation in the Brain after Experimental Subarachnoid—Hemorrhage,” Neurosurgery, Vol. 56, No. 5, 2005, pp. 1082-1092.
- M. Bavbek, R. Polin, A. L. Kwan, A. S. Arthur, N. F. Kassell and K. S. Lee, “Monoclonal Antibodies against ICAM-1 and CD18 Attenuate Cerebral Vasospasm after Experimental Subarachnoid Hemorrhage in Rabbits,” Stroke, Vol. 29, No. 9, 1998, pp. 1930-1935. http://dx.doi.org/10.1161/01.STR.29.9.1930
- R. S. Polin, M. Bavbek, M. E. Shaffrey, et al., “Detection of Soluble E-Selectin, ICAM-1, VCAM-1, and L-Selectin in the Cerebrospinal Fluid of Patients after Subarachnoid Hemorrhage,” Journal of Neurosurgery, Vol. 89, No. 4, 1998, pp. 559-567. http://dx.doi.org/10.3171/jns.1998.89.4.0559
- J. Cahill, J. W. Calvert, I. Solaroglu and J. H. Zhang, “Vasospasm and p53-Induced Apoptosis in an Experimental Model of Subarachnoid Hemorrhage,” Stroke, Vol. 37, No. 7, 2006, pp. 1868-1874. http://dx.doi.org/10.1161/01.STR.0000226995.27230.96
- E. Kozniewska, R. Michalik, J. Rafalowska, et al., “Mechanisms of Vascular Dysfunction after Subarachnoid Hemorrhage,” Journal of Physiology and Pharmacology, Vol. 57, Suppl. 11, 2006, pp. 145-160.
- E. Grote and W. Hassler, “The Critical First Minutes after Subarachnoid Hemorrhage,” Neurosurgery, Vol. 22, No. 4, 1988, pp. 654-661. http://dx.doi.org/10.1227/00006123-198804000-00006
- W. E. Hoffman, P. Wheeler, G. Edelman, F. T. Charbel, N. J. Torres and J. I. Ausman, “Hypoxic Brain Tissue Following Subarachnoid Hemorrhage,” Anesthesiology, Vol. 92, No. 2, 2000, pp. 442-446. http://dx.doi.org/10.1097/00000542-200002000-00026
- T. Hishikawa, S. Ono, T. Ogawa, K. Tokunaga, K. Sugiu and I. Date, “Effects of Deferoxamine-Activated Hypoxia-Inducible Factor-1 on the Brainstem after Subarachnoid Hemorrhage in Rats,” Neurosurgery, Vol. 62, No. 1, 2008, pp. 232-240. http://dx.doi.org/10.1227/01.NEU.0000311082.88766.33
- G. L. Semenza, “Regulation of Oxygen Homeostasis by Hypoxia-Inducible Factor 1,” Physiology Bethesda, Vol. 24, 2009, pp. 97-106. http://dx.doi.org/10.1152/physiol.00045.2008
- R. Schmidt-Kastner, C. Aguirre-Chen, T. Kietzmann, I. Saul, R. Busto and M. D. Ginsberg, “Nuclear Localization of the Hypoxia-Regulated Pro-Apoptotic Protein BNIP3 after Global Brain Ischemia in the Rat Hippocampus,” Brain Research, Vol. 1001, No. 1-2, 2004, pp. 133-142. http://dx.doi.org/10.1016/j.brainres.2003.11.065
- H. Jeon, J. Ai, M. Sabri, et al., “Neurological and Neurobehavioral Assessment of Experimental Subarachnoid Hemorrhage,” BMC Neuroscience, Vol. 10, 2009, p. 103. http://dx.doi.org/10.1186/1471-2202-10-103
- J. Cahill, J. W. Calvert, S. Marcantonio and J. H. Zhang, “p53 May Play an Orchestrating Role in Apoptotic Cell Death after Experimental Subarachnoid Hemorrhage,” Neurosurgery, Vol. 60, No. 3, 2007, pp. 531-545. http://dx.doi.org/10.1227/01.NEU.0000249287.99878.9B
- J. Yan, C. Chen, J. Lei, et al., “2-Methoxyestradiol Reduces Cerebral Vasospasm after 48 Hours of Experimental Subarachnoid Hemorrhage in Rats,” Experimental Neurology, Vol. 202, No. 2, 2006, pp. 348-356. http://dx.doi.org/10.1016/j.expneurol.2006.06.009
