Open Journal of Pathology
Vol.06 No.03(2016), Article ID:68158,8 pages
10.4236/ojpathology.2016.63018
Gα12 Regulates Interleukin-8 Expression after Epithelial Cell Injury
Alexandra K. Kim1, Barrett Richard1, Ilene Boucher1, Wanfeng Yu1, Tianqing Kong2, Bradley M. Denker1*
1Renal Division, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA, USA
2Brigham and Women’s Hospital, Boston, MA, USA

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 22 April 2016; accepted 8 July 2016; published 11 July 2016
ABSTRACT
Acute kidney injury (AKI) is common in hospitalized patients and is strongly correlated with increased morbidity, mortality, and prolonged hospitalization. However, signals that determine whether injured tissues following AKI will repair or fibrose and lead to chronic kidney disease (CKD) are not well defined. Numerous cytokines are activated at various times after injury and recruit inflammatory cells. Interleukin-8 (IL-8) is upregulated following activation of Gα12 by H2O2, a reactive oxygen species (ROS). Herein, we study this occurrence in vitro and in vivo. IL-8 was measured by ELISA in Gα12-silenced (si-Gα12) and inducible QLα12 (constitutively active Gα12) Madin-Darby Canine Kidney (QLα12-MDCK) cell lines after H2O2/catalase cell injury. QLα12- and si-Gα12 MDCK cells showed time-, agonist- and Gα12-dependent increases in IL-8 mRNA and protein. Gα12-silenced MDCK cells demonstrated lower IL-8 expression and blunted IL-8 increases. In transgenic mice (QLα12γGTCre+, proximal tubule Qα12 expression) ischemia reperfusion injury led to significant upregulation of CXCL-1 (IL-8 homologue) at 48 hours that was not observed in Gα12 knockout mice. Macrophages in renal cells from these mice were imaged by immunofluorescent microscopy and QLα12γGTCre+ showed increased macrophage infiltration. We demonstrate that IL-8 is a critical link between H2O2 stimulated Gα12 and renal injury. Gα12 activation led to increased IL-8 expression, a potent mediator of inflammation after injury. Future studies targeting Gα12 for inhibition after injury may blunt the IL-8 response and allow for organ recovery.
Keywords:
Gα12, Interleukin-8, Acute Kidney Injury, Inflammation, Fibrosis

1. Introduction
Acute kidney injury (AKI) is common in hospitalized patients, reporting to affect 1% - 25% of intensive care unit (ICU) patients, and is frequently not recognized in the outpatient setting [1] . AKI in hospitalized patients is strongly correlated with prolonged hospitalization and increased morbidity and mortality, with mortality rates ranging from 15% to 60% in ICU patients [2] - [4] . Etiologies of AKI include ischemic and toxic injury (drugs), obstruction, and delayed graft function after renal transplantation. Clinical features include oliguria, fluid overload, and electrolyte disturbances and often require dialysis for correction of these abnormalities [1] [5] .
Importantly, mechanisms initiated with acute kidney injury can lead to renal tubular fibrosis and progressive chronic kidney disease (CKD) [6] - [8] . The amount of interstitial fibrosis is the best predictor of long-term renal outcome, regardless of the etiology [9] . Although significant progress has been made in defining the complexities of AKI (reviewed in [10] ), little is known about the molecular switches that determine whether injured epithelia will repair or progress to fibrosis. Reactive oxygen species (ROS) are increased with tissue injury and contribute to progressive fibrosis, diabetic nephropathy and hypertensive nephrosclerosis [11] - [13] . ROS remains elevated for more than 16 days after 30 min of ischemia [14] leading to sustained activation of numerous signaling pathways. Oxidative stress through ROS leads to proximal tubule cell detachment, actin cytoskeleton disruption and TJ disruption [15] ; all processes are linked to G protein signaling and specifically, Gα12 [16] - [18] .
The heterotrimeric G protein family is comprised of Gαs, Gαi/o, Gαq, and Gα12/13. The α subunits of these signal transduction proteins bind GTP with activation and utilize a conformational switch to promote interactions with downstream effectors. G proteins are essential to cellular function and regulate numerous processes including: proliferation, apoptosis, differentiation, cell attachment and migration and many others. The Gα12 family regulates cell migration and attachment in addition to apoptosis in epithelial cells [16] [19] . We previously demonstrated an essential role for Gα12 in regulating the epithelial cell tight junction and barrier function [17] [20] [21] . Recently, we showed that Gα12 is directly activated by the reactive oxygen species (ROS), H2O2, a major signaling molecule mediating oxidative injury seen in ischemia reperfusion (I/R). We found that silencing Gα12 protected epithelial cells from injury in the H2O2/catalase model of reversible injury. Furthermore, Gα12 knockout mice (Gα12 KO) were highly protected from ischemia reperfusion injury (IRI) and mice with proximal tubule targeted expression of constitutively activated Gα12 (QLα12γGTCre+) showed more severe injury [22] .
Numerous cytokines are activated at various times after injury and these molecules play critical roles in recruitment of inflammatory cells and activation of other pro-inflammatory/pro-fibrotic factors. Now we demonstrate that IL-8 is a critical link between H2O2 stimulated Gα12 and renal injury. We show that Gα12 stimulates IL-8 production in cell culture and activated Gα12 (QLα12) enhances IL-8 expression and macrophage infiltration after injury. IL-8 is significantly upregulated in human acute kidney injury [23] and these results suggest that Gα12 is an important proximal mediator of IL-8 production. Taken together, these studies extend previous findings indicating an important role of Gα12 in propagating injury signals and link Gα12 activation to increased IL-8 expression, a potent mediator of inflammation after injury. Targeting Gα12 for inhibition after injury may blunt the IL-8 response and contribute to more rapid and complete organ recovery.
2. Methods
2.1. MDCK Cell Lines, Cell Culture and H2O2/Catalase Injury Model
Tet-off inducible Gα12- and QLα12-MDCK cell lines (as previously described in [17] ) were cultured at 37˚C in 5% (vol/vol) CO2 and maintained in DMEM (Cellgro) containing 5% (vol/vol) FBS (Clontech) (DMEM) and 100 μg/mL G418 and 40 ng/mL doxycycline. Gα12 expression was induced by dox removal. Si-Gα12- and Si-GFP-MDCK cells were previously described [16] . Monolayers were serum starved for 24 h and then incubated with 10 U/mL thrombin or 2.5 mM H2O2 as described in [24] . Recovery at T = 0 was induced by the addition of the ROS scavenger catalase (5000 U/mL) and cells removed at various times for analysis of IL-8 concentration (T = 0, 16, 20, 24 hours).
2.2. IL-8 ELISA
Canine IL-8 from MDCK cells was measured by ELISA in 96-well plates (Linbro/Titertek; ICN Biochemicals, Costa Mesa, CA) coated overnight with 1 g/mL anti-rabbit IL-8 monoclonal antibody and detected with rabbit anti-canine IL-8 polyclonal antibody. Concentrations of IL-8 in si-Gα12, si-GFP, and QLα12 cells at t = 0, 16, 20, and 24 hours after exposure to thrombin or H2O2 were determined by a curve of optical density vs. concentration.
2.3. Real-Time PCR
Kidneys were obtained from QLα12γGTCre+ mice, Gα12 knockout mice, and C57/B6 control mice 48 h following ischemia reperfusion (murine models previously described in [22] ). cDNA was isolated from whole kidneys and TaqMan Gene Expression assays (Applied Biosystems, Foster City, CA, USA) were performed on the cDNA using an ABI 7300 (Applied Biosystems) with the following conditions: 2 min at 50˚C and 10 min at 95˚C, followed by 50 cycles of 95˚C for 15 s and 60˚C for 1 min. The CXCL-1 5’ sense primer was ACCCGCTCGCTTCTCTGT and the 3’ antisense primer was AAGGGAGCTTCAGGGTCAAG. Data analysis used the ΔΔCt method where the Ct was normalized to the CXCL-1 expression in the C57/B6 mice.
2.4. Immunofluorescence Microscopy
Macrophages in kidney sections from transgenic mice obtained 48 h after I/R were stained with F4/80 antibody and co-stained with DAPI. Coverslips were mounted in Fluoromount (Southern Biotechnology Associates) and viewed by using a Nikon Labophot-2 microscope with digital camera. Images were processed by using Adobe Photoshop and assembled in Adobe Illustrator (Adobe Systems Incorporated).
2.5. Statistics
Data are expressed as medians or means ± SEM as indicated. Statistical analysis was performed by using Excel using the two-tailed t test. Statistical significance was identified at P < 0.05.
3. Results
To identify potential targets of Gα12 activation, inducible QLα12 MDCK cells (previously described in [17] ) were utilized in a microarray analysis comparing baseline (no QLα12 expression (+dox)) to induced QLα12 expression (−dox for 3 days). In addition to changes in integrin expression [25] , it was noted that within the cytokine family, IL-8 was highly induced. Relative message levels for the IL-6, IL-10, and others showed no significant change (defined as <2-fold with p < 0.05; not shown), but IL-8 was significantly upregulated from ~30 to 120 fold. Based on the importance of IL-8 in inflammation and our recent findings showing that Gα12 knockout mice were protected from injury, we further examined a possible link between Gα12 activation and IL-8 expression.
To link increased IL-8 mRNA levels to increased secreted IL-8 protein, Gα12- and QLα12-MDCK cells (+/−dox) were assayed for IL-8 expression by ELISA. Figure 1 shows a time course of induced IL-8 protein levels in Gα12- and QLα12-MDCK cells (+/−dox). Within 24 h of dox removal (−dox), Gα12 and QLα12 proteins are induced and plateau by days 2 - 3 (see [17] ). IL-8 protein levels increased in parallel with induced Gα12 expression in both Gα12- and QLα12-MDCK cell lines. However, QLα12 expression (−dox) led to >20 fold higher IL-8 levels at day 1 when compared to Gα12 +/−dox or QLα12-MDCK maintained in +dox (Figure 1). Levels of IL-8 further increased at day 2 and plateaued by day 3 of −dox exposure correlating with the known time course of QLα12 protein expression in these cells. At day 3, IL-8 levels were >5 fold higher in QLα12 expressing cells when compared with the +dox control. Similar findings were seen with Gα12-MDCK cells but lower levels of IL-8 were induced consistent with the previously reported low level activation of Gα12 effectors seen with higher Gα12 expression. Figure 1(a) (inset) shows IL-8 protein expression in QLα12+dox and Gα12-MDCK cells (+/−dox) on an expanded scale. There is a small increase in IL-8 protein levels with induced Gα12 expression (−dox vs +dox). QLα12-MDCK+dox reveal subtle phenotypes due to leaky expression and increased IL-8 was observed.
Next, Gα12-MDCK cells +/−dox were stimulated with thrombin (a Gα12 agonist) for 24 h. Prior to inducing Gα12 expression with −dox, baseline IL-8 levels were 400 ± 300 pg/mL (n = 4) (Figure 1(b)). Gα12-MDCK cells (−dox) show a 4-fold increase in IL-8 production when stimulated with thrombin (Figure 1(b)). To extend these findings, baseline IL-8 was measured in previously characterized Gα12-silenced MDCK cells (si-Gα12) and controls (si-GFP) [26] . Figure 1(c) shows significantly lower IL-8 expression in the si-Gα12 cells in comparison to the controls.
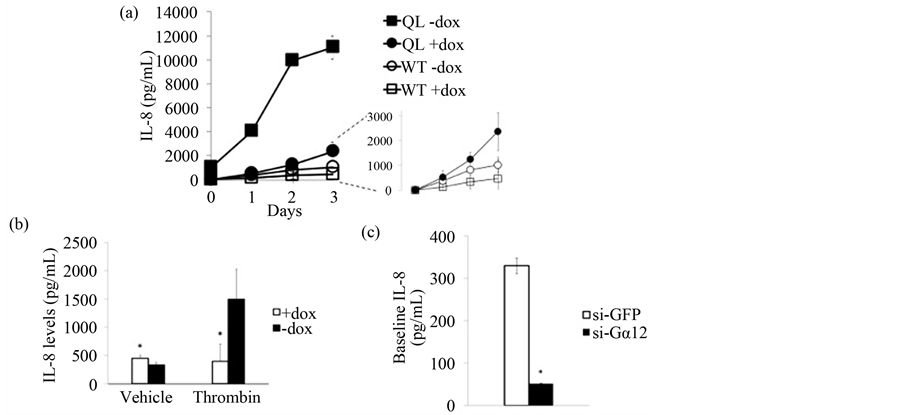
Figure 1. IL-8 is upregulated by Gα12 activation. (a) Time course of increased IL-8 release with expression of both wild-type and constitutively active (QL) Gα12. Gα12 and QLα12-MDCK cells were incubated +/−dox (−dox = Gα12 expression; +dox = control) for 3 days (n = 8 for each set). Supernatants were collected and assayed by ELISA for IL-8 levels; (b) Gα12-MDCK cells −dox showed greater IL-8 production than +dox cells (n = 4) 1 day after thrombin stimulation (P < 0.0118); (c) Baseline IL-8 levels in the si-Gα12 in comparison to the si-GFP MDCK cells (P < 0.0001).
H2O2 is a key ROS mediator of injury and directly activates Gα12 [22] . To determine if H2O2 stimulated Gα12 regulates IL-8 expression, we utilized the well-established ROS model of reversible epithelial injury with H2O2/catalase [22] [24] in si-Gα12 and si-GFP MDCK cells (Figure 2). si-Gα12 and si-GFP MDCK cells were compared +/− catalase at baseline, and at multiple times up to 24 h after exposure to 2.5 mM H2O2 (Figure 2). Control cells (si-GFP) have significantly higher baseline IL-8 expression levels than si-Gα12-cells (0 h in Figure 2) and similar to what is shown in Figure 1(c). At T = 0, cells are exposed to H2O2 +/− catalase and si-GFP cells were more prone to barrier disruption (see [22] ). With H2O2 injury, si-Gα12 MDCK are protected and secrete significantly lower amounts of IL-8 in comparison with controls.
The link between Gα12 activation and IL-8 expression was further investigated in in vivo using ischemia reperfusion injury in two transgenic models; QLα12γGTCre+ mice and Gα12 KO mice [22] . The functional homologue of IL-8 in mice is CXCL-1/KC and CXCL-1/KC was quantified utilizing real-time PCR in previously reported Gα12 KO mice (protected from injury)and QLα12γGTCre+ mice (show accelerated injury) [22] . Figure 3(a) shows that CXCL-1 gene expression was significantly upregulated in the QLα12γGTCre+ mice 2 days following ischemia reperfusion injury, corresponding to a ~4 fold increase (Figure 3(a)). The CXCL-1 relative gene expression in the Gα12 KO mice was indistinguishable from the controls. Thus, the more severe injury seen in QLα12γGTCre+ mice may reflect enhanced CXCL-1 expression and increased inflammation. IL-8 is released from inflammatory macrophages following injury [27] . To determine whether increased IL-8 also demonstrated increased macrophage (M1) infiltration, Gα12 KO and QLα12γGTCre+mice were stained at 48 hours following IRI. Analysis of macrophage staining demonstrated that the infiltration of macrophages was enhanced in the QLα12γGTCre+ mice (Figure 3(b)). This suggests that activated Gα12 enhances macrophage infiltration 2 days following ischemia reperfusion injury and similar findings were seen in 3 mice.
4. Discussion
IL-8 is an important mediator of the inflammation process. IL-8 is highly specific to CXCR1, a canonical seven-helical transmembrane G-protein receptor. When oxidative stress disturbs the permeability barrier of epithelial cells, IL-8 is secreted and binds to CXCR1 and CXCR2 expressed on neutrophils [28] . This results in rapid changes in cell morphology, activation of integrins, and the release the granule contents of neutrophils. IL-8 acts as a leukocyte chemotactic activating cytokine recruiting T lymphocytes and basophils to induce inflammation to the site of injury [29] . In renal injury, urinary IL-8 levels are associated with sustained renal allograft dysfunction due to ischemia-reperfusion injury [30] . Serial plasma IL-8 levels have been shown to predict
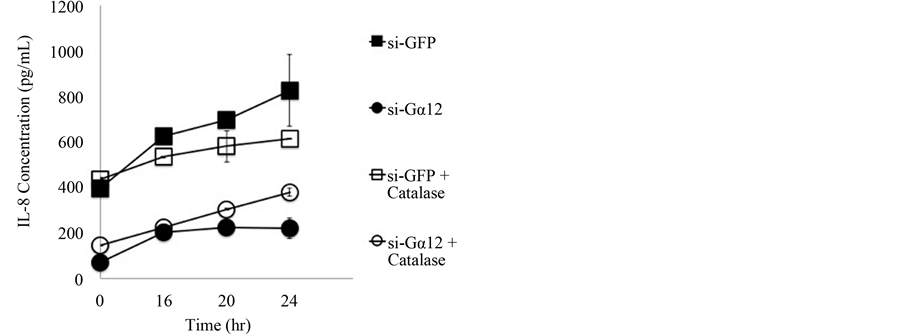
Figure 2. H2O2/catalase reversible injury model shows blunted IL-8 stimulation in si-Gα12 MDCK cells. Time course of IL-8 levels in si-GFP and si-Gα12 MDCK cells after exposure to 2.5 mM H2O2 +/− catalase at T = 0 hours. The effect of IL-8 increase was blunted with respect to exposure to 2.5 mM H2O2 without catalase.

Figure 3. Gα12 stimulates the IL-8 mouse homologue, CXCL1. (a) Relative expression of CXCL1 in QLα12γGTCre+ mice and Gα12 KO mice shown relative to the control mice 48 hours following ischemia reperfusion. CXCL1 was increased in QLα12γGTCre+ mice in comparison to the Gα12 KO mice (P < 0.0001). Data is based on 5 wildtype mice, 8 QLα12γGTCre+, and 2 Gα12 KO mice; (b) Macrophages were imaged by immunofluorescent microscopy after staining with F4/80 antibody and co-stained with DAPI. Kidneys were obtained at sacrifice 48 h after IRI. There was less macrophage staining in the Gα12 KO mice and increased staining in the QLα12γGTCre+ compared to the WT controls. Similar findings were seen in 3 mice.
the development in AKI in patients with sepsis [31] . Plasma IL-8 levels have also been shown to be elevated among critically ill patients with AKI that did not survive during hospitalization [27] .
We have demonstrated that activation of Gα12 by H2O2, a reactive oxygen species (ROS), promotes increased IL-8 expression in vivo and in vitro. As Gα12 activation is critical in the pro-inflammatory pathway, this study links Gα12 activation to IL-8 expression. However, numerous cytokines and pathways are stimulated with I/R injury and the definitive experiment of blocking IL-8 activity in QLα12γGTCre+ mice remains to be performed. Despite this, our findings further establishes Gα12 as a potential therapeutic target for ameliorating ROS mediated injury and add to the narrative of Gα12’s role in IL-8 activation and in injury responses. It has been found that Gα12/13 regulates NF-κB activation [30] and NF-κB bound to IL-8 acts as a transcriptional activator at the IL-8 promoter in all cell types [32] . Furthermore, Gα12 specifically regulates NF-κB-mediated Cyclooxygenase-2 (COX-2), a critical gene in the inflammatory responses during platelet aggregation and thrombosis [33] . Gα12 has also been suggested to stimulate IL-6 and IL-8 activation in the oral squamous carcinoma cell, which in turn promotes the oral squamous carcinoma cell’s invasive behavior characteristic of cancerous cells [34] .
Thus we believe targeting Gα12 for inhibition after injury may blunt the IL-8 response and permit engagement of recovery pathways through a mechanism that reduces inflammation after injury and thus prevent AKI evolving into CKD. Although exposure to H2O2 activates Gα12 and leads to barrier disruption, silencing Gα12 protected cells from tight junction disassembly despite H2O2 exposure [22] . Additionally, it has been previously found that Gα12 knockout mice are protected in bilateral ischemia reperfusion [22] . Since Gα12 knockout mice are phenotypically normal (indicating that the absence of Gα12 signaling is tolerated), we believe future studies investigating inhibitors of Gα12 as a potential drug treatment to prevent progressive injury following acute injury will be fruitful and well-tolerated. Although there are many downstream signaling pathways linked to Gα12, targeting the activated conformation of Gα12 would limit drug effects only to cells at the site of injury. Future studies include investigating the pathway(s) involved in the activation of IL-8, as well as studies pursuing the identification of molecules that inhibit activated Gα12 to promote repair processes following AKI.
5. Conclusion
Signals that determine whether injured tissues following AKI will repair or fibrose and lead to chronic kidney disease (CKD) are not well defined. We demonstrate that Gα12 activation by H2O2, a reactive oxygen species (ROS), leads to increased IL-8 expression in vivo and in vitro. Future studies inhibiting Gα12 after injury may reduce the IL-8 response and pro-fibrotic pathway, and permit more complete organ recovery.
Cite this paper
Alexandra K. Kim,Barrett Richard,Ilene Boucher,Wanfeng Yu,Tianqing Kong,Bradley M. Denker, (2016) Gα12 Regulates Interleukin-8 Expression after Epithelial Cell Injury. Open Journal of Pathology,06,154-161. doi: 10.4236/ojpathology.2016.63018
References
- 1. Kellum, J.A. (2008) Acute Kidney Injury. Critical Care Medicine, 36, S141-S145.
http://dx.doi.org/10.1097/ccm.0b013e318168c4a4 - 2. Levy, E.M., Viscoli, C.M. and Horwitz, R.I. (1996) The Effect of Acute Renal Failure on Mortality. A Cohort Analysis. JAMA, 275, 1489-1494.
http://dx.doi.org/10.1001/jama.1996.03530430033035 - 3. Thadