Open Journal of Blood Diseases
Vol.3 No.3A(2013), Article ID:38951,11 pages DOI:10.4236/ojbd.2013.33A005
Distinct Transforming Activity of ABL Family Tyrosine Kinase Oncogenes Is Induced by Their C-Terminal Domain*
![]()
1Department of Molecular Diagnostics and Therapeutics, Kyoto Prefectural University of Medicine Graduates School of Medical Science, Kyoto, Japan; 2Department of Transfusion Medicine and Cell Therapy, Kyoto University Hospital, Kyoto, Japan.
Email: #keiko_o@koto.kpu-m.ac.jp
Copyright © 2013 Keiko Okuda, Hideyo Hirai. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received September 10th, 2013; revised October 10th, 2013; accepted October 18th, 2013
Keywords: Leukemia; Chimeric Oncogene; ABL Family Tyrosine Kinase; Signal Transduction
ABSTRACT
The TEL/ARG oncogene is similar in structure to the TEL/ABL fusion found in human leukemia, however, we have demonstrated previously that the expression of TEL/ARG in Ba/F3 cells does not sustain strong activity of proliferation, whereas, that of TEL/ABL appeared to induce immediate cell proliferation. To study the molecular basis of the difference in the transforming activity of TEL/ARG and TEL/ABL, TEL/ARG mutants that swapped the kinase domain or C-terminus of ARG with the corresponding domain in ABL were generated, and each mutant was expressed in Ba/F3 cells. A TEL/ARG mutant containing the ABL kinase domain was similar to TEL/ARG in this study, but replacing the ARG C-terminal domain with that of ABL resulted in accelerated proliferation that was similar to that of TEL/ABL. When expressed in primary mouse bone marrow cells by retroviral transduction, spontaneous colony formation in methylcellulose culture was observed, in a fashion dependent on the C-terminal portion of ABL. These results indicate that distinct bio-phenotypes associated with these oncogenes are likely to be regulated by their C-termini, and the C-terminus of ARG contains a functional subdomain that impairs the growth signal induced by ABL family tyrosine kinase.
1. Introduction
ARG (ABL2) is a member of the ABL tyrosine kinase gene family and is highly homologous to ABL (ABL1) in overall domain structure (SH3-SH2-SH1) and amino acid sequence [1-3]. ARG has recently been implicated in the pathogenesis of human acute leukemia through a t (1; 12) (q25; p13) reciprocal translocation that fuses a transcription factor gene, ETV6/TEL, to ARG. The TEL/ARG fusion transcript has been reported in a small number of patients with leukemia, some who had (AML) M3, (AML) M4, or T-cell acute lymphoblastic leukemia, with an additional chromosomal abnormality of t (15; 17) (q12; q21), inv(16) (p13; q12), or t (1; 10; 12) (q25; q23; p13) translocation, respectively [4-6]. Although ABL fusion proteins, such as BCR/ABL and TEL/ABL, are well-established oncogenes in humans, the TEL/ARG translocation is the first instance in which ARG has been implicated in human cancers. The resulting TEL/ARG fusion tyrosine kinase, which contains the same portion of TEL fused to ARG rather than ABL, is similar in structure to the TEL/ABL fusion found in some acute leukemias of B-lymphoid, T-lymphoid, or myeloid origin and atypical or typical CML patients [7-10]. TEL has been identified previously as a frequent fusion partner of tyrosine kinase oncogenes in patents with acute leukemia and myeloproliferative syndrome with a suggestion that the PNT domain in TEL mediates homo-oligomerizaion of TEL/tyrosine kinase fusion oncogenes that generally function as constitutively activated tyrosine kinases, prolonging viability and enhancing proliferation as demonstrated in TEL/ABL [11-14]. However, the exact role of TEL/ARG in leukemogenesis is not well understood yet. Through our previous characterization of these oncogenes, we have reported that the stable expression of TEL/ARG induces ARG tyrosine kinase activation, and, like TEL/ABL, activates a similar set of intercellular signaling pathways then eventually transforms Ba/F3 cells and fibroblasts in vitro in a manner indistinguishable from that of TEL/ABL [15]. We have also demonstrated that ARG kinase activity in TEL/ARG is inhibited by the ABL tyrosine kinase inhibitor (TKI) STI571 [16].
However, these transformed cells can be associated with mutations in other genes, which may not accurately reflect the biological activities of tyrosine kinases, especially if the cell lines were selected for certain properties, such as factor-independent proliferation. In a stringent assay to study the immediate consequences of these oncogenes, sublines of Ba/F3 cells were generated in which a TEL/ARG complementary DNA was expressed under the control of a tetracycline-inducible promoter [17]. The biological effects of activating TEL/ARG using doxycycline are striking changes in viability, hyper-responsiveness to growth factors, and enhanced integrin-mediated short-term adhesion to fibronectin. Although it shows equally high levels of kinase activity, TEL/ARG induces only weak and short-term factor-independent proliferation, in contrast that of TEL/ABL is sufficient, by itself, to induce significant factor-independent proliferation as an immediate consequence.
Given the similarities and differences between TEL/ ARG with TEL/ABL, we examined the domains of ARG and ABL required for biological effects on transforming activity in the context of the TEL-ABL family tyrosine kinase fusion oncogenes by generating mutants with exchanged kinase domains or C-termini of ARG and ABL, and demonstrated that the difference in TEL/ARG and TEL/ABL transforming activity was mediated by the structurally distinct C-terminus of the two proteins.
2. Material and Methods
2.1. Plasmid Constructs
A complementary DNA (cDNA) encoding TEL/ARG was generated by RT-PCR reactions from HT93A cells with FLAG epitope tags at their 3’ ends, as described previously [4]. A TEL/ABL cDNA in a pSRα vector was obtained from Dr TR Golub (Harvard Medical School, Boston MA, USA), and used to generate chimeric cDNAs. Site-directed mutagenesis was carried out using a KOD Plus mutagenesis kit (TOYOBO, Osaka, Japan) in accordance with the manufacturer’s instructions. All the constructs were confirmed by DNA sequencing. The complete individual cDNAs were ligated into either pTRE (Clontech, Palo Alto, CA, USA), which contains a tet-responsive promoter, or the retroviral expression vector pMSCV ires GFP (MIG) [18] at a position 5’ to the internal ribosomal entry site followed by an EGFP gene.
2.2. Cell Lines and Cell Culture
Ba/F3 is an IL-3 dependent murine hematopoietic pro-B cell line [19]. Both Ba/F3 parental and sublines expressing the reverse Tet transactivator pUHD172-1 (TonB1) were maintained in RPMI 1640 medium supplemented with 10% (vol/vol) fetal calf serum (FCS) and 10% conditioned medium from WEHI-3B cells as a source of murine IL-3. Sublines of TonB1 that expressed the mutant TEL/ARG or TEL/ABL by induction, were generated by electroporation using a gene pulser (BioRad, Hercules, CA, USA) as described previously [17].
2.3. Generation of Retrovirus Stocks and Bone Marrow Transduction
High-titer, helper-free retroviral stocks were prepared by transient transfection of Plat-E cells [20] using FuGENE 6 transfection reagent (Roche Diagnostics Corp, Indianapolis, IN, USA) in accordance with the manufacturer’s instructions.
Balb/c mice from 6 to 12 week of age were primed by intraperitoneal injection with 5-fluorouracil (5-FU; 200 mg/kg) 3 days before bone marrow harvest. Femurs and tibias from sacrificed mice were collected, and bone marrow was harvested by flushing with a syringe and 23-gauge needle. Cells were counted and plated in prestimulation medium of DME, 15% FCS, 5% WEHI conditioned medium, penicillin/streptomycin, 200 µM Lglutamine, 6 ng/ml recombinant murine IL-3, 10 ng/ml recombinant murine IL-6, and 100 ng/ml recombinant murine stem cell factor (SCF). After pre-stimulation for 48 h at 37˚C, viable cells were counted and transduced with retroviral stocks in the same medium containing 50% retroviral supernatant, 10 mM Hepes, pH 7.4, and 2 µg/ml polybrene. The medium was changed after a 2 - 4 h adsorption period. At 24 h, a second round of transduction was performed, and the cells were collected 24 h later [21].
2.4. Flow Cytometric Analysis and Cell Sorting
Cell sorting for the transduced cells was performed on a FACS Vantage (Becton Dickinson). Sorting windows were established for populations with an equivalent intensity of GFP fluorescence.
2.5. Immunoblotting and Immunoprecipitation
Cells were deprived of growth factors by culturing them in medium containing 10% FCS in RPMI1640, and were then stimulated with 0.5 µg/ml doxycycline. Immunoblotting and immunoprecipitation were performed using standard chemiluminescence technique as described previously [22].
2.6. Antibodies
Anti-phosphotyrosine monoclonal antibody (4G10), antiPI3-kinase antibody and anti-ARG antibody were purchased from Upstate Biotechnology Inc. (Lake Placed, NY, USA). The anti-ABL, anti-CBL, anti-Doc, and antiStat5 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-Shc antibody was purchased from Transduction Laboratories Inc. (Lexington, KY, USA). The anti-FLAG M2 antibody was purchased from SIGMA-ALDRICH (St. Louis, M0, USA). The anti-ERK1/2 antibody was from Cell Signaling Technology (Beverly, MA, USA).
2.7. Proliferation and Viability Assays
The number of viable cells was determined using trypan blue, and viable cells were counted using a hemocytometer. Cell concentrations were calculated as the number of cells × 10,000/ml. Cell viability was reported as a percentage of total cells.
2.8. Migration Assay
Migration assays were performed using Transwell plates (8 µm pore-size polycarbonate membrane, Corning Coster Corp., Cambridge, MA, USA). Both sides of the membrane were coated with human fibronectin (5 µg/ml). The lower chamber contained 600 µl of RPMI 1640 containing 0.5% BSA. 0.2 × 106 cells in 100 µl of RPMI 1640 containing 0.5% BSA with or without doxycycline were placed into the upper chamber and allowed to migrate into the lower chamber for 5 h at 37˚C.
2.9. Clonal Bone Marrow Cell Culture
Methylcellulose culture cocktail MethoCult (Stem Cell Technology Inc., Vancouver, BC, Canada), with or without murine colony stimulating factors, was used for the culture. The primary murine bone marrow from donors pretreated with 5-FU was transduced with retroviral stocks expressing each indicated oncogene or with empty MIG vector as described already. The transduced bone marrow cells were sorted and suspended in MethoCult, then plated 1 ml each in 35-mm Lux suspension culture dishes (Nunc Inc., Naperville, IL, USA). Dishes were incubated at 37˚C in a fully humidified atmosphere with 5% CO2. On days 14 - 16 of incubation, all colonies were scored under an inverted microscope according to their typical morphologic appearance.
3. Results
3.1. Doxycycline-Dependent Activation of Tyrosine Kinase Activity in TonB Cells
The IL-3 dependent TonB cell line was transfected with cDNAs (Figure 1(a)), and more than six polyclonal, independently derived sublines from each transfection were generated. Three or more lines with an approximately equal expression of the appropriate oncogene, as measured by immunoblotting with either anti-ABL or anti-ARG antibodies, were then selected for further study. Treatment of these cells with doxycycline induced the expression of each mutant, as shown in Figure 1(b) (bottom). The results showed that each of the mutants was auto-phosphorylated (Figure 1(b), top). The tyrosine phosphorylation of other cellular proteins was also induced in those sublines treated with doxycycline (Figure 2).
Several specific proteins are known to be tyrosine phosphorylated by either wild-type TEL/ABL or TEL/ ARG [15,23]. Therefore, the ability of the chimeric mutants to phosphorylate these proteins was tested (Figure 3). The overall level and pattern of tyrosine-phosphorylated proteins were similar in TonB cells expressing the different catalytically active chimeric proteins, and there were no significant differences in the constitutive activation of MAPK/ERK, PI3K, or STAT5, as assessed by Western blotting.
 (a)
(a) (b)
(b)
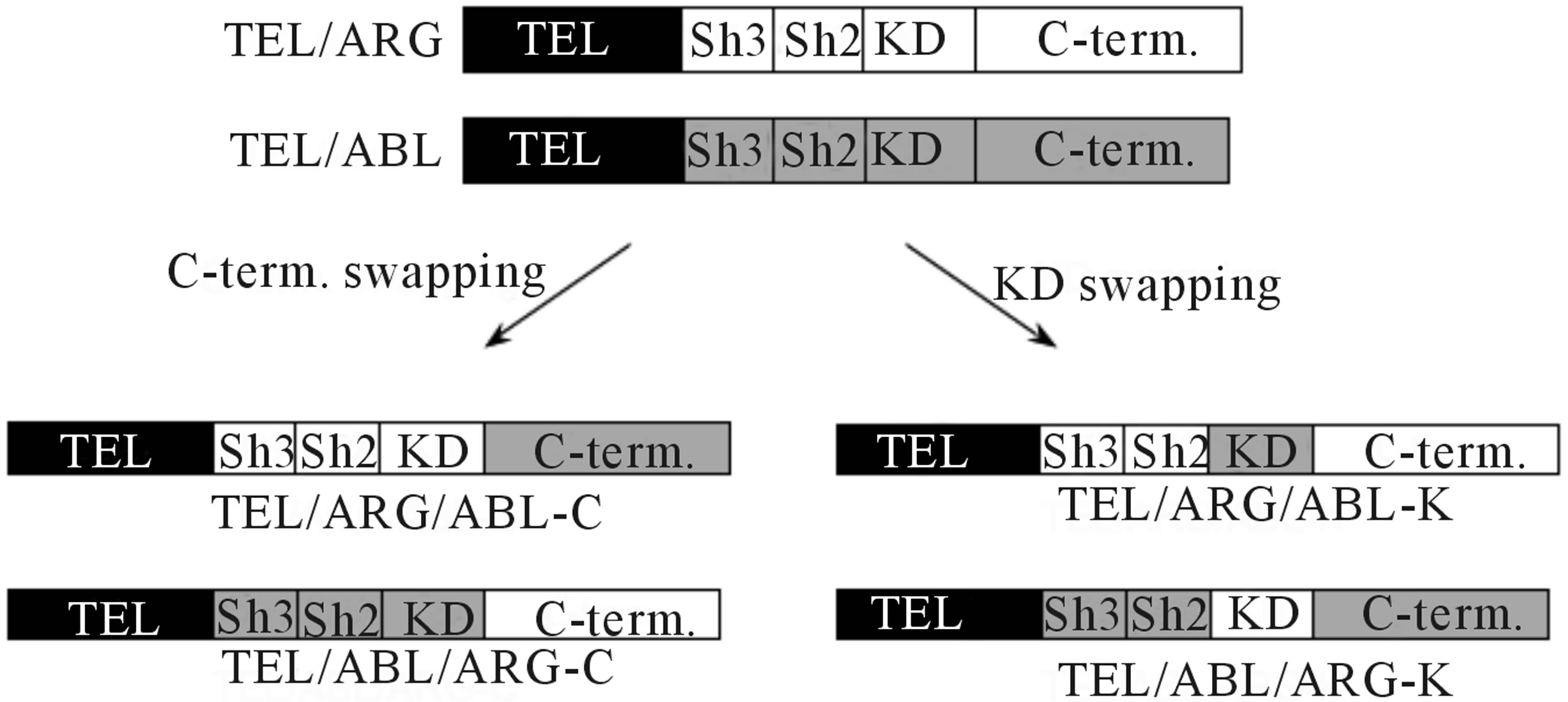
Figure 1. Schematic representation of the mutant genes and their protein expression in TonB cells. (a) Diagrams of chimeric mutant constructs used in this study. KD: tyrosine kinase domain, SH: Src homology domain, C-term: C-terminal domain. Each domain belonging to ARG or ABL is indicated by white and gray regions, respectively; (b) Doxycycline-induced TEL/ARG expression in each mutant subline. TonB TEL/ARG cells were incubated with 0.5 µg/ml of doxycycline for 6 h. Expression and auto-tyrosine phosphorylation of the mutant TEL/ARG was confirmed by immunoblotting using an anti-phosphotyrosine antibody after immunoprecipitation with anti-FLAG antibody.

Figure 2. Effect of doxycycline-induced oncogene expression on tyrosine phosphorylation of total cellular proteins. Cells were factor deprived for 3 h then incubated with doxycycline 0.5 µg/ml for 6 h. Changes in protein tyrosine phosphorylation were detected by antiphosphotyrosine antibody immunoprecipitation followed by immunoblotting with an antiphosphotyrosine antibody. Molecular masses are shown in kilodaltons. A juxtaposing image from the same experiment is shown for comparison and the boundary between the gels is delineating with black line in the figure.
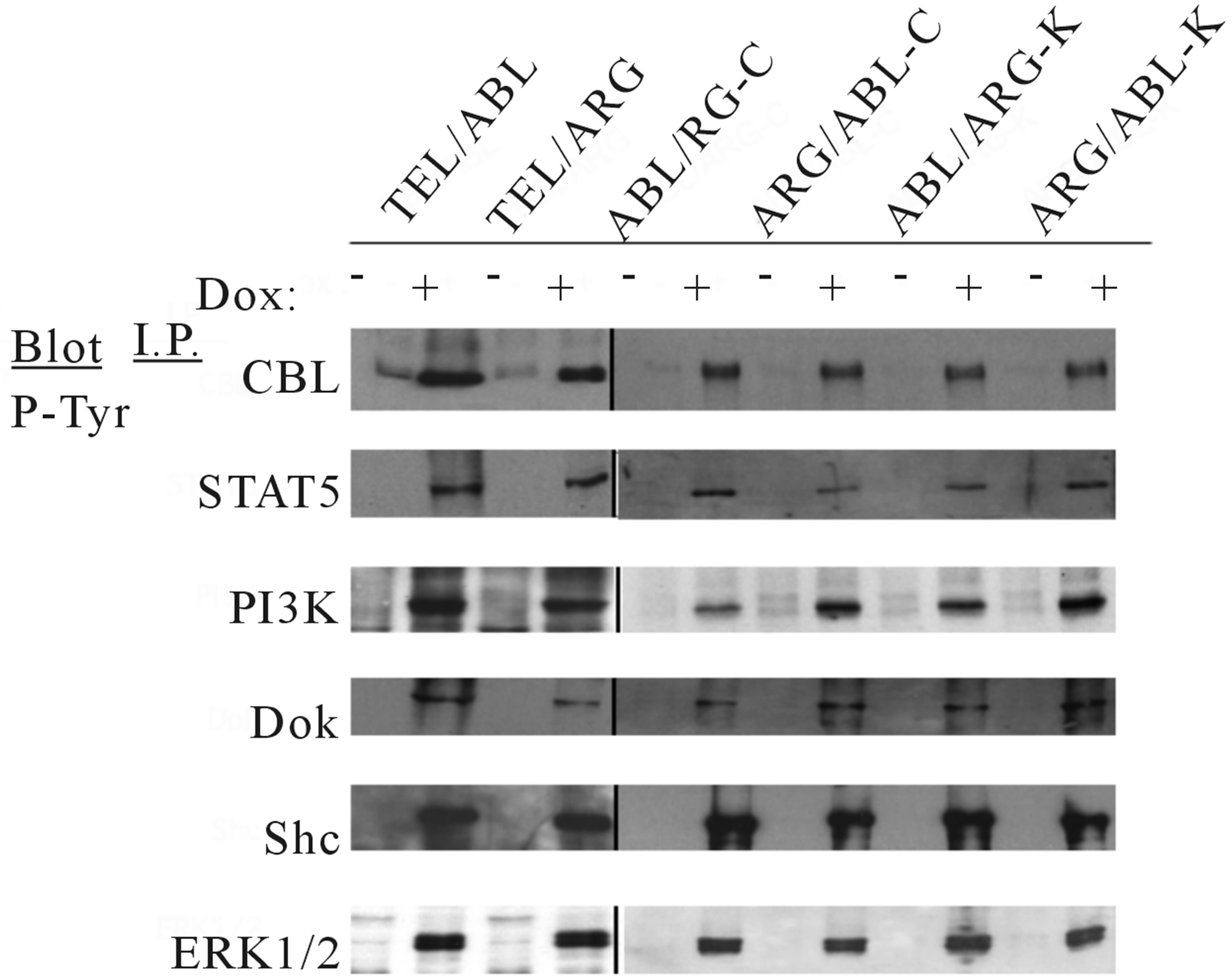
Figure 3. Protein expression and tyrosine phosphorylation of cellular proteins in the mutant oncogenes induced by doxycycline. The indicated TonB mutant cells were incubated in the absence or presence of 0.5 µg/ml doxycycline for 6 h at 37˚C. Cell lysates were immunoprecipitated with antibodies as indicated, followed by antiphosphotyrosine immunoblotting with 4G10. The juxtaposing images from the same experiment are shown for comparison and the each boundary between the gels is delineating with black line in the figure.