- J. D. Pearl and R. L. Macdonald, “Vasospasm after Aneurysmal Subarachnoid Hemorrhage: Need for Further Study,” Acta Neurochirurgica Supplement, Vol. 105, 2008, pp. 207-210. http://dx.doi.org/10.1007/978-3-211-09469-3_39
- J. P. Dreier, S. Major, A. Manning, et al., “Cortical Spreading Ischaemia Is a Novel Process Involved in Ischaemic Damage in Patients with Aneurysmal Subarachnoid Haemorrhage,” Brain, Vol. 132, 2009, pp. 1866- 1881. http://dx.doi.org/10.1093/brain/awp102
- J. Kleeberg, G. C. Petzold, S. Major, U. Dirnagl and J. P. Dreier, “ET-1 Induces Cortical Spreading Depression via Activation of the ETA Receptor/Phospholipase C Pathway in Vivo,” American Journal of Physiology—Heart and Circulatory Physiology, Vol. 286, No. 4, 2004, pp. H1339-H1346. http://dx.doi.org/10.1152/ajpheart.00227.2003
- M. Balestrino, J. Young and P. Aitken, “Block of Na+, K+ ATPase with Ouabain Induces Spreading DepressionLike Depolarization in Hippocampal Slices,” Brain Research, Vol. 838, No. 1-2, 1999, pp. 37-44. http://dx.doi.org/10.1016/S0006-8993(99)01674-1
- K. Yufu, T. Itoh, R. Edamatsu, A. Mori and M. Hirakawa, “Effect of Hyperbaric Oxygenation on the Na+, K+-ATPase and Membrane Fluidity of Cerebrocortical Membranes after Experimental Subarachnoid Hemorrhage,” Neurochemical Research, Vol. 18, No. 9, 1993, pp. 1033- 1039. http://dx.doi.org/10.1007/BF00966765
- G. D. Murray, “Surgical Bleeding and Calcium Antagonists. British Aneurysm Nimodipine Trial Reported Improved Clinical Outcome with Nimodipine,” British Medical Journal, Vol. 311, No. 7001, 1995, pp. 388-389. http://dx.doi.org/10.1136/bmj.311.7001.388c
- J. D. Pickard, G. D. Murray, R. Illingworth, M. D. Shaw, G. M. Teasdale, P. M. Foy, et al., “Effect of Oral Nimodipine on Cerebral Infarction and Outcome after subarachnoid Haemorrhage: British Aneurysm Nimodipine Trial,” British Medical Journal, Vol. 298, 1989, pp. 636- 642. http://dx.doi.org/10.1136/bmj.298.6674.636
- F. G. Barker II and C. S. Ogilvy, “Efficacy of Prophylactic Nimodipine for Delayed Ischemic Deficit after Subarachnoid Hemorrhage: A Metaanalysis,” Journal of Neurosurgery, Vol. 84, No. 3, 1996, pp. 405-414. http://dx.doi.org/10.3171/jns.1996.84.3.0405
- P. Karinen, P. Koivukangas, A. Ohinmaa, J. Koivukangas and J. Ohman, “Cost-Effectiveness Analysis of Nimodipine Treatment after Aneurysmal Subarachnoid Hemorrhage and Surgery,” Neurosurgery, Vol. 45, No. 4, 1999, pp. 780-785. http://dx.doi.org/10.1097/00006123-199910000-00009
- F. A. Schanne, A. B. Kane, E. E. Young and J. L. Farber, “Calcium Dependence of Toxic Cell Death: A Final Common Pathway,” Science, Vol. 206, No. 4419, 1979, pp. 700-702.
- J. Dale, K. H. Landmark and E. Myhre, “The Effects of Nifedipine, a Calcium Antagonist, on Platelet Function,” American Heart Journal, Vol. 105, No. 1, 1983, pp. 103-105. http://dx.doi.org/10.1016/0002-8703(83)90285-5
- L. M. Auer, “Pial Arterial Vasodilation by Intravenous Nimodipine in Cats,” Arzneimittelforschung, Vol. 31, No. 9, 1981, pp. 1423-1425.
- N. F. Kassell, E. C. Haley Jr., C. Apperson-Hansen and W. M. Alves, “Randomized, Double-Blind, Vehicle-Controlled Trial of Tirilazad Mesylate in Patients with Aneurysmal Subarachnoid Hemorrhage: A Cooperative Study in Europe, Australia, and New Zealand,” Journal of Neurosurgery, Vol. 84, No. 2, 1996, pp. 221-228. http://dx.doi.org/10.3171/jns.1996.84.2.0221
- A Munakata, H Ohkuma and N. Shimamura, “Effect of a Free Radical Scavenger, Edaravone, on Free Radical Reactions: Related Signal Transduction and Cerebral Vasospasm in the Rabbit Subarachnoid Hemorrhage Model,” Acta Neurochirurgica Supplement, Vol. 110, 2011, pp. 17-22.
- D. M. Pollock, T. L. Keith and R. F. Highsmith, “Endothelin Receptors and Calcium Signaling,” The FASEB Journal, Vol. 9, No. 12, 1995, pp. 1196-1204.
- R. L. Macdonald, R. T. Higashida, E. Keller, et al., “Clazosentan, an Endothelin Receptor Antagonist, in Patients with Aneurysmal Subarachnoid Haemorrhage Undergoing Surgical Clipping: A randomised, Double-Blind, Placebo-Controlled Phase 3 Trial (CONSCIOUS-2),” The Lancet Neurology, Vol. 10, No. 7, 2011, pp. 618-625. http://dx.doi.org/10.1016/S1474-4422(11)70108-9
- R. L. Macdonald, R. T. Higashida, E. Keller, et al., “Preventing Vasospasm Improves Outcome after Aneurysmal Subarachnoid Hemorrhage: Rationale and Design of CONSCIOUS-2 and CONSCIOUS-3 Trials,” Neurocritical Care, Vol. 13, No. 3, 2010, pp. 416-427. http://dx.doi.org/10.1007/s12028-010-9433-3
- T. Kerz, S. Boor, C. Beyer, S. Welschehold, A. Schuessler and J. Oertel, “Effect of Intraarterial Papaverine or Nimodipine on Vessel Diameter in Patients with Cerebral Vasospasm after Subarachnoid Hemorrhage,” British Journal of Neurosurgery, Vol. 26, No. 4, 2012, pp. 517-524. http://dx.doi.org/10.3109/02688697.2011.650737
- M. Lannes, J. Teitelbaum, M. del Pilar Cortés, M. Cardoso and M. Angle, “Milrinone and Homeostasis to Treat Cerebral Vasospasm Associated With Subarachnoid Hemorrhage: The Montreal Neurological Hospital Protocol,” Neurocritical Care, Vol. 16, No. 3, 2012, pp. 354-362. http://dx.doi.org/10.1007/s12028-012-9701-5
- A. Baumann, A. L. Derelle, P. M. Mertes and G. Audibert, “Seeking New Approaches: Milrinone in the Treatment of Cerebral Vasospasm,” Neurocritical Care, Vol. 16, No. 3, 2012, pp. 351-353. http://dx.doi.org/10.1007/s12028-012-9718-9
- M. Yamaguchi, C. Zhou, A. Nanda and J. H. Zhang, “Ras Protein Contributes to Cerebral Vasospasm in a Canine Double-Hemorrhage Model,” Stroke, Vol. 35, No. 7, 2004, pp. 1750-1755. http://dx.doi.org/10.1161/01.STR.0000129898.68350.9f
- C. Thome, M. Seiz, G. A. Schubert, et al., “Nicardipine Pellets for the Prevention of Cerebral Vasospasm,” Acta Neurochirurgica Supplement, Vol. 110, 2011, pp. 209- 211.
- H. Kasuya, “Clinical Trial of Nicardipine Prolonged-Release Implants for Preventing Cerebral Vasospasm: Multicenter Cooperative Study in Tokyo,” Acta Neurochirurgica Supplement, Vol. 110, 2011, pp. 165-167.
- M. Barth, P. Pena, M. Seiz, et al., “Feasibility of Intraventricular Nicardipine Prolonged Release Implants in Patients Following Aneurysmal Subarachnoid Haemorrhage,” British Journal of Neurosurgery, Vol. 25, No. 6, 2011, pp. 677-683. http://dx.doi.org/10.3109/02688697.2010.548878
- M. M. Treggiari, J. A. Romand, J. B. Martin, A. Reverdin, D. A. Rüfenacht and N. de Tribolet, “Cervical sympathetic Block to Reverse Delayed Ischemic Neurological Deficits after Aneurysmal Subarachnoid Hemorrhage,” Stroke, Vol. 34, No. 4, 2003, pp. 961-967. http://dx.doi.org/10.1161/01.STR.0000060893.72098.80
- [Y. Wu, K. Tang, R. Q. Huang, Z. Zhuang, H. L. Cheng, H. X. Yin and J. X. Shi, “Therapeutic Potential of peroxisome Proliferator-Activated Receptor γ Agonist Rosiglitazone in Cerebral Vasospasm after a Rat Experimental Subarachnoid Hemorrhage Model,” Journal of the Neurological Sciences, Vol. 305, No. 1-2, 2011, pp. 85-91. http://dx.doi.org/10.1016/j.jns.2011.03.006
- R. Sercombe, Y. R. Dinh and P. Gomis, “Cerebrovascular Inflammation Following Subarachnoid Hemorrhage,” Japanese Journal of Pharmacology, Vol. 88, No. 3, 2002, pp. 227-249.
- A. R. Fathi, K. D. Bakhtian and R. M. Pluta, “The Role of Nitric Oxide Donors in Treating Cerebral Vasospasm after Subarachnoid Hemorrhage,” Acta Neurochirurgica Supplement, Vol. 110, 2011, pp. 93-97.
- A. R. Fathi, R. M. Pluta, K. D. Bakhtian, et al., “Reversal of Cerebral Vasospasm via Intravenous Sodium Nitrite after Subarachnoid Hemorrhage in Primates,” Journal of Neurosurgery, Vol. 115, No. 6, 2011, pp. 1213-1220. http://dx.doi.org/10.3171/2011.7.JNS11390
- R. M. Pluta, E. H. Oldfield, K. D. Bakhtian, et al., “Safety and Feasibility of Long-Term Intravenous Sodium Nitrite Infusion in Healthy Volunteers,” PLoS One, Vol. 6, No. 1, 2011, Article ID: e14504. http://dx.doi.org/10.1371/journal.pone.0014504
- K. Satoh, Y. Fukumoto and H. Shimokawa, “Rho-Kinase: Important New Therapeutic Target in Cardiovascular Diseases,” American Journal of Physiology—Heart and Circulatory Physiology, Vol. 301, No. 2, 2011, pp. H287- H296. http://dx.doi.org/10.1152/ajpheart.00327.2011
- K. Ito, Y. Hirooka, K. Sakai, T. Kishi, K. Kaibuchi, H. Shimokawa and A. Takeshita, “Rho/Rho-Kinase Pathway in Brain Stem Contributes to Blood Pressure Regulation via Sympathetic Nervous System: Possible Involvement in Neural Mechanisms of Hypertension,” Circulation Research, Vol. 92, No. 12, 2003, pp. 1337-1343. http://dx.doi.org/10.1161/01.RES.0000079941.59846.D4
- T. Zoerle, D. C. Ilodigwe, H. Wan, K. Lakovic, M. Sabri, J. Ai and R. L. Macdonald, “Pharmacologic Reduction of Angiographic Vasospasm in Experimental Subarachnoid Hemorrhage: Systematic Review and Meta-Analysis,” Journal of Cerebral Blood Flow & Metabolism, Vol. 32, No. 9, 2012, pp. 1645-1658. http://dx.doi.org/10.1038/jcbfm.2012.57
- S. Satoh, M. Takayasu, K. Kawasaki, I. Ikegaki, A. Hitomi, K. Yano, M. Shibuya and T. Asano, “Antivasospastic Effects of Hydroxyfasudil, a Rho-Kinase Inhibitor, after Subarachnoid Hemorrhage,” Journal of Pharmacological Sciences, Vol. 118, No. 1, 2012, pp. 92-98. http://dx.doi.org/10.1254/jphs.11075FP
- G. J. Velat, M. M. Kimball, J. D. Mocco and B. L. Hoh, “Vasospasm after Aneurysmal Subarachnoid Hemorrhage: Review of Randomized Controlled Trials and Meta-Analyses in the Literature,” World Neurosurgery, Vol. 76, No. 5, 2011, pp. 446-454. http://dx.doi.org/10.1016/j.wneu.2011.02.030
- J. Zhao, D. Zhou, J. Guo, Z. Ren, L. Zhou, S. Wang, Y. Zhang, B. Xu, K. Zhao, R. Wang, Y. Mao, B. Xu, X. Zhang; Fasudil Aneurysmal Subarachnoid Hemorrhage Study Group, “Efficacy and Safety of Fasudil in Patients with Subarachnoid Hemorrhage: Final Results of a randomized Trial of Fasudil versus Nimodipine,” Neurologia Medico-Chirurgica (Tokyo), Vol. 51, No. 10, 2011, pp. 679-683. http://dx.doi.org/10.2176/nmc.51.679
- T. Nakamura, T. Matsui, A. Hosono, A. Okano, N. Fujisawa, T. Tsuchiya, M. Indo, Y. Suzuki, S. Oya and H. S. Chang, “Beneficial Effect of Selective Intra-Arterial Infusion of Fasudil Hydrochloride as a Treatment of Symptomatic Vasospasm Following SAH,” Acta Neurochirurgica Supplement, Vol. 115, 2013, pp. 81-85.
- G. J. Liu, Z. J. Wang, Y. F. Wang, L. L. Xu, X. L. Wang, Y. Liu, G. J. Luo, G. H. He and Y. J. Zeng, “Systematic Assessment and Meta-Analysis of the Efficacy and Safety of Fasudil in the Treatment of Cerebral Vasospasm in Patients with Subarachnoid Hemorrhage,” European Journal of Clinical Pharmacology, Vol. 68, No. 2, 2012, pp. 131-139. http://dx.doi.org/10.1007/s00228-011-1100-x
- S. Wolf, “Routine Management of Volume Status after Aneurysmal Subarachnoid Hemorrhage,” Neurocritical Care, Vol. 15, No. 2, 2011, pp. 275-280. http://dx.doi.org/10.1007/s12028-011-9593-9
- M. N. Diringer, T. P. Bleck, J. Claude Hemphill 3rd, et al., “Critical Care Management of Patients Following Aneurysmal Subarachnoid Hemorrhage: Recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference,” Neurocritical Care, Vol. 15, No. 2, 2011, pp. 211-240. http://dx.doi.org/10.1007/s12028-011-9605-9
- P. Chittiboina, S. Conrad, P. McCarthy, et al., “The Evolving Role of Hemodilution in Treatment of Cerebral Vasospasm: A Historical Perspective,” World Neurosurgery, Vol. 75, No. 5-6, 2011, pp. 660-664. http://dx.doi.org/10.1016/j.wneu.2011.02.019
- A. M. Naidech, A. Shaibani, R. K. Garg, et al., “Prospective, Randomized Trial of Higher Goal Hemoglobin after Subarachnoid Hemorrhage,” Neurocritical Care, Vol. 13, No. 3, 2010, pp. 313-320. http://dx.doi.org/10.1007/s12028-010-9424-4
- G. Appelboom, D. Strozyk, B. Y. Hwang, et al., “Bedside Use of a Dual Aortic Balloon Occlusion for the Treatment of Cerebral Vasospasm,” Neurocritical Care, Vol. 13, No. 3, 2010, 385-388. http://dx.doi.org/10.1007/s12028-010-9442-2
- T. Mori, Y. Katayama, T. Igarashi, N. Moro, J. Kojima and T. Hirayama, “Is the Circulating Plasma Volume Sufficiently Maintained? Fluid Management of an Aneurysmal Subarachnoid Hemorrhage in the Acute Phase,” Neurological Research, Vol. 34, No. 10, 2012, pp. 1016- 1019. http://dx.doi.org/10.1179/1743132812Y.0000000093
- L. T. Iseri and J. H. French, “Magnesium: Nature’s Physiologic Calcium Blocker,” American Heart Journal, Vol. 108, No. 1, 1984, pp. 188-193. http://dx.doi.org/10.1016/0002-8703(84)90572-6
- J. I. Suarez, “Magnesium Sulfate Administration in Subarachnoid Hemorrhage,” Neurocritical Care, Vol. 15, No. 2, 2011, pp. 302-307. http://dx.doi.org/10.1007/s12028-011-9603-y
- G. K. Wong, W. S. Poon, M. T. Chan, et al., “Intravenous Magnesium Sulphate for Aneurysmal Subarachnoid Hemorrhage (IMASH): A Randomized, Double-Blinded, Placebo-Controlled, Multicenter Phase III Trial,” Stroke, Vol. 41, No. 5, 2010, pp. 921-926. http://dx.doi.org/10.1161/STROKEAHA.109.571125
- M. D. Vergouwen, “Magnesium Sulfate for Aneurysmal Subarachnoid Hemorrhage: The End of the Road or More Trials?” Critical Care, Vol. 15, No. 2, 2011, p. 140. http://dx.doi.org/10.1186/cc10055
- K. Mori, T. Yamamoto, M. Miyazaki, et al., “Optimal Cerebrospinal Fluid Magnesium Ion Concentration for Vasodilatory Effect and Duration after Intracisternal Injection of Magnesium Sulfate Solution in a Canine Subarachnoid Hemorrhage Model,” Journal of Neurosurgery, Vol. 114, No. 4, 2011, pp. 1168-1175. http://dx.doi.org/10.3171/2010.10.JNS10866
- M. Y. Tseng, “Summary of Evidence on Immediate Statins Therapy Following Aneurysmal Subarachnoid Hemorrhage,” Neurocritical Care, Vol. 15, No. 2, 2011, pp. 298-301. http://dx.doi.org/10.1007/s12028-011-9596-6
- K. K. Mukherjee, S. K. Singh, V. K. Khosla, S. Mohindra and P. Salunke, “Safety and Efficacy of Sildenafil Citrate in Reversal of Cerebral Vasospasm: A Feasibility Study,” Surgical Neurology International, Vol. 3, 2012, p. 3. http://dx.doi.org/10.4103/2152-7806.92164
- C. Kruuse, S. Gupta, E. Nilsson, L. Kruse and L. Edvinsson, “Differential Vasoactive Effects of Sildenafil and Tadalafil on Cerebral Arteries,” European Journal of Pharmacology, Vol. 674, No. 2-3, 2012, pp. 345-351. http://dx.doi.org/10.1016/j.ejphar.2011.10.037
- J. Bardutzky, J. Witsch, E. Juttler, et al., “EARLYDRAIN-Outcome after Early Lumbar CSF-Drainage in Aneurysmal Subarachnoid Hemorrhage: Study Protocol for a Randomized Controlled Trial,” Trials, Vol. 12, 2011, p. 203. http://dx.doi.org/10.1186/1745-6215-12-203
- R. J. Komotar, D. K. Hahn, G. H. Kim, et al., “Efficacy of Lamina Terminalis Fenestration in Reducing ShuntDependent Hydrocephalus Following Aneurysmal Subarachnoid Hemorrhage: A Systematic Review,” Journal of Neurosurgery, Vol. 111, No. 1, 2009, pp. 147-154. http://dx.doi.org/10.3171/2009.1.JNS0821
- D. C. Widenka, S. Wolf, L. Schurer, D. V. Plev and C. B. Lumenta, “Factors Leading to Hydrocephalus after Aneurysmal Subarachnoid Hemorrhage,” Neurologia i Neurochirurgia polska, Vol. 34, No. 6, 2000, pp. 56-60.
- P. Jun, N. U. Ko, J. D. English, et al., “Endovascular Treatment of Medically Refractory Cerebral Vasospasm Following Aneurysmal Subarachnoid Hemorrhage,” American Journal of Neuroradiology, Vol. 31, No. 10, 2010, pp. 1911-1916. http://dx.doi.org/10.3174/ajnr.A2183
- M. M. Kimball, G. J. Velat and B. L. Hoh, “Critical Care Guidelines on the Endovascular Management of Cerebral Vasospasm,” Neurocritical Care, Vol. 15, No. 2, 2011, pp. 336-341. http://dx.doi.org/10.1007/s12028-011-9600-1
- J. V. Sehy, W. E. Holloway, S. P. Lin, et al., “Improvement in Angiographic Cerebral Vasospasm after IntraArterial Verapamil Administration,” American Journal of Neuroradiology, Vol. 31, No. 9, 2010, pp. 1923-1928. http://dx.doi.org/10.3174/ajnr.A2215
- E. Albanese, A. Russo, M. Quiroga, et al., “UltrahighDose Intraarterial Infusion of Verapamil through an Indwelling Microcatheter for Medically Refractory Severe Vasospasm: Initial Experience,” Journal of Neurosurgery, Vol. 113, No. 4, 2010, pp. 913-922. http://dx.doi.org/10.3171/2009.9.JNS0997
- J. T. Miley, N. Tariq, F. G. Souslian, et al., “Comparison between Angioplasty Using Compliant and Noncompliant Balloons for Treatment of Cerebral Vasospasm Associated with Subarachnoid Hemorrhage,” Neurosurgery, Vol. 69, Suppl. 2, 2011, pp. ons161-ons168.
- A. A. Rabinstein, M. A. Pichelmann, J. A. Friedman, et al., “Symptomatic Vasospasm and Outcomes Following Aneurysmal Subarachnoid Hemorrhage: A Comparison between Surgical Repair and Endovascular Coil Occlusion,” Journal of Neurosurgery, Vol. 98, No. 2, 2003, pp. 319-325. http://dx.doi.org/10.3171/jns.2003.98.2.0319
- A. R. Dehdashti, B. Mermillod, D. A. Rufenacht, et al., “Does Treatment Modality of Intracranial Ruptured Aneurysms Influence the Incidence of Cerebral Vasospasm and Clinical Outcome?” Cerebrovascular Diseases, Vol. 17, No. 1, 2004, pp. 53-60. http://dx.doi.org/10.1159/000073898
- A. J. Goddard, P. P. Raju and A. Gholkar “Does the Method of Treatment of Acutely Ruptured Intracranial Aneurysms Influence the Incidence and Duration of Cerebral Vasospasm and Clinical Outcome?” Journal of Neurology, Neurosurgery & Psychiatry, Vol. 75, No. 6, 2004, pp. 868-872. http://dx.doi.org/10.1136/jnnp.2003.033068
- D. Hanggi and H. J. Steiger, “The Influence of Cisternal and Ventricular Lavage on Cerebral Vasospasm in Patients Suffering from Subarachnoid Hemorrhage: Analysis of Effectiveness,” Acta Neurochirurgica Supplement, Vol. 110, Pt. 2, 2011, pp. 95-98.
- T. Nakagomi, K. Furuya, H. Nagashima, et al., “Surgical Procedure and Results of Cisternal Washing Therapy for the Prevention of Cerebral Vasospasm Following SAH,” Acta Neurochirurgica Supplement, Vol. 110, Pt. 2, 2011, pp. 105-109.
- S. Muehlschlegel, G. Rordorf and J. Sims, “Effects of a Single Dose of Dantrolene in Patients with Cerebral Vasospasm after Subarachnoid Hemorrhage: A Prospective Pilot Study,” Stroke, Vol. 42, No. 5, 2011, pp. 1301-1306. http://dx.doi.org/10.1161/STROKEAHA.110.603159
- M. Y. Tseng, P. J. Hutchinson and P. J. Kirkpatrick, “Interaction of Neurovascular Protection of Erythropoietin with Age, Sepsis, and Statin Therapy Following Aneurysmal Subarachnoid Hemorrhage,” Journal of Neurosurgery, Vol. 112, No. 6, 2010, pp. 1235-1239. http://dx.doi.org/10.3171/2009.10.JNS09954
- H. C. Lee, C. L. Hsieh, C. C. Chen, et al., “A Pilot Study in Acute Subarachnoid Haemorrhagic Patients after Aneurysm Clipping with Complementary Therapies of Chinese Medicine,” Complementary Therapies in Medicine, Vol. 18, No. 5, 2010, pp. 191-198. http://dx.doi.org/10.1016/j.ctim.2010.07.002
- L. H. Calabrese, D. W. Dodick, T. J. Schwedt and A. B. Singhal, “Narrative Review: Reversible Cerebral Vasoconstriction Syndromes,” Annals of Internal Medicine, Vol. 146, No. 1, 2007, pp. 34-44. http://dx.doi.org/10.7326/0003-4819-146-1-200701020-00007
- A. B. Singhal and R. A. Bernstein, “Postpartum Angiopathy and Other Cerebral Vasoconstriction Syndromes,” Neurocritical Care, Vol. 3, No. 1, 2005, pp. 91-97. http://dx.doi.org/10.1385/NCC:3:1:091
- A. B. Singhal, V. S. Caviness, A. F. Begleiter, E. J. Mark, G. Rordorf and W. J. Koroshetz, “Cerebral Vasoconstriction and Stroke after Use of Serotonergic Drugs,” Neurology, Vol. 58, No. 1, 2002, pp. 130-133. http://dx.doi.org/10.1212/WNL.58.1.130
- D. A. Nowak, S. O. Rodiek, S. Henneken, J. Zinner, R. Schreiner, H. H. Fuchs and H. Topka, “Reversible Segmental Cerebral Vasoconstriction (Call-Fleming Syndrome): Are Calcium Channel Inhibitors a Potential Treatment Option?” Cephalalgia, Vol. 23, No. 3, 2003, pp. 218-222. http://dx.doi.org/10.1046/j.1468-2982.2003.00507.x
- A. B. Singhal, “Cerebral Vasoconstriction Syndromes,” Topics in Stroke Rehabilitation, Vol. 11, No. 2, 2004, pp. 1-6. http://dx.doi.org/10.1310/ATK7-QTP7-7NE2-5G8X
- S. P. Chen, J. L. Fuh, J. F. Lirng, F. C. Chang and S. J. Wang, “Recurrent Primary Thunderclap Headache and Benign CNS Angiopathy: Spectra of the Same Disorder?” Neurology, Vol. 67, No. 12, 2006, pp. 2164-2169. http://dx.doi.org/10.1212/01.wnl.0000249115.63436.6d
- A. Ducros, U. Fiedler, R. Porcher, M. Boukobza, C. Stapf and M. G. Bousser, “Hemorrhagic Manifestations of reversible Cerebral Vasoconstriction Syndrome: Frequency, Features, and Risk Factors,” Stroke, Vol. 41, No. 11, 2010, pp. 2505-2011. http://dx.doi.org/10.1161/STROKEAHA.109.572313
- S. P. Chen, J. L. Fuh, F. C. Chang, J. F. Lirng, B. C. Shia and S. J. Wang, “Transcranial Color Doppler Study for Reversible Cerebral Vasoconstriction Syndromes,” Annals of Neurology, Vol. 63, No. 6, 2008, pp. 751-757. http://dx.doi.org/10.1002/ana.21384
- S. P. Chen, J. L. Fuh and S. J.Wang, “Reversible Cerebral Vasoconstriction Syndrome: An Under-Recognized Clinical Emergency,” Therapeutic Advances in Neurological Disorders, Vol. 3, No. 3, 2010, pp. 161-171. http://dx.doi.org/10.1177/1756285610361795
- M. Elstner, J. Linn, S. Müller-Schunk and A. Straube, “Reversible Cerebral Vasoconstriction Syndrome: A Complicated Clinical Course Treated with Intra-Arterial Application of Nimodipine,” Cephalalgia, Vol. 29, No. 6, 2009, pp. 677-682. http://dx.doi.org/10.1111/j.1468-2982.2008.01768.x
- M. Klein, G. Fesl, H. W. Pfister, A. Straube, H. Brückmann, L. A. Hoffmann and T. Pfefferkorn, “Intra-Arterial Nimodipine in Progressive Postpartum Cerebral Angiopathy,” Cephalalgia, Vol. 29, No. 2, 2009, pp. 279-282. http://dx.doi.org/10.1111/j.1468-2982.2008.01728.x
- M. Bouchard, S. Verreault, J. L. Gariépy and N. Dupré, “Intra-Arterial Milrinone for Reversible Cerebral Vasoconstriction Syndrome,” Headache, Vol. 49, No. 1, 2009, pp. 142-145. http://dx.doi.org/10.1111/j.1526-4610.2008.01211.x
NOTES
*Corresponding author.