Open Journal of Rheumatology and Autoimmune Diseases
Vol. 2 No. 2 (2012) , Article ID: 18965 , 6 pages DOI:10.4236/ojra.2012.22006
Osteoprotegerin Secretion by Mevastatin via p38MAPK and NF-kB
![]()
Institute of Science Techoology and Medicine, Keele University, Staffordshire, UK.
Email: Helenl.smith@rjah.nhs.uk
Received February 23rd, 2012; revised March 25th, 2012; accepted March 30th, 2012
Keywords: Osteoprotegerin; Osteoporosis; Statin; Mevastatin
ABSTRACT
Osteoprotegerin (OPG) is a protein produced by many cell types that has the remarkable property of inhibiting bone loss. It does this by binding to the key bone resorptive cytokine, receptor activator of NF-kB ligand (RANKL). This cytokine is produced mainly by osteoblastic cells and is instrumental in osteoclast differentiation. If the ratio of RANKL: OPG increases, bone resorption increases and results in bone loss in diseases such osteoporosis, rheumatoid arthritis and hypercalcaemia of malignancy. Hence, if drugs can be found that increase OPG, this will decrease the activity of osteoclasts and therefore bone resorption. Statins are cholesterol lowering drugs that have recently been shown to increase bone formation in rodents. It was hypothesised from this finding that this could be due to an increase in OPG production. If these commonly prescribed drugs could be used to prevent bone loss or to increase bone formation then this may prove a useful means of reducing fracture risk in patients. Treating Saos-2 osteoblast-like cells in vitro with mevastatin increased OPG production and secretion through the mevalonate pathway. A failure of geranylgeranylation of Rho and/or farnesylation of Ras proteins leads to an increase in PI-3K activation then AKT activation leading to several different signaling pathways such as MAPK’s and NF-kB. NF-kB and p38MAPK inhibitors prevented the statin stimulation of OPG but not the decrease in cell number, suggesting that statins regulate OPG secretion via PI-3K, p38MAPK and NF-kB.
1. Introduction
As well as lowering LDL and cholesterol levels which statins were originally marketed for, other non-lipid lowering beneficial “pleiotropic” effects of statins have become evident. Positive effects of statins include, decreased incidence of dementia [1], anti-tumour properties [2,3], reduced fracture risk [4], increased bone formation [5,6], and anti-atherosclerotic effects [7].
Statins have also been reported to increase bone formation. If this result can be translated into a useful clinical effect then statins may be an alternative to bone anabolic agents like teriparatide. Statins may have additional advantages, such as they would be cheaper than teriparatide and they are taken orally rather than subcutaneously. Mundy et al. [5] examined the effect of 30,000 natural compounds in search of an agent that increases osteoblast differentiation by increasing the activity of the promoter of bone morphogenic protein-2 (BMP-2). BMP- 2 is a cytokine secreted by osteoblastic progenitor cells which stimulates the differentiation of osteoblastic cells [8]. Lovastatin was the only compound that increased BMP-2 activity. This finding was then repeated using simvastatin, mevastatin and fluvastatin to treat murine (2T3) and human (MG63) bone cells. The result was an increase in BMP-2 mRNA. Following these results Mundy et al. [5] went on to investigate the biological effects of mevastatin and lovastatin on mouse calvarial bones in culture. After 72 hours bone formation had increased two fold compared to the control, there was also an increase in osteoblast number. In order to look at the effect of statins on bone formation in vivo lovastatin and simvastatin were injected three times a day for 5 days into the subcutaneous tissue overlying the calvaria. The mice were then killed on day 21. Results showed that there was approximately a 50% increase in bone formation. The final experiment carried out by Mundy et al. [5] was to determine the effect of statins when given systemically. Three month old ovariectomised rats were treated orally with varying doses of simvastatin for 35 days. Compared to controls simvastatin increased trabecular bone volume and bone formation rate and also decreased osteoclast cell number [5].
Following this in vivo work in rats by Mundy et al. [5] the experiments have been reproduced by other research groups including Skogland, Forslund and Aspenberg [9] and Oxlund and Andreassen [10]. Skogland, Forslund and Aspenberg [9] looked at BALB/c mice with fractured femurs, 41 of these mice were fed food prepared with simvastatin and the other 41 mice were given food excluding the simvastatin. On examining the fracture callus at day 14 the callus in treated mice was 53% larger than controls and the force needed to break the bone was 63% greater than the controls. Oxlund and Andreassen [10] carried out an investigation using 60 4-month old Wistar female rats. The rats were divided into four groups; baseline control, sham + placebo group, ovariectomy (OVX) + placebo, and OVX + simvastatin. Simvaststin or placebo were given twice daily by a gastric tube for three months. Findings showed that the bone formation rate increased by two fold, a reduced rate of cancellous bone loss and a decrease in TRAP activity (indicating decreased osteoclast activity) in the OVX + simvastatin group compared to the OVX + placebo. It was concluded that simvastatin induces an increase in bone formation at the periosteal surface and reduces the loss of cancellous bone induced by ovariectomy [10].
Using primary human osteoblastic cells cultured from iliac crest bone fragments Viereck et al. [11] investigated the effect of atorvastatin on markers of osteoblastic differentiation. They found a dose dependent increase in osteoprotegerin (OPG) protein and OPG mRNA production with a maximum of three fold increases at a concentration of atorvastatin of one micromolar. These results were time dependent with atorvastatin increasing OPG mRNA levels from 24 hours and OPG protein secretion between 48 and 72 hours. They also found atorvastatin to increase the expression of the two osteoblastic differentiation markers alkaline phosphatase and osteocalcin. In summary, treatment of primary human osteoblasts with atorvastatin increases osteoblastic differentiation and the production of OPG. This offers a possible explanation of how statins result in increased bone formation through OPG induced inhibition of bone resorption. Hence, statins may be a useful future treatment for osteoporosis [11].
Here we have focused on the general effect of statins on OPG secretion from cultured osteoblastic cells and the detailed signalling mechanisms affected.
2. Methods
Saos-2 osteoblast like cells immortalised from a human osteosarcoma [12] were from the European Collection of Cell Cultures, they were routinely cultured using α-MEM medium, containing 10% foetal bovine serum (FCS), 100 IU/ml benzylpenicillin, 100 μg/ml streptomycin and 2 mM L-glutamine (complete medium) [13]. Cells were maintained in a humidified environment at 37˚C, 5% carbon dioxide in air, unless stated otherwise.
For each experiment looking at the effects of statins on OPG secretion and the signalling mechanisms involved, Saos-2 cells were seeded into 96 well tissue culture plates at a density of 10,000 cells per well in 200 μl of complete medium until confluent, the medium was then changed and effectors added. Following the treatment periods culture media from each well was removed and stored at –20˚C. 100 μl of fresh culture medium and an additional 10 μl of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxypheyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (MTS) and 10 μl of phenazine ethosulfate (PES) were added to each well to give a final concentration of 150 μg/ml MTS and 30 μM PES to determine an index of cell metabolic activity [14]. After 60 minutes the optical densities were measured at 490 nm with a 700 nm reference filter using a Bio-Tek Synergy HT plate reader. The MTS assay was routinely used to ensure that cell number did not vary across tissue culture plates, and to determine the OPG concentration secreted per million cells. MTS standard curves were produced for each experiment; cells were seeded at varying densities from 0 - 100,000 and allowed to adhere for 1 hour. The MTS assay was then performed on each well, giving optical densities for specific cell numbers allowing unknown cell numbers to be calculated from MTS optical density.
The concentration of secreted OPG by the Saos-2 cells was measured in the culture media by enzyme linked immuno-sorbant assay (ELISA); briefly a 96 well ELISA plate was coated with 100 μl of 200 ng/ml anti-human OPG (R&D Systems, AF805) in 0.1 M sodium bicarbonate pH 8.6 and incubated overnight at 4˚C. Excess primary antibody was discarded and non-specific binding to the polystyrene plate blocked with 200 μl of milk per well (8 ml of skimmed milk in 100 ml of 0.1 M Tris HCl buffer pH 7.4), incubated at 4˚C for 1 hour. The plate was washed three times each for 2 minutes with 200 μl of 0.2% Tween 20 in 0.1 M Tris HCl pH 7.4. Samples and recombinant human OPG standards (R&D Systems, 805- 05-100) (100 μl) were added to the plate and incubated for a further 1 hour at 4˚C. Samples were discarded and the plate washed as previously described. A biotinylated anti-human OPG antibody (R&D Systems, BAF805) (100 μl) at a concentration of 200 ng/ml was added to each well and the plate incubated for 1 hour at 4˚C. This secondary antibody solution was then discarded and the plate washed again (as above). Avidin peroxidase (Sigma, E2886) was diluted in milk to a concentration of 2 μg/ml and 100 μl added to each well and the plate incubated for a final 1 hour at 4˚C. The plate was washed and 100 μl per well of 3, 3’5, 5 Tetramethylbenzidine (TMB) was added to each well, and incubated at room temperature for 30 minutes. This reaction was then stopped using 10% sulphuric acid. Optical densities were read at 450 nm and 570 nm was used as a reference wavelength on a Bio-Tek Synergy HT plate reader (Bio-Tek® Instruments, Inc. Vermont USA).