3.2. The C-Terminus Domain of ABL Is Required for Growth Signaling, while That of ARG Is Required for Hyper-Responsiveness to IL-3 in TonB Sublines
Each mutant was tested for the ability to support the viability and proliferation of TonB cells (Figure 4). The expression of wild-type TEL/ARG in TonB cells had a substantial effect on viability and some activity to induce short-term proliferation (left lower panel), however, TEL/ARG by itself induced much slower cell proliferation compared to that with wild-type TEL/ABL (left upper), despite maintaining high levels of kinase activity in both. A TEL/ARG mutant with the ARG C-terminal domain replacing that of ABL increased proliferation to levels equivalent to TEL/ABL (middle lower). Similarly, a TEL/ABL mutant with the ABL C-terminal domain replacing that of ARG decreased proliferation to levels equivalent to TEL/ARG (middle upper). Substitution of the kinase domains did not affect their activity in viability and proliferation (right upper and lower). The results indicated that the C-terminal domain of ABL is required for growth signaling and the C-terminal domain of ARG is required for viability signaling of the chimeric tyrosine kinase.
As we reported previously, expression of TEL/ARG enhanced the proliferative response to IL-3 in TonB cells, and the C-terminal domain of ARG is required for viable signaling and for hyper-responsiveness to IL-3 in TonB TEL/ARG cells [17,23]. So, we next asked if the presence of the mutant oncogene affected the dose-response of these cells to IL-3 (Figure 5). The cells expressing TEL/ARG proliferated in response to significantly lower concentrations of IL-3 (0.01 ng/ml) than control cells without doxycycline (0.1 ng/ml). The maximum rate of proliferation induced by IL-3 was also higher in cells expressing TEL/ARG (left lower panel). On the other hand, proliferation of TEL/ABL-expressing cells was already observed without IL-3, and addition of IL-3 did not enhance their growth rate especially (left upper). There was no significant change in the maximum rate of proliferation induced by IL-3 with and without TEL/ABL, indicating that substantial effects in hyper-responsiveness to IL-3 were induced by TEL/ARG but not by TEL/ABL. As shown here, replacing the ARG C-terminal domain with that of ABL in TEL/ARG lost the ability to induce hyper-responsiveness to IL-3 (middle lower), while the TEL/ABL mutant replacing the ABL C-terminal domain with that of ARG led to obtain the ability (middle upper). Substitution of the kinase domain was silent in this function also (right upper and lower).
Both TEL/ABL and TEL/ARG expression enhanced spontaneous migration as measured by migration assay, and this activity was not impaired in all of the engineered molecules (Figure 6).
3.3. The C-Terminus Domain of ABL Contributes to Rapid Transformation of Ba/F3 Cells by the Chimeric Oncogene
An alternative approach to evaluate the quantitative transforming activity of the oncogenes in Ba/F3 cells is by measuring the number of days required to achieve measurable cell growth after IL-3 deprivation. The mutant cDNAs were stably expressed in the parental Ba/F3 cells by retroviral transduction, and the transduced cells
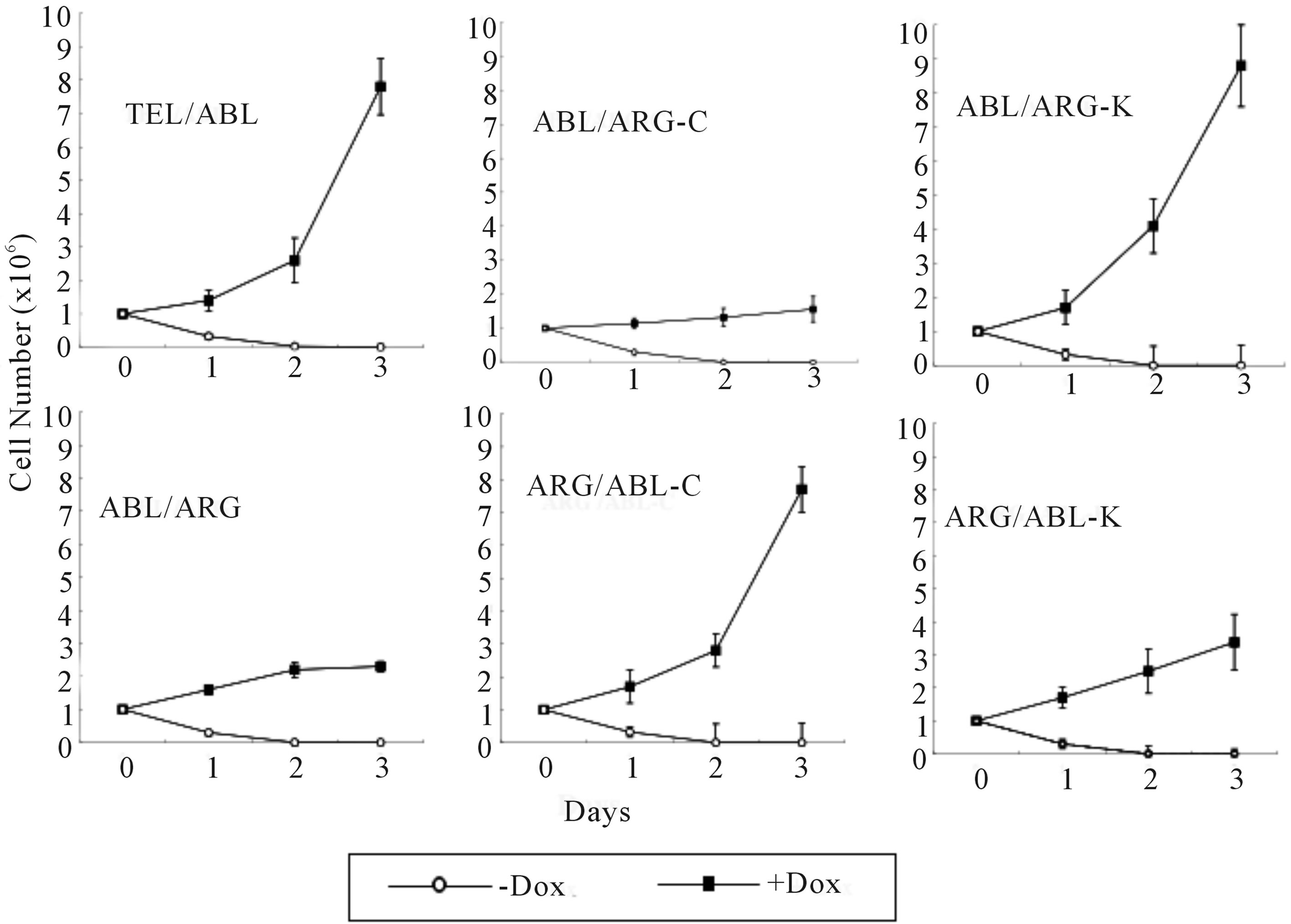
Figure 4. Proliferation assay in the absence of IL-3. Cells growing in the presence of IL-3 were washed 3 times and re-susspended at a density of 0.5 × 106 cells/ml × 2 ml each in RPMI supplemented with 10% FCS. Cells were then cultured at 37˚C in the absence or presence of 0.5 µg/ml doxycycline. The total number of viable cells was evaluated every 24 h. The data presented are the average of five separate experiments.
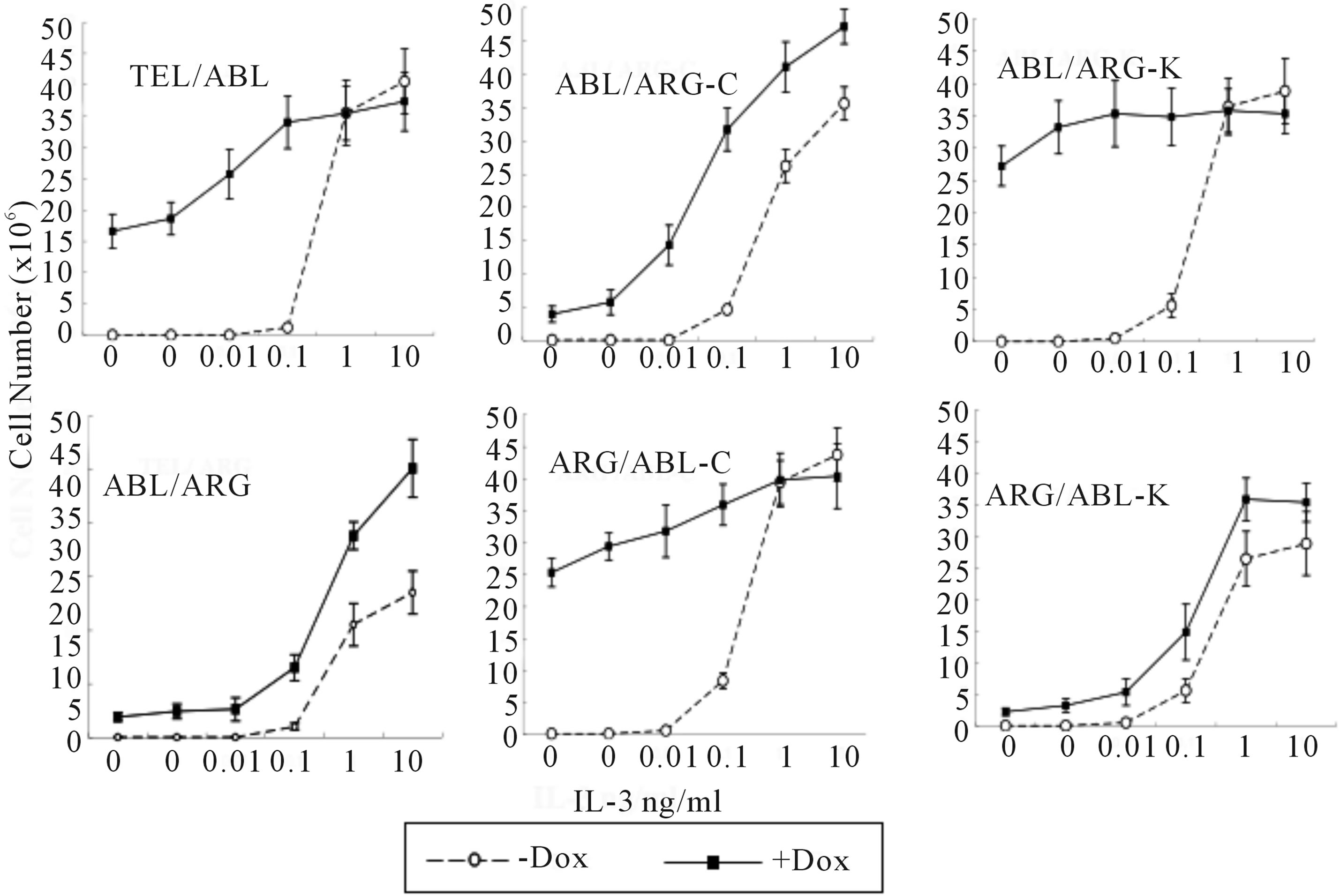
Figure 5. Dose response of TonB sublines to IL-3.Cells growing in the presence of IL-3 were washed 3 times with RPMI and re-suspended at a density of 0.5 × 106 cells/ml × 2 ml each in RPMI supplemented with 10% FCS. Cells were then incubated with the indicated concentration of recombinant IL-3, in the absence or presence of the each oncoprotein by doxycycline induction. The total number of viable cells was evaluated every 24 h, and the cell number on Day 4 is reported in the figure. The data presented are the average of five separate experiments.
were selected within 3 days after infection for equal expression of the fusion protein by flow sorting for populations with equivalent intensity of GFP fluorescence. The sorted cells were cultured in 10% FCS-RPMI medium without IL-3, and the viable cell count was determined (Figure 7).
The detection of similar expression levels of each protein by western blotting analysis, as shown in Figure

Figure 6. Effect on cell migration with swapped mutants. Cells were used for Trasnwell migration assays, and the number of viable cells in the lower chamber was determined after 5 h by trypan blue exclusion. The migration index was expressed as the ratio of the number of doxycycline-stimulated cells migrating to a number of unstimulated cells migrating over the same time period.
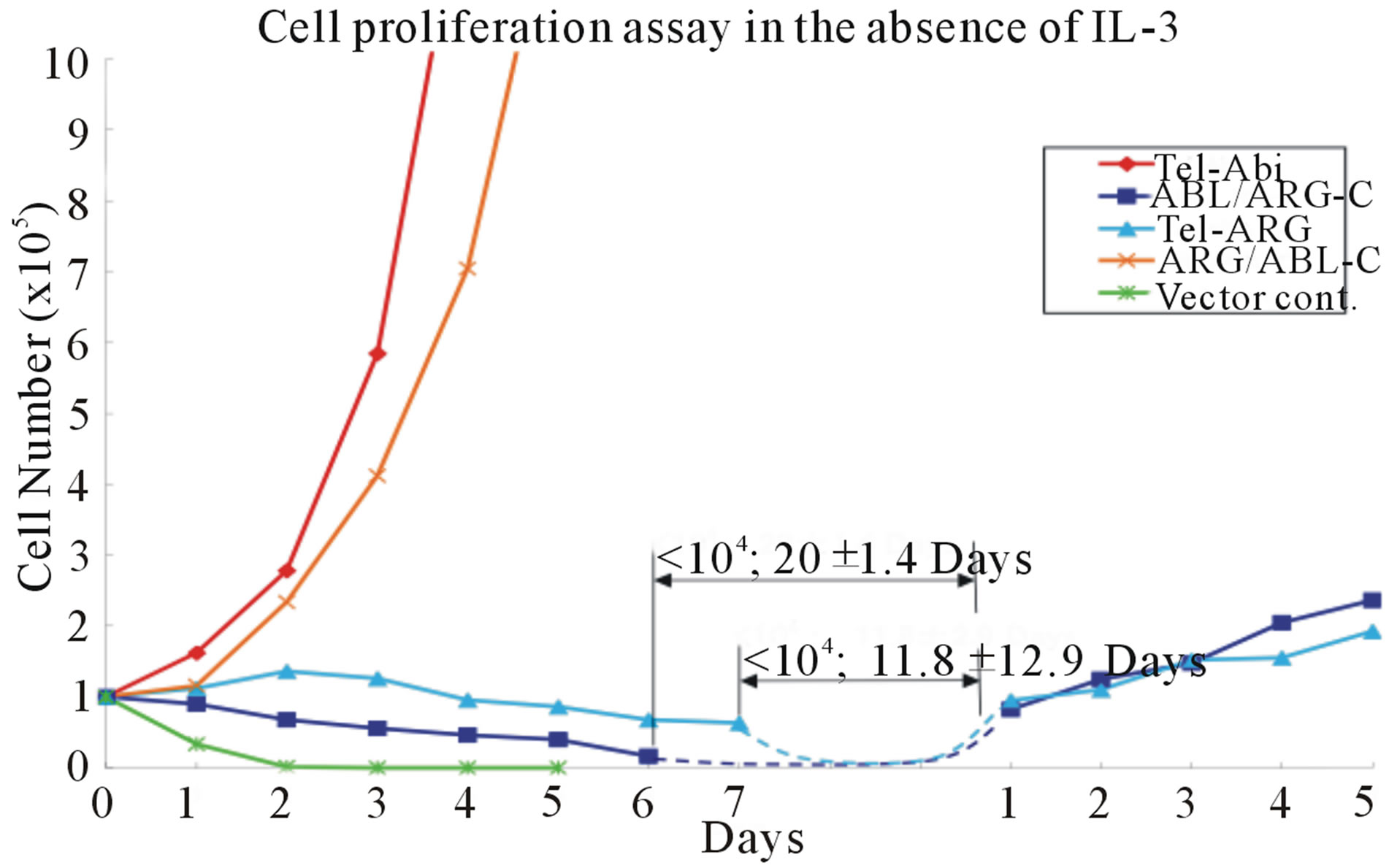
7(b), suggested that the difference in activity was not based on substantial differences in expression levels. All chimeric proteins showed prominent auto-phosphorylation and induced tyrosine phosphorylation on a similar set of substrates in the sorted cells (Figure 7(c)). Nevertheless, TEL/ARG had significantly decreased transforming activity compared with TEL/ABL. The quantitative transforming activity, measured by the time in days to start factor-independent growth after the infection, was 1 to 2 days for TEL/ABL but 25 ± 4.3 days for TEL/ ARG. The latency was not altered by swapping the kinase domains (data not shown), but was completely restored by swapping their C-termini (Figure 7(a)). The transforming activity in this assay was significantly higher using the chimeric oncogene with the ABL Cterminus than that with the ARG C-terminus, suggesting that the ARG C-terminus is unsuitable for in vitro cell transformation.
3.4. In Vitro Transformation of Primary Bone Marrow Stem Cells Requires the ABL C-Terminus Domain
We compared the ability of the different ABL/ARG fusion oncogenes to stimulate the growth of hematopoietic stem cells in methylcellulose culture in the retroviral bone marrow transduction system. In this assay, bone marrow from donors pretreated with 5-flurouracil was transduced with retroviruses expressing TEL/ABL, TEL/ ARG, or their mutants with swapped C-terminal domains. Cells successfully transduced were selected after infection by flow sorting for the expression of GFP fluorescence. The efficiency of virus transduction (% GFP positive cells in total bone marrow cells) was about 20% in each experiment. The sorted cells were immediately cultured in methylcellulose with or without murine colony stimulating factors. Each colony type was distinguished based on a distinct morphology. The capacity of the re-
 (a)
(a)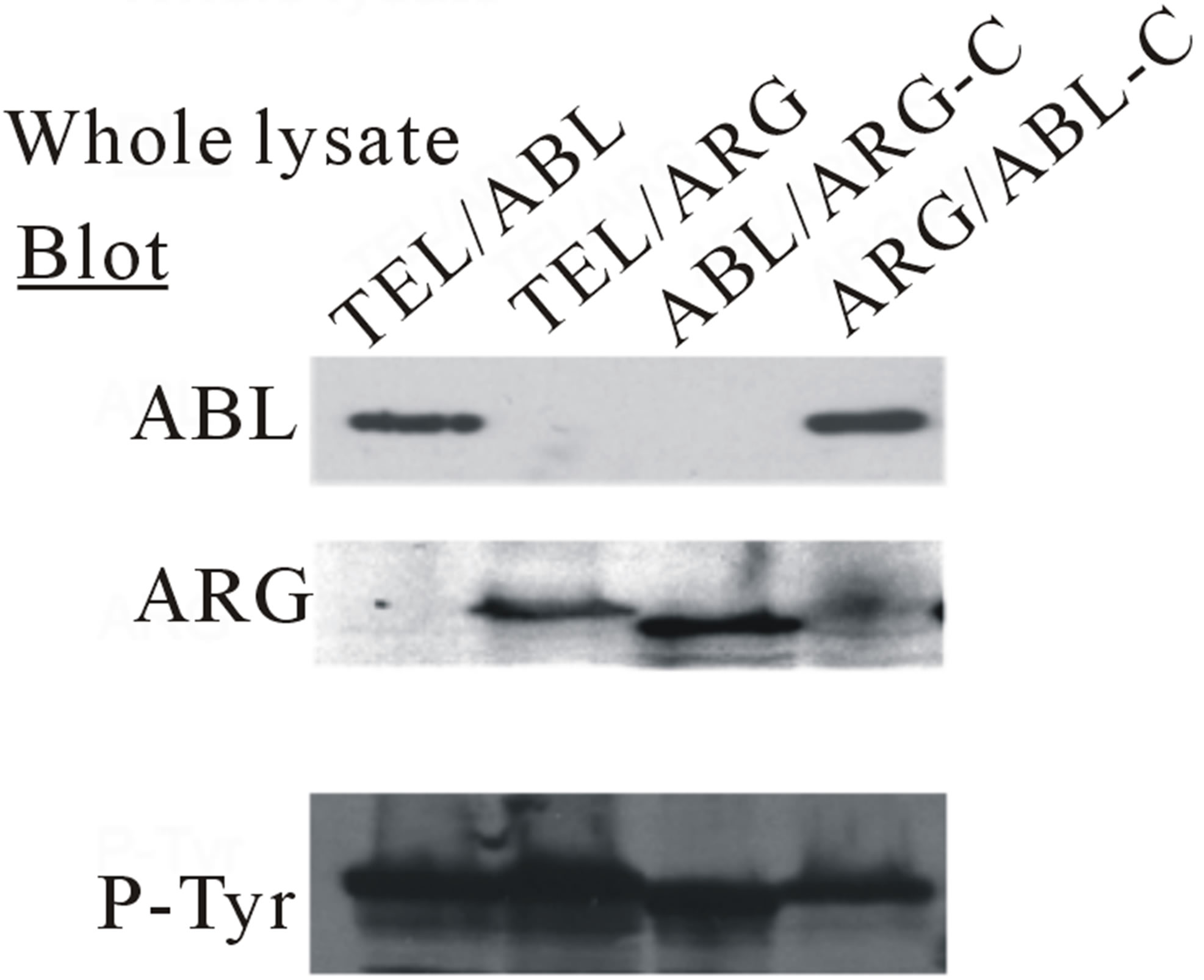 (b)
(b)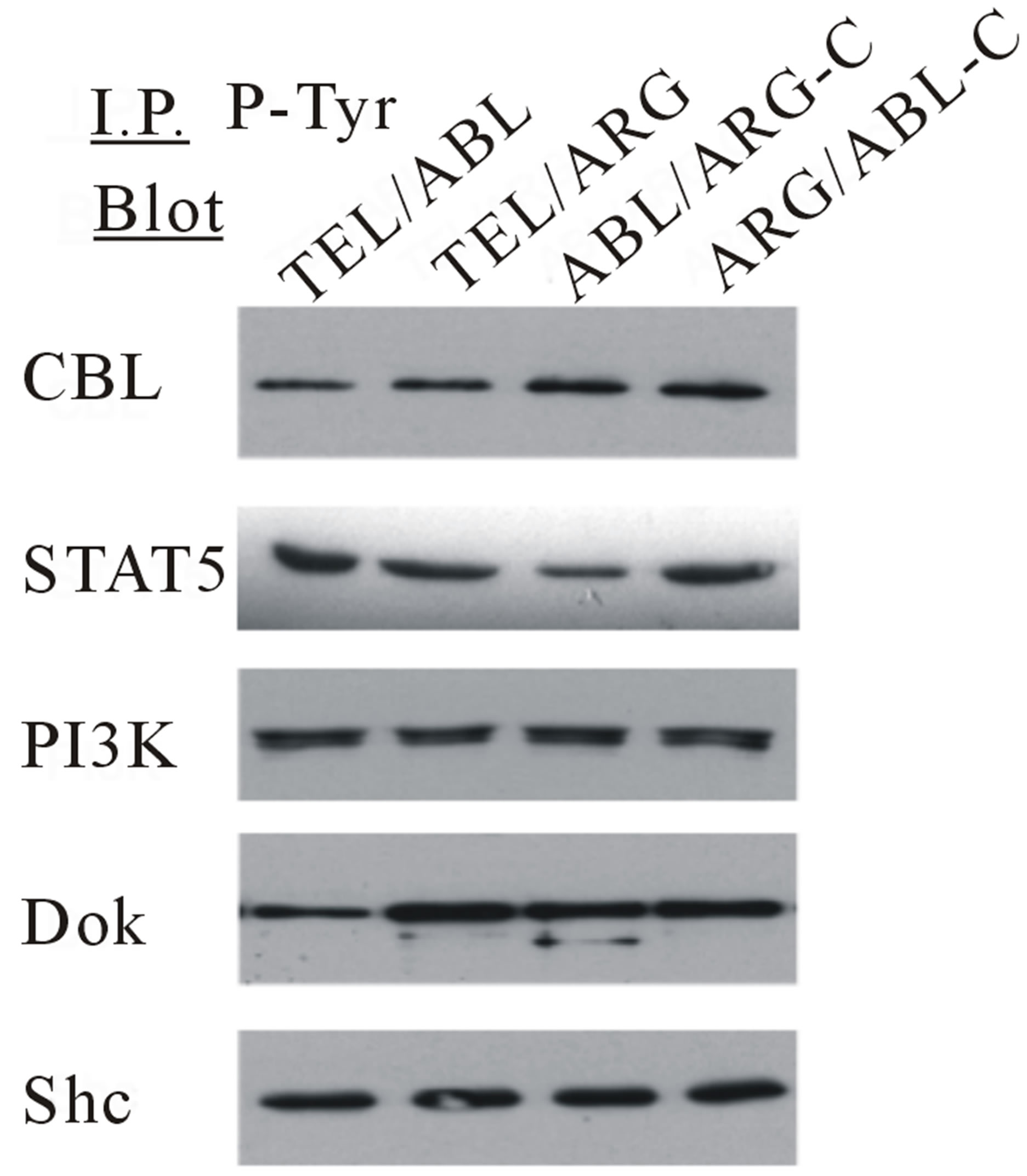 (c)
(c)
Figure 7. Transformation of cytokine-dependent Ba/F3 cell line. (a) A cell proliferation assay was performed for the chimeric mutants in the absence of IL-3. In each case, the mutant cDNAs were introduced into Ba/F3 cells by retroviral transduction and selected for GFP expression by sorting within 2 - 3 days after the transduction. The transduced cells, selected for equal expression of the fusion protein by flow sorting for populations with equivalent intensity of GFP fluorescence, were counted and plated in RPMI medium without IL-3 in triplicate wells of a 24-well plate. Cells were incubated at 37˚C, and the viable cell count was determined daily as indicated. The difference between cell numbers for constructs with the C-terminal regions of ARG and ABL was significant; (b) Ba/F3 swapped mutant sublines were generated by retroviral transduction. Cells were lysed after factor deprivation for 6 h. Protein expression was confirmed by Western blotting with the indicated antibodies; (c) Tyrosine phosphorylation of signaling proteins in Ba/F3 swapped mutants. The tyrosine phosphorylation state of a series of signaling proteins was evaluated by anti-phosphotyrosine antibody immunoprecipitation followed by blotting with the specified antibodies.
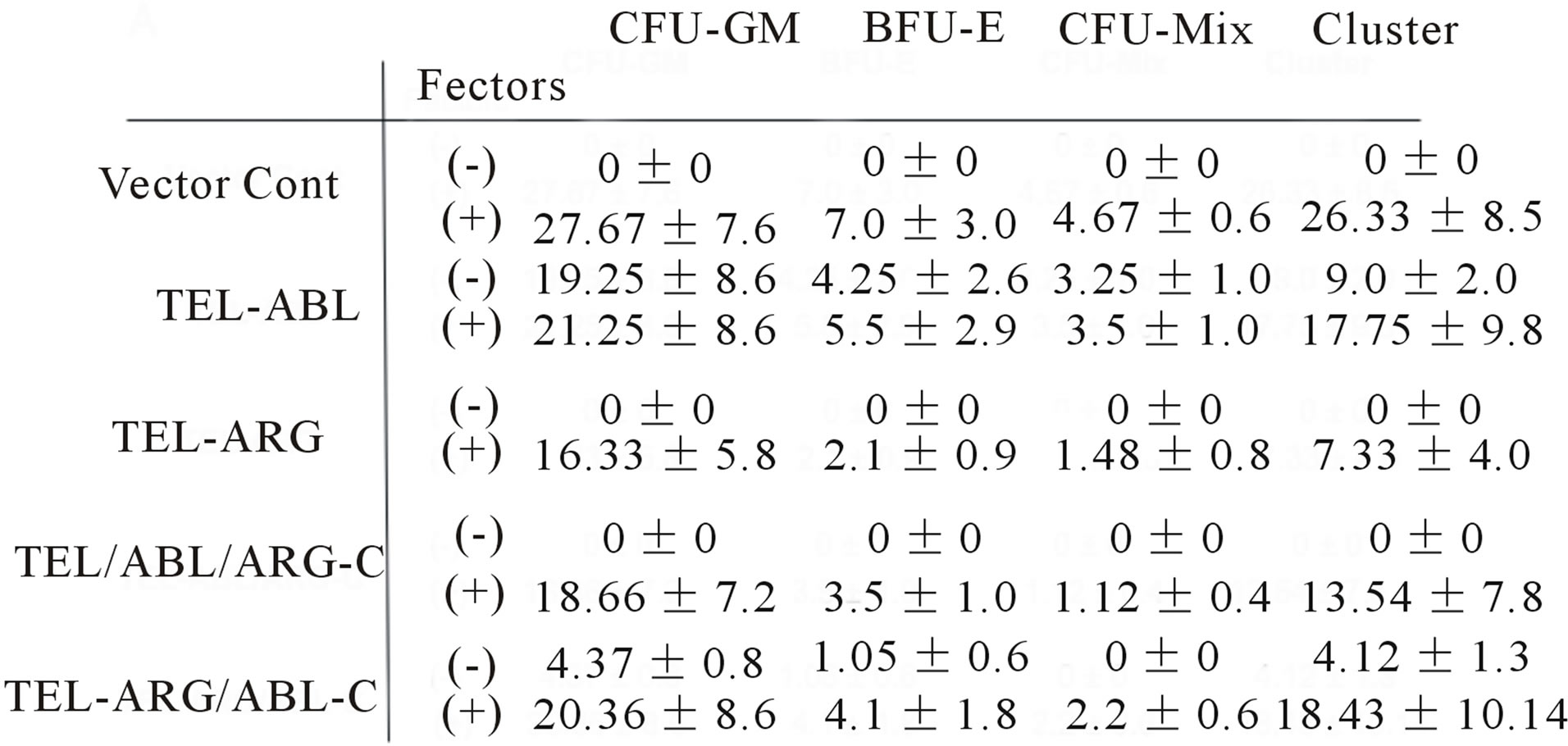
sulting cells to form colonies in methylcellulose is summarized in Figure 8(a). All colonies in which the fusion proteins were expressed were visualized by means of tagged EGFP using an inverted confocal laser microscope, as shown in Figure 8(b). After in about 2 weeks of growth, roughly equivalent numbers of macroscopic colonies were observed for every bone marrow subjected to culture with colony stimulating factors. By comparison, in a culture without colony stimulating factors, substantial numbers of spontaneous colonies were observed for TEL/ABL-expressing bone marrow but not for the TEL/ARG-expressing or the background of vectortransduced cells. The TEL/ARG chimera containing a substitution of the ABL C-terminus exhibited robust activity in colony formation, similar to that of TEL/ABLconferred transforming activity. Microscopic examination of spontaneous colonies displayed an appearance indistinguishable from TEL/ABL colonies. However, substitution of the ABL C-terminus in TEL/ARG completely abolished the activity for spontaneous colony formation of primary marrow cells.
Thus, the results indicated that substitution of the C-terminus conferred the activity of the parent molecule from which the domain was derived, and the ABL C-terminus was required for in vitro transformation of primary bone marrow cells by the chimeric oncogene.
 (a)
(a) (b)
(b)
Figure 8. Factor-independent colony formation in bone marrow cells was induced by the ABL C-terminal region. (a) All colonies arising from sorted bone marrow cells were scored on days 14 - 16 of culture. Scores represent cultures containing 1000 bone marrow cells/dish. Data represent the mean ± SD of three to four independent experiments. Colony types identified in situ are; CFU-GM: granulocytemacrophage colonies, BFU-E: burst-forming units, CFUMix: erythrocyte-containing mixed colonies, cluster: mass with less than 30 cells without typical morphologic appearance as others; (b) Chimeric protein expression visualized by tagged EGFP was detected in all colonies. Representative colony images obtained using inverted and inverted confocal laser scanning microscopes are shown.
4. Discussion
Although TEL/ARG and TEL/ABL have essentially identical transforming properties in cultured cells, an immediate comparison of the transforming properties of the two kinases in our study revealed important differences between them. Thus, the difference in transforming activity led to a structure-function analysis of oncogenic activity based on the analysis of chimeric TEL/ARG and TEL/ABL molecules.
In the present study, this was explored by generating specific mutants that contained TEL fused to ARG and ABL constructs, in which the kinase or C-terminal domains were swapped. Because ARG and ABL share a striking degree of structural resemblance in the SH2, SH3, and tyrosine kinase domains, which are highly conserved (89%, 90%, and 93% identity, respectively), their distinct C-terminal domains, which share only 29% amino acid identity, likely account for the difference in the ability to induce their proper function, if any. As anticipated, our analysis of chimeric molecules revealed that substitution of the C-terminus dramatically altered transforming activity between the two chimeric proteins, whereas substitutions of the tyrosine kinase domains were silent. This indicated that the distinct ARG and ABL C-termini account for the different transforming activities, and further suggested that the ARG C-terminus may exert a prominent negative influence on activity.
However, the large C-terminus, which constitutes about half the length of ABL and ARG, contains several functional sub-domains, and its role in specific activity is complex and not well understood. Nuclear localization, DNA binding, and Fand G-actin binding are mediated by sequences within the ABL C-terminal domain [24-26], whereas, Fand G-actin and microtubule binding domains and the RNA polymerase binding sequence have been reported as a functional subdomain in ARG [2,3, 27-29]. From the conserved Fand G-actin binding domains at the C-terminus of ABL and ARG, we have demonstrated direct evidence that TEL/ARG is localized at least in part to the actin cytoskeleton though a domain in the C-terminus of ARG, whereby it regulates cytoskeletal dynamics via its kinase activity, as is the case for TEL/ABL [7,23]. This is apparently involved as a positive role in oncogenic signaling, because focal adhesion proteins normally receive and transmit signals through phosphorylation and dephosphorylation on tyrosine residues in response to several tyrosine kinase oncoproteins, including v-Src, v-Crk, v-Abl and chimeric ABL oncogenes (BCR/ABL, TEL/ABL) [30]. In accordance with this idea, we have reported previously that the actin binding domain in the C-terminus of ABL is required for proliferation and viability signaling in the context of an ABL/erythropoietin receptor fusion protein [31]. However, previous studies have not consistently found a role for the actin binding function of BCR/ABL in transformation [32-34]. With regard to ARG, we have reported previously that the C-terminus domain is required for all the biological functions of TEL/ARG, such as prolonged viability, hyper-responsiveness to IL-3, and enhanced mobility, using the same doxycycline-based induction system [23]. All biological functions were profoundly impaired with the deletion of the ARG C-terminus in TEL/ARG, despite maintaining high levels of its kinase activity, indicating that there is at least a sequence or domain, if not the actin binding domain, essential for transforming activity.
In another approach to evaluate the structure-based transforming activity of ARG, a Gag/ARG fusion protein, which is analogous to the oncogenic Gag/ABL fusion protein of Abelson Murine Leukemia Virus (v-abl), was generated and shown that, in contrast to Gag/ABL, it lacked transforming activity in a focus formation assay using NIH3T3 cells [35]. An engineered Gag/ARG molecule containing a truncation of almost the entire C-terminus was associated with a gain of oncogenic activity of Gag/ARG similar to that of Gag/ABL in the same experiment, indicating that loss of the C-terminal domain was associated with a gain of oncogenic activity. The activation of ARG by fusion with either Gag or TEL resulted in different transforming events, although we had close observations indicating that the distinct transforming activity mediated by ARG and ABL is accounted for their C-terminal domains. One prominent difference that may account for these results is in the subcellular localization of the fusion proteins. The subcellular localization of Gag/ARG has not been demonstrated clearly, but is likely to be anchored to the cell membrane through myristylation, probably similar to v-abl. This is in contrast to c-ARG and TEL/ARG, which are normally located in the cytoplasm and cytoskeleton [3,23]. Gag/ARG containing a truncation of almost the entire C-terminus could induce biological activity, possibly because localization to the cell membrane reduced the requirement of the actin binding domain for direct interaction with actin filaments to express its own function, which was similar to previous studies demonstrating that the C-terminus of v-abl has not been shown to be required for transformation [36]. On the other hand, TEL can also contribute signaling motifs, such as a direct binding site of the GRB2 adapter protein, tyrosine 314 (Y314), similarly to the tyrosine 177 GRB2 binding site of BCR as one potentially unique contribution of BCR to the transformation of BCR/ABL [37,38], and this is also true for TEL/ABL [39] and TEL/ARG [23]. Thus, in this study, it was not proven whether the clustering of the ARG molecule itself is sufficient to activate ARG tyrosine kinase activity to mimic the biological properties of TEL/ARG, as we have reported previously for ABL using a receptor/ABL fusion protein [40]. Gag/ARG retroviral gag sequences fused to the ARG SH3/SH2 junction, such that the ARG amino terminus and SH3 domains are absent in the fusion protein, were analogous to Gag/ABL. Although TEL contributes an N-terminal domain to the TEL/ARG fusion, the TEL/ARG transcript contains almost the complete ARG protein and would not be expected to alter the structure of ARG, it is likely that the comparison in molecular functions of ARG is still evaluated accurately. Our results suggest that the C-terminus of ARG contains a functional subdomain that impairs the transforming activity induced by ABL family tyrosine kinase, including the associated enzymatic and signaling functions of the actin-binding domain and the additional TEL sequence. Further studies are necessary to identify the functional subdomain in the ARG C-terminus in order to define the biochemical basis of the differences in transforming activity of TEL/ARG and TEL/ABL.
Its lack of transformability in bone marrow in methylcellulose culture does not necessarily imply that the TEL/ARG is not sufficient to cause leukemia in vivo. It is possible that in vivo there are sufficient growth factors, such as IL-3, in the marrow and spleen microenvironments to keep these cells alive, whereas the signal alone is insufficient in vitro. In a murine retroviral bone marrow transduction-transplantation model, TEL/ABL has been demonstrated to induce two distinct myeloproliferative diseases, CML-like leukemia similar to that induced by BCR/ABL and a novel fetal syndrome characterized by small-bowel myeloid cell infiltration and necrosis, and fulminant hepatic and renal failure [41]. Whether TEL/ARG has leukemogenic properties in mice is currently being examined using the same retroviral bone marrow transduction-transplantation strategy. However, the degree of clinical phenotype in recipients of TEL/ ARG-transduced marrow, if any, is anticipated to be weaker in magnitude than those seen in recipients of TEL/ABL. Therefore, it will be of interest to determine if the ARG C-terminus inhibitory activity is also evident in the leukemogenic properties of ABL oncogenes in mice.
Because the kinase activity of TEL/ARG is inhibited by STI571 (imatinib mesylate) [16], any disease or tumor associated with ARG or TEL/ARG could be a good candidate for treatment with this drug as well as that of BCR/ABL and TEL/ABL. The use of small molecule drugs to inhibit specific oncogenes is currently the most prominent therapy to improve outcomes and reduce sideeffects in many type of cancer, and there have been many recent advances in the development of TKIs as drugs. As a representative, STI571 and the second-generation compounds, which were developed shortly thereafter, have been used as frontline treatment for patients with CML and B-ALL induced by BCR/ABL. Despite the success of TKI-based therapies in treating CML patients, TKI resistance, particularly that conferred by the T315I mutation in the ABL kinase domain of BCR/ABL, now represents a major concern in the treatment of CML [42, 43]. The poor prognosis for patients who failed or acquire resistance to TKI therapy highlights the need to develop effective treatments for this subset of patients. To investigate the mechanism for how the ARG C-terminus attenuates the transforming activity induced by ABL tyrosine kinase without turning off its kinase activity is important and of interest, because the mechanism, which is clearly different from TKIs, may offer an important new strategy for treatment. An intensive study to characterize the subdomain in the ARG C-terminus may contribute to a clear medical need by offering targets for the development of rational therapeutics for ABL-induced leukemia, and especially an alternative therapy for the population of CML patients who fail TKI therapy.
5. Acknowledgements
The authors would like to thank Dr. Nari Harakawa, Dr. Naochika Domae, Osaka Dental University, and Dr. Richard A. Van Etten, Tufts-New England Medical Center, USA, for their encouragement and helpful discussions in carrying out this study.
K. O. designed the research, performed experiments, analysed results and wrote the paper; H. H. performed experiments, analysed results and edited the paper.
REFERENCES
- G. D. Kruh, C. R. King, M. H. Kraus, N. C. Popescu, S. C. Amsbaugh, W. O. McBride and S. A. Aaronson, “A Novel Human Gene Closely Related to the Abl Proto-Oncogene,” Science, Vol. 234, No. 4783, 1986, pp. 1545- 1548. http://dx.doi.org/10.1126/science.3787260
- G. D. Kruh, R. Perego, T. Miki and S. A. Aaronson, “The Complete Coding Sequence of Arg Defines the Abelson Subfamily of Cytoplasmic Tyrosine Kinases,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 87, No. 15, 1990, pp. 5802-5806. http://dx.doi.org/10.1073/pnas.87.15.5802
- R. Perego, D. Ron and G. D. Kruh, “Arg Encodes a Widely Expressed 145-kDa Protein-Tyrosine Kinase,” Oncogene, Vol. 6, 1991, pp. 1899-1902.
- Y. Iijima, T. Ito, T. Oikawa, M. Eguchi, M. EguchiIshimae, N. Kamada, K. Kishi, S. Asano, Y. Sakaki, and Y. Sato, “A New ETV6/TEL Partner Gene ARG(ABLRelated Gene or ABL2), Identified in an AML-M3 Cell Line with a t(1;12)(q25;p13) Translocation,” Blood, Vol. 95, 2000, pp. 2126-2131.
- G. Cazzaniga, S. Tosi, A. Aloisi, G. Giudici, M. Daniotti, P. Pioltelli, L. Kearney and A. Biondi, “The Tyrosine Kinase Abl-Related Gene ARG Is Fused to ETV6 in an AML-M4Eo Patient with a t(1;12)(q25;p13): Molecular Cloning of Both Reciprocal Transcripts,” Blood, Vol. 94, 1999, pp. 4370-4373.
- J. Griesinger, A. Janke, M. Podleschny and S. K. Boglander, “Identification of an ETV6-ABL2 Fusion Transcript in Combination with an ETV6 Point Mutation in a T-Cell Acute Lymphoblastic Leukaemia Cell Line,” British Journal of Haematology, Vol. 119, No. 2, 2002, pp. 454-458. http://dx.doi.org/10.1046/j.1365-2141.2002.03850.x
- T. R. Golub, A. Goga, G. F. Barker, D. E. H. Afar, J. McLaughlin, S. K. Bohlander, J. D. Rowley, O. N. Witte and G. D. Gilliland, “Oligomerization of the ABL Tyrosine Kinase by the Ets Protein TEL in Human Leukemia,” Molecular and Cellular Biology, Vol. 16, 1996, pp. 4107- 4116.
- H. Van Limbergen, H. B. Beverloo, E. Van Drunen, A. Janssens, K. Hahlen, B. Poppe, N. Van Roy, P. Marynen, A. de Paepe, R. Slater and F. Speleman, “Molecular Cytogenetic and Clinical Findings in ETV6/ABL1-Positive Leukemia,” Genes Chromosomes and Cancer, Vol. 30, No. 3, 2001, pp. 274-282. http://dx.doi.org/10.1002/1098-2264(2000)9999:9999<1::AID-GCC1089>3.0.CO;2-1
- V. Brunel, D. Sainty, N. Carbuccia, M. Mozzicolacci, F. Fernandez, J. Simonetti, J. Gabert, P. Dubreuil, M. Lafage-Pochitaloff and F. Birg, “A TEL/ABL Fusion Gene on Chromosome 12p13 in a Case of Ph-, BCR-Atypical CML,” Leukemia, Vol. 10, 1996.
- P. Andreasson, B. Johansson, M. Carlsson, I. Jarlsfelt, T. Fioretos, F. Mitelman and M. Hoglund, “BCR/ABL-Negative Chronic Myeloid Leukemia with ETV6/ABL Fusion,” Genes Chromosomes and Cancer, Vol. 20, No. 3, 1997, pp. 299-304. http://dx.doi.org/10.1002/(SICI)1098-2264(199711)20:3<299::AID-GCC11>3.0.CO;2-K
- M. Carroll, M. H. Tomasson, G. F. Barker, T. R. Golub and D. G. Gilliland, “The TEL/Platelet-Derived Growth Factor Beta Receptor (PDGF Beta R) Fusion in Chronic Myelomonocytic Leukemia Is a Transforming Protein That Self-Associates and Activates PDGF Beta R KinaseDependent Signaling Pathways,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 93, No. 25, 1996, pp. 14845-14850. http://dx.doi.org/10.1073/pnas.93.25.14845
- V. Lacronique, A. Boureux, V. D. Valle, H. Poirel, C. T. Quang, M. Mauchauffe, C. Berthou, M. Lessard, R. Berger, J. Ghysdael and O. A. Bernard, “A TEL-JAK2 Fusion Protein with Constitutive Kinase Activity in Human Leukemia,” Science, Vol. 278, No. 5341, 1997, pp. 1309- 1312. http://dx.doi.org/10.1126/science.278.5341.1309
- Q. Liu, J. Schwaller, J. Kutok, D. Cain, J. C. Aster, I. R. Williams and D. G. Gilliland, “Signal Transduction and Transforming Properties of the TEL-TRKC Fusions Associated with t(12;15)(p13;q25) in Congenital Fibrosarcoma and Cute Myelogenous Leukemia,” EMBO Journal, Vol. 19, 2000, pp. 1827-1838. http://dx.doi.org/10.1093/emboj/19.8.1827
- Y. Kuno, A. Abe, N. Emi, M. Iida, T. Yokozawa, M. Towatari, M. Tanimoto and H. Saito, “Constitutive Kinase Activation of the TEL-Syk Fusion Gene in Myelodysplastic Syndrome with t(9;12)(q22:p12),” Blood, Vol. 97, No. 4, 2001, pp. 1050-1055. http://dx.doi.org/10.1182/blood.V97.4.1050
- Y. Iijima, K. Okuda, N. K. Tri, A. Tojo, M. Setoyama, G. D. Kruh, Y. Sakaki, S. Asano and Y. Sato, “Transformation of Ba/F3 Cells and Rat-1 Cells by ETV6/ARG,” Oncogene, Vol. 21, No. 28, 2002, pp. 4374-4383. http://dx.doi.org/10.1038/sj.onc.1205544
- K. Okuda, E. Weisberg, D. G. Gilliland and J. D. Griffin, “ARG Tyrosine Kinase Activity Is Inhibited by STI571,” Blood, Vol. 15, 2001, pp. 2440-2448. http://dx.doi.org/10.1182/blood.V97.8.2440
- K. Okuda, Y. Sato, Y. Sonoda and J. D. Grifffin, The TEL/ARG leukemia oncogene promotes viability and hyper-responsiveness to hematopoietic growth factors. International Journal of Hematology, Vol. 79, No. 2, 2004, pp. 138-146. http://dx.doi.org/10.1532/IJH97.03125
- R. G. Hawley, F. H. Lieu, A. Z. Fong and T. S. Hawley, “Versatile Retroviral Vectors for Potential Us in Gene Therapy,” Gene Therapy, Vol. 1, 1994, pp. 136-138.
- R. Palacios and M. Steinmetz, “IL-3-Dependent Mouse Clones that Express B-220 Surface Antigen, Contain Ig Genes in Germ-Line Configuration, and Generate B Lymphocytes in Vivo,” Cell, Vol. 41, 1985, pp. 727-734. http://dx.doi.org/10.1016/S0092-8674(85)80053-2
- S. Morita, T. Kojima and T. Kitamura, “Plat-E: An Efficient and Stable System for Transient Packaging of Retroviruses,” Gene Therapy, Vol. 7, No. 12, 2000, pp. 1063- 1066. http://dx.doi.org/10.1038/sj.gt.3301206
- Y. Hayashi, H. Hirai, N. Kamio, H. Yao, S. Yoshioka, Y. Miura, E. Ashihara, Y. Fujiyama, D. G. Tenen and T. Maekawa, “C/EBP Promotes BCR-ABL-Mediated Myeloid Expansion and Leukemic Stem Cell Exhaustion,” Leukemia, Vol. 27, 2013, pp. 619-628. http://dx.doi.org/10.1038/leu.2012.258
- K. Okuda, T. R. Golub, D. G. Gilliland and J. D. Griffin, “P210BCR/ABL, p190BCR/ABL, and TEL/ABL Activate Similar Signal Transduction Pathways in Hematopoietic Cell Lines,” Oncogene, Vol. 13, 1996, pp. 1147- 1152.
- K. Okuda, A. Oda, Y. Sato, A. Nakayama, H. Fujita, Y. Sonoda and J. D. Griffin, “Signal Transduction and Cellular Functions of the TEL/ARG Oncoprotein,” Leukemia, Vol. 19, 2005, pp. 603-610.
- R. A. Van Etten, P. Jackson and D. Baltimore, “The Mouse Type IV c-Abl Gene Product Is a Nuclear Protein, and Activation of Transforming Ability Is Associated with Cytoplasmic Localization,” Cell, Vol. 58, 1989, pp. 669- 678. http://dx.doi.org/10.1016/0092-8674(89)90102-5
- E. T. Kipreos and J. Y. Wang, “Cell Cycle-Regulated Binding of c-Abl Tyrosine Kinase to DNA,” Science, Vol. 256, No. 5055, 1992, pp. 382-385. http://dx.doi.org/10.1126/science.256.5055.382
- R. A. Van Etten, P. Fackson, D. Baltimore, M. C. Sanders, P. T. Matsudaira and P. A. Janmey, “The COOH Terminus of the c-Abl Tyrosine Kinase Contains Distinct Fand G-Actin Binding Domains with Bundling Activity,” Journal of Cellular Biology, Vol. 124, 1994, pp. 325-340. http://dx.doi.org/10.1083/jcb.124.3.325
- B. Wang and G. D. Kruh, “Subcellular Localization of the Arg Protein Tyrosine Kinase,” Oncogene, Vol. 13, 1996, pp. 193-197.
- Y. Wang, A. L. Miller, M. S. Mooseker and A. J. Koleske, “The Abl-Related Gene (Arg) Nonreceptor Tyrosine Kinase Uses Two F-Actin-Binding Domains to Bundle FActin,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 98, No. 26, 2001, pp. 14865-14870. http://dx.doi.org/10.1073/pnas.251249298
- A. L. Miller, Y. Wang, M. S. Mooseker and A. J. Koleske, “The Abl-Related Gene (Arg) Requires Its F-Actin-Microtubule Cross-Linking Activity to Regulate Lamelipodial Dynamics during Fibroblast Adhesion,” Journal of Cellular Biology, Vol. 165, No. 3, 2004, pp. 407-419. http://dx.doi.org/10.1083/jcb.200308055
- R. Salgia, J. L. Li, D. S. Ewaniuk, W. Pear, E. Pisick, S. A. Burky, T. Ernst, M. Sattler, L. B. Chen and J. D. Griffin, “BCR/ABL Induces Multiple Abnormalities of Cytoskeletal Function,” The Journal of Clinical Investigation, Vol. 100, 1997, pp. 46-57. http://dx.doi.org/10.1172/JCI119520
- K. Okuda, A. D’Andrea, R. A. Van Etten and J. D. Griffin, “The c-Terminus of c-Abl Is Required for Proliferation and Viability Signaling in a c-Abl/Erythropoietin Receptor Fusion Protein,” Blood, Vol. 92, 1998, pp. 3848- 3856.
- N. Heisterkamp, J. W. Voncken, D. Senadheera, I. Gonzalez-Gomez, A. Reichert, L. Haataja, A. Reinkainen, P. K. Pattengale and J. Groffen, “Reduced Oncogenicity of p190Bcr/Abl F-Actin-Binding Domain Mutants,” Blood, Vol. 96, 2000, pp. 2226-2232.
- J. A. Wertheim, S. A. Perera, D. A. Hammer, R. Ren, D. Boettiger and W. S. Pear, “Localization of BCR-ABL to F-Actin Regulates Cell Adhesion but Does Not Attenuate CML Development,” Blood, Vol. 102, No. 6, 2003, pp. 2220-2228. http://dx.doi.org/10.1182/blood-2003-01-0062
- P. J. Woodring, T. Hunter and J. Y. J. Wang, “Regulation of F-Actin-Dependent Processes by the Abl Family of Tyrosine Kinases,” Journal of Cell Science, Vol. 116, 2003, pp. 2613-2626. http://dx.doi.org/10.1242/jcs.00622
- T. Mysliwiec, R. Perego and G. D. Kruh, “Analysis of Chimeric Gag-Arg/Abl Molecules Indicates a Distinct Negative Regulatory Role for the Arg C-Terminal Domain,” Oncogene, Vol. 12, 1996, pp. 631-640.
- J. R. McWhirter and J. Y.Wang, “An Actin-Binding Function Contributes to Transformation by the Bcr-Abl Oncoprotein of Philadelphia Chromosome-Positive Human Leukemias,” EMBO Journal, Vol. 12, 1993, pp. 1533-1546.
- A. M. Pendergast, L. A. Quillian, L. D. Cripe, C. H. Bassing, Z. Dai, N. Li, A. Batzer, K. M. Rabun, C. J. Der, J. Schlessinger and M. L. Gishizky, “BCR-ABL Induced Oncogenesis Is Mediated by Direct Interaction with the SH2 Domain of the GRB2 Adaptor Protein,” Cell, Vol. 75, 1993, pp. 175-185.
- R. P. Million and R. A. Van Etten, “The Grb2 Binding Site Is Required for the Induction of Chronic Myeloid Leukemia-Like Disease in Mice by the Bcr/Abl Tyrosine Kinase,” Blood, Vol. 96, 2000, pp. 664-670.
- R. P. Million, N. Harakawa, S. Roumiantsev, L. Varticovski and R. A. Van Etten, “A Direct Binding Site for Grb2 Contributes to Transformation and Leukemogenesis by the Tel-Abl (ETV6-Abl) Tyrosine Kinase,” Molecular and Cellular Biology, Vol. 24, No. 11, 2004, pp. 4685- 4695. http://dx.doi.org/10.1128/MCB.24.11.4685-4695.2004
- K. Okuda, A. D’Andrea, R. A. Van Etten and J. D. Griffin, “A Chimeric Receptor/Oncogene that Can Be Regulated by a Ligand in Vitro and in Vivo,” The Journal of Clinical Investigation, Vol. 100, 1997, pp. 1708-1715. http://dx.doi.org/10.1172/JCI119695
- R. P. Million, J. Aster, G. D. Gilliland and R. A. Van Etten, “The Tel-Abl (ETV6-Abl) Tyrosine Kinase, Product of Complex (9;12) Translocations in Human Leukemia, Induces Distinct Myeloproliferative Disease in Mice,” Blood, Vol. 99, No. 12, 2002, pp. 4568-4577. http://dx.doi.org/10.1182/blood-2001-12-0244
- C. B. Gambacorti-Passerini, R. H. Gunby, R. Piazza, A. Galietta, R. Rostagno and L. Scapozza, “Molecular Mechanisms of Resistance to Imatinib in Philadelphia-Chromosome-Positive Leukaemias,” Lancet Oncology, Vol. 4, No. 2, 2003, pp. 75-85. http://dx.doi.org/10.1016/S1470-2045(03)00979-3
- F. E. Nicolini, M. J. Mauro, G. Martinelli, D. W. Kim, S. Soverini, M. C. Muller, A. Hochhaus, J. Cortes, C. Chuah, I. H. Dufva, J. F. Apperley, F. Yagasaki, J. D. Pearson, S. Peter, C. S. Rodriguez, C. Preudhomme, F. Giles, J. M. Goldman and W. Zhou, “Epidemiologic Study on Survival of Chronic Myeloid Leukemia and Ph(+) Acute Lymphoblastic Leukemia Patients with BCR-ABL T315I Mutation,” Blood, Vol. 114, No. 26, 2009, pp. 5271-5278. http://dx.doi.org/10.1182/blood-2009-04-219410
NOTES
*Disclosure of potential conflicts of interest: No potential conflicts of interest were disclosed.
#Corresponding author.