3. Results and Discussion
Statins are prescribed to patients for their cholesterol lowering properties [15,16]. Therefore osteoblastic cells were co-incubated with cholesterol and mevastatin to determine if the statin was having its effects by lowering cell membrane cholesterol. The results showed that 10 μM mevastatin significantly increased OPG secretion (**P < 0.001) to approximately 425 nanograms/million cells (±50 SD) compared to a control (no mevastatin and no cholesterol) level of approximately 100 nanograms/ million cells (±10 SD), addition of cholesterol (0 - 32 μg/ml) did not inhibit the stimulation of OPG secretion (Figure 1). This suggests that effects on OPG secretion are not as a result of the statin reducing the cholesterol concentration in the cell.
Further down the mevalonate pathway intermediates are produced which are used by the cell to modify target proteins (prenylation). This involves the addition of a farnesyl (C15) or geranylgeranyl (C20) group to appropriate cysteine residues and is important in determining the activities of proteins and in targeting them to specific subcellular membrane domains [17]. Many signalling proteins such as small GTP binding proteins are modified by prenylation; such as Ras which is only active after farnesylation and members of the Rho subfamily (RhoA, RhoC, Rac1 and Cdc42) which are geranylgeranylated [18]. To determine whether the statin effect on OPG secretion was occurring due to a lack of farnesylated or geranylgeranylated proteins the isoprenoids involved in
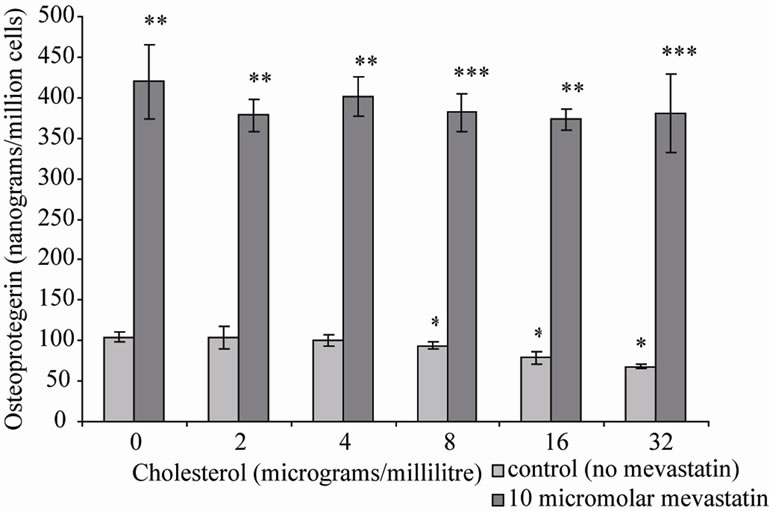
Figure 1. Osteoprtegerin (OPG) secretion from Saos-2 cells and the effect of mevastatin with and without cholesterol. Saos-2 cells were incubated with cholesterol with or without 10 micromolar mevastatin for 48 hours. Results significantly different from the control (no mevastatin and no cholesterol) are shown by *P < 0.05, **P < 0.01 and ***P < 0.001. Data is presented as mean ± SD.
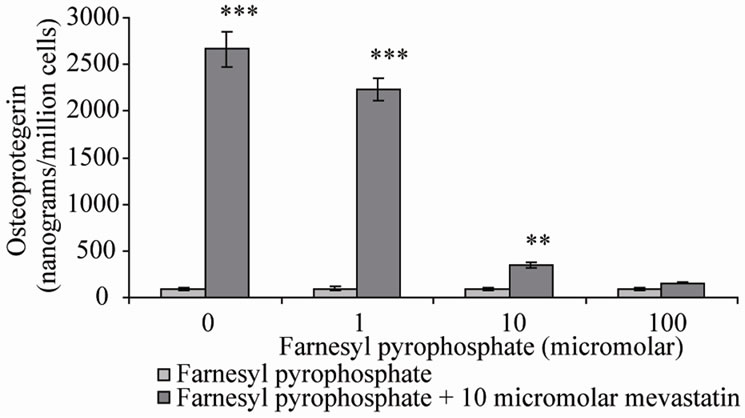
these reactions, farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP) were co-incubated with mevastatin on Saos-2 cells. Note that in the presence of statin, GGPP can not be formed by adding FPP despite being down stream of FPP in the mevalonate pathway. This is because of the requirement for a five carbon isoprene from earlier in the pathway. The results showed that as previously 10 μM mevastain significantly increased OPG secretion (***P < 0.001), addition of FPP dose-dependently inhibited the statin stimulation of OPG secretion, with full prevention at 100 μM (Figure 2(a)), 10 μM GGPP also prevented the increase in OPG secretion (Figure 2(b)). These results suggest that both farnesylation and geranylgeranylation play a part in OPG regulation.
It is important to be clear about the relative effects of FPP and GGPP because these two intermediates are used to modify different families of proteins such as the Ras and Rho sub families respectively. This could help to identify the prenylated proteins involved in the action of statins on cells. The above findings with FPP and GGPP were therefore investigated further using various effec-
 (a)
(a)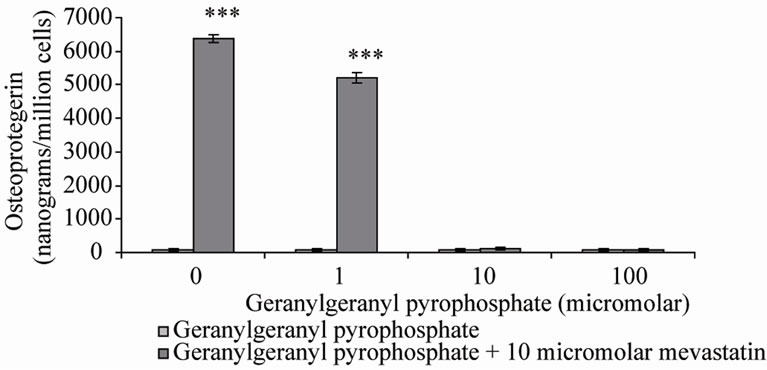 (b)
(b)
Figure 2. Osteoprotegerin (OPG) secretion from Saos-2 cells and the effect of mevastatin with and without (a) Farnesyl pyrophosphate (FPP) and (b) Geranylgeranyl pyrophosphate (GGPP). Saos-2 cells were incubated for 48 hours with 10 micromolar mevastatin and increasing concentrations of FPP or GGPP. Results significantly different from the control (no mevastatin and no FPP or GGPP) are shown by *P < 0.05, **P < 0.01 and ***P < 0.001. Data is presented as mean ± SD.
tors that inhibit downstream proteins.
The phosphotidylinositide-3 kinase (PI-3K/AKT) pathway is essential for cell growth, proliferation, survival and nitric oxide production. These effects have been shown to occur through the transcription factor, AKT, which is a downstream target of PI-3K. Activation of the PI-3K pathway requires a ligand such as VEGF (vascular endothelial growth factor) [19] to bind to a HER kinase receptor. This binding results in HER kinase activation and auto-trans-phosphorylation of intracellular domains and activation of PI-3K [20]. PI-3K catalyses the phosphorylation of phosphatidylinositol (PI) lipids resulting in the release of PI(3,4)P2 and PI(3,4,5)P3. These cause the phosphotidylinositide dependent kinase (PDK1) to translocate to the cell membrane where it is phosphorylated. PDK1 binds directly to AKT and phosphorylates it at Thr-308. This results in autophosphorylation of Ser-473 and hence increased AKT activity [19]. PI-3K contains a Src homology domain and two proline rich regions that flank a breakpoint cluster region homology (BH) domain. This BH domain is structurally similar to GTPase activating proteins for the Rho family of small G proteins. Rac and Cdc42 can bind to this domain and be activated [21].
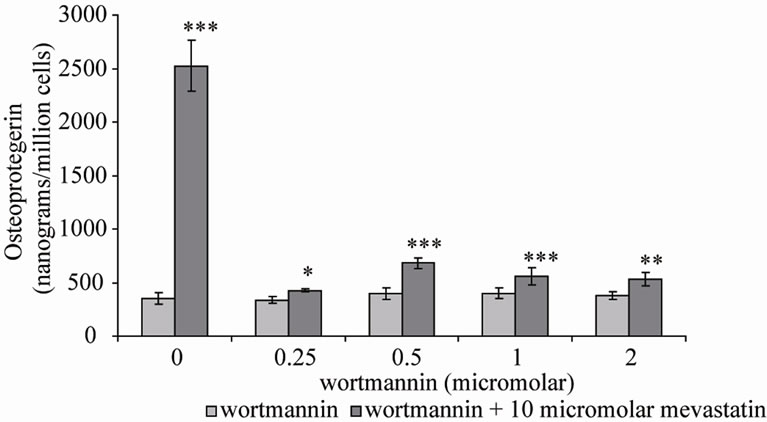
Two inhibitors of PI-3K were co-incubated with mevastatin on saos-2 cells; wortmannin and LY294002. Figure 3(a) shows 10 μM mevastatin significantly increased OPG secretion by approximately 6 fold from 400 nanograms/million cells to 2500 nanograms/million cells (***P < 0.001), this stimulation by mevastatin was reduced to less than a 1 fold increase by co-incubation with concentrations of wortmannin as little as 0.25 μM, however concentrations of secreted OPG were still significantly raised compared to control (no mevastatin and no wortmannin). Figure 3(b) shows that LY294002 dose dependently inhibited the mevastatin stimulated increase in OPG, with it being totally prevented at 50 nM. These observations indicate that statins have their effect by increasing PI-3K activity and that this is an early intermediary in the effects of mevastatin. Statins appear to act to increase PI-3K activity through their inhibition of the production of farnesyl and geranylgeranyl pyrophosphates and subsequent modification of Rho family proteins. In the absence of this modification these proteins will not be able to localise to the cell membrane but how this deficiency results in the activation of PI-3K is not clear. Downstream of PI-3K, AKT activates mitogen activated protein kinases (MAPK) pathways and the NF-kB pathway.
NF-kB is a transcription factor that promotes cell survival and protects cells from apoptosis and its activation is thought to be part of a stress response. BAY 11-7082, a specific inhibitor of NF-kB, dose dependently inhibited the statin stimulation of OPG, although did not quite re-
 (a)
(a)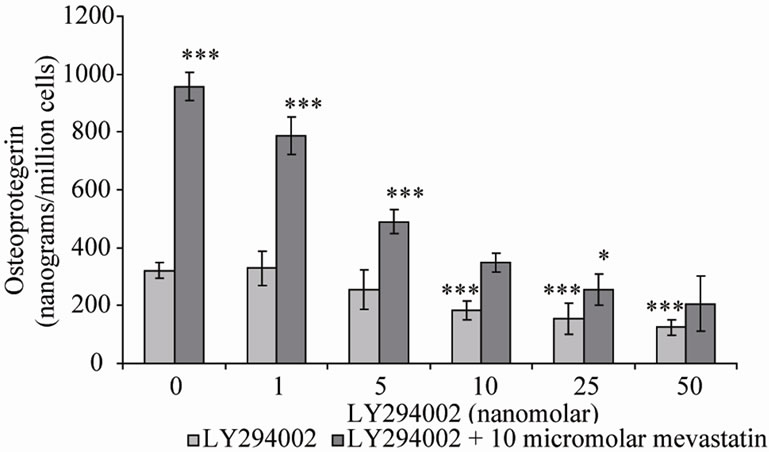 (b)
(b)
Figure 3. Osteoprotegerin (OPG) secretion from Saos-2 cells and the effect of mevastatin with and without (a) Wortmannin and (b) LY294002. Saos-2 cells were incubated with 10micromolar mevastatin and increasing concentrations of wortmannin or LY294002 for 48 hours. Results significantly different from the control (no mevastatin and no wortmannin or LY294002) are shown by *P < 0.05, **P < 0.01 and ***P < 0.001. Data is presented as mean ± SD.
duce it back to control levels. 10 μM mevastatin alone significantly increased the OPG concentration (***P < 0.001) from roughly 100 nanograms/million cells (±15 SD) to 1100 nanograms/million cells (±50 SD). 100 μM BAY 11-7082 reduced this increase in OPG secretion to roughly 300 nanograms/million cells (±75 SD) this however was still significantly greater (*P < 0.05) than the control (no mevastatin and no BAY 11-7082) (Data not shown).
Further inhibitors were used as co-treatments in order to elucidate the role of mitogen activated protein kinases (MAPK’s), p38MAPK, ERK and JNK, in the response of Saos-2 cells to statin treatment. MAPK’s regulate cell cycle progression through G1 phase, regulation of embryonic development, cell movement, apoptosis and differentiation. These three different MAPK signalling cascades all contain the same series of three kinases. A MEK kinase (MEKK) phosphorylates and activates a MAP kinase kinase (MEK), this MEK then phosphorylates and activates a MAP kinase (MAPK) [22].
SB202190, a p38MAPK inhibitor, produced similar results to BAY 11-7082, the stimulation in OPG secretion by mevastatin was inhibited by 100nM SB202190 by approximately 50%. However the concentration of secreted OPG remained significantly raised (***P < 0.001) compared to control level (no mevastatin and no SB202190) (Figure 4). In contrast further inhibitors; U 0126 and SP 600125 for ERK and JNK respectively did not prevent the statin stimulation of OPG.
ERK activation is independent of PI-3K and is instead activated by the farnesylated Ras directly via the MEK kinase Raf-1. To further confirm that the statin effects were not as a result of ERK activation, Saos-2 cells were treated with a Raf-1 kinase inhibitor. This did not have any effect on OPG secretion (data not shown). This suggests along with earlier findings that elevation of OPG is not as a result of mechanisms downstream of Ras farnesylation or as a result of ERK inhibition.
4. Conclusions
The effects of one member of the statin family, mevastatin, were overcome by mevalonate. This showed that mevastatin was acting to block the mevalonate pathway. Farnesyl and geranylgeranyl pyrophosphate intermediates down stream of mevalonate could also overcome the effects of mevastatin, however, addition of cholesterol could not. These results suggest that intermediates in the mevalonate pathway, probably farnesyl and geranylgeranyl pyrophosphate, were crucial in the mechanism of action of statins on Saos-2 cells. Farnesyl and geranylgeranyl pyrophosphate are used by cells to modify specific proteins so that they adhere to lipid membranes on the inner surface of cell membranes. The most ubiquitous
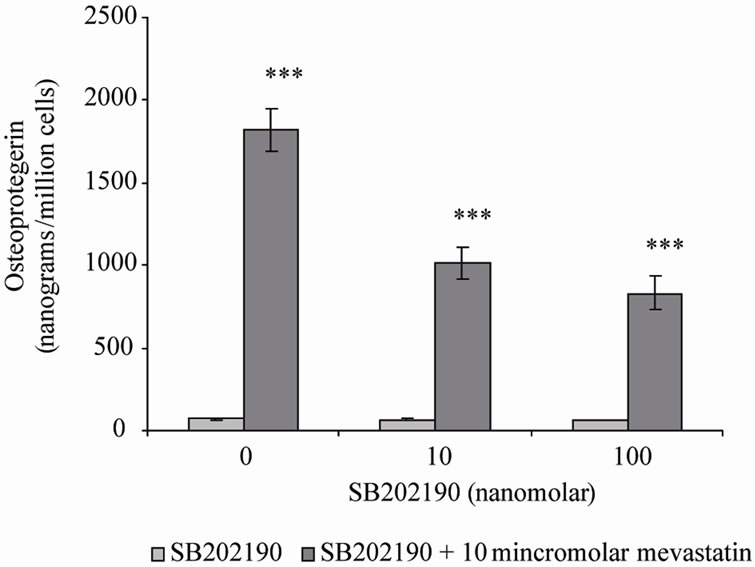
Figure 4. Osteoprotegerin (OPG) secretion from Saos-2 cells and the effect of mevastatin with and without SB202190. Saos-2 cells were incubated with 10 micromolar mevastatin and either 10 or 100 nanomolar SB202190 for 48 hours. Results significantly different from the control (no mevastatin and no SB202190) are shown by *P < 0.05, **P < 0.01 and ***P < 0.001. Data is presented as mean ± SD.
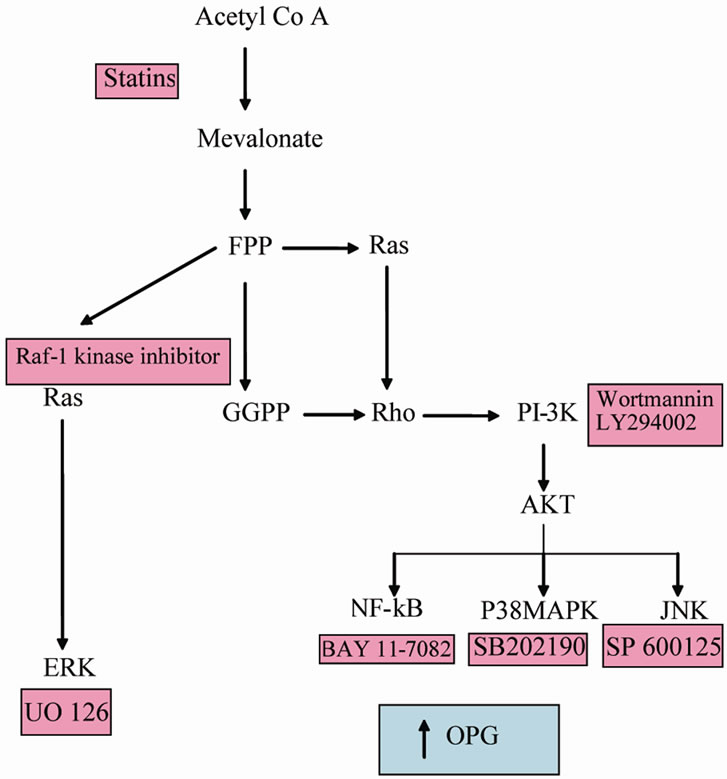
Figure 5. The mevalonate pathway indicating where signalling inhibitors function which pathways result in increased OPG secretion.
of these proteins belong to the Ras and Rho family which are GTP binding molecular switches associated with a wide range of protein tyrosine kinase receptors. Also widely associated with these receptors is the enzyme PI-3K. Inhibtion of this enzyme with two unrelated inhibitors blocks the effects of statins in these cells. It appears likely therefore that in the presence of statin, a failure to properly modify Rho and/or Ras proteins leads to an activation of PI-3K or alternatively aberrant Rho and/or Ras fail to down regulate PI-3K. Active PI-3K leads to the transcription factor, phosphorylated AKT which leads to a range of signal transduction pathways including the three MAPK pathways, NF-kB activation and cytoskeletal rearrangements. Inhibitor studies showed that while JNK and ERK were not involved in mediating the effects of statins, P38 and NF-kB pathways were responsible for OPG elevation. These findings are diagrammatically summarised in Figure 5.
Thus, one can postulate that statins increase PI-3K which activates AKT leading to elevated OPG via p38- MAPK and NF-kB. If this mechanism occurs in patients treated with statins then the elevated OPG might inhibit bone loss that occurs with ageing and thus, diminish fracture prevalence.
5. Acknowledgements
Michael Marshall from the Robert Jones and Agnus Hunt Orthopaedic Hospital, Oswestry, UK for his support and advice during this research.
REFERENCES
- S. T. Dekosky, “Statin Therapy in the Treatment of Alzheimer Disease: What Is the Rationale?” American Journal of Medicine, Vol. 118, No. 12, 2001, pp. 48-53.
- M. Z. Katz, “Therapy Insight: Potential of Statins for Cancer Chemoprevention and Therapy,” Nature Clinical Practice Oncology, Vol. 2, No. 2, 2005, pp. 82-89. doi:10.1038/ncponc0097
- A. O. Muck, H. Seeger and D. Wallwiener, “Inhibitory Effect of Statins on the Proliferation of Human Breast Cancer Cells,” International Journal of Clinical Pharmacology and Therapeutics, Vol. 42, No. 12, 2004, pp. 695-700.
- R. E. Scranton, M. Young, E. Lawler, D. Solomon, D. Gagnon and J. M. Gaziano, “Statin Use and Fracture Risk: Study of a Us Veterans Population,” Archives of Internal Medicine, Vol. 165, No. 17, 2005, pp. 2007-2012. doi:10.1001/archinte.165.17.2007
- G. Mundy, R. Garrett, S. Harris, J. Chan, D. Chen, G. Rosini, B. Boyce, M. Zhao and G. Gutierrez, “Stimulation of Bone Formation in Vitro and in Rodents by Statins,” Science, Vol. 286, No. 5446, pp. 1946-1949.
- F. von Knoch, C. Wedemeyer, A. Heckelei, G. Saxler, G. Hilken, J. Brankamp, T. Sterner, S. Landgraeber, F. Henschke, F. Loer and M. von Knoch, “Promotion of Bone Formation by Simvastatin in Polyethylene Particle-Induced Osteolysis,” Biomaterials, Vol. 26, No. 29, 2005, pp. 5783-5789. doi:10.1016/j.biomaterials.2005.02.008
- P. Riboldi, M. Gerosa and P. L. Meroni, “Statins and Autoimmune Disease,” Lupus, Vol. 14, No. 9, 2005, pp. 765-768. doi:10.1191/0961203305lu2217oa
- M. Koida, R. Fukuyama and H. Nakamuta, “Osteoporosis Requires Bone-Specific Statins,” Current Pharmaceutical Design, Vol. 10, No. 21, 2004, pp. 2605-2613. doi:10.2174/1381612043383827
- B. Skoglund, C. Forslund and P. Aspenberg, “Simvastatin Improves Fracture Healing in Mice,” Journal of Bone and Mineral Research, Vol. 17, No. 11, 2002, pp. 2004-2008. doi:10.1359/jbmr.2002.17.11.2004
- H. Oxlund and T. T. Andreassen, “Simvastatin Treatment Partially Prevents Ovariectomy-Induced Bone Loss While Increasing Cortical Bone Formation,” Bone, Vol. 34, No. 4, 2004, pp. 609-618. doi:10.1016/j.bone.2003.12.014
- V. Viereck, C. Grundker, S. Blaschke, K.-H. Frosch, M. Schopper, G. Emons and L. C. Hofbauer, “Atorvastatin Stimulates the Production of Osteoprotegerin by Human Osteoblasts,” Journal of Cellular Biochemistry, Vol. 96, No. 6, 2005, pp. 1244-1253. doi:10.1002/jcb.20598
- J. Fogh, J. M. Fogh and T. Orfeo, “One Hundred and Twenty-Seven Cultured Human Tumour Cell Lines Producing Tumours in Nude Mice,” Journal of the National Cancer Institute, Vol. 59, No. 1, 1977, pp. 221-226.
- C. D. Helgason and C. L. Miller, “Basic Cell Culture,” 3rd Edition, Humana Press, England, pp. 1-371.
- J. Kumi-Diaki, S. Saddler-Shawnette, A. Aller and J. Brown, “Potential Mechanism of Phytochemical-Induced Apoptosis in Human Prostate Adenocarcinoma Cells: Therapeutic Synergy in Genistein and B-Lapachone Combination Treatment,” Cancer Cell International, Vol. 4, No. 5, 2004, pp. 1475-2867. doi:10.1186/1475-2867-4-5
- M. A. Omar and J. P. Wilson, “FDA Adverse Event Report on Statin-Associated Rhabdomyolysis,” The Annals of Pharmacotherapy, Vol. 36, No. 2, 2002, pp. 288-295. doi:10.1345/aph.1A289
- J. Shepherd, “Lipid Lowering: Statins and the Future,” Heart, Vol. 84, 2000, pp. 46-47. doi:10.1136/heart.84.suppl_1.i46
- T. Magee and M. C. Seabra, “Fatty Acylation and Prenylation of Proteins: What’s Hot in Fat,” Current Opinion in Cell Biology, Vol. 17, No. 2, 2005, pp. 190-196. doi:10.1016/j.ceb.2005.02.003
- M. R. Graaf, D. J. Richel, C. J. F. Noorden and H.-J. Guchelaar, “Effects of Statins and Farnesyltransrease Inhibitors on the Development and Progression of Cancer,” Cancer Treatment Review, Vol. 30, No. 7, 2004, pp. 609- 641. doi:10.1016/j.ctrv.2004.06.010
- J. P. Gratton, M. Morales-Ruiz, Y. Kureishi, D. Fulton, K. Walsh and W. C. Sessa, “Akt Down-Regulation of P38 Signalling Provides a Novel Mechanism of Vascular Endothelial Growth Factor-Mediated Cytoprotection in Endothelial Cells,” The Journal of Biological Chemistry, Vol. 276, No. 32, 2001, pp. 30359-30365. doi:10.1074/jbc.M009698200
- Q. B. She, D. B. Solit, K. E. O’Reilly, J. Lobo and N. Rosen, “The Bad Protein Integrates Survival Signaling by Egfr/Mapk and Pi3k/Akt Kinase Pathways in Pten-Deficient Tumour Cells,” Cancer Cell, Vol. 8, No. 4, 2005, pp. 287-297. doi:10.1016/j.ccr.2005.09.006
- A. C. Donahue and D. A. Fruman, “Pi3K Signaling Controls Cell Fate at Many Points in B Lymphocyte Development and Activation,” Seminars in Cell and Developmental Biology, Vol. 15, No. 2, 2004, pp. 183-197. doi:10.1016/j.semcdb.2003.12.024
- T. Ravingerova, M. Barancik and M. Strniskova, “Mitogen-Activated Protein Kinases: A New Therapeutic Target in Cardiac Pathology,” Molecular and Cellular Biochemistry, Vol. 247, No. 1-2, 2003, pp. 127-138. doi:10.1023/A:1024119224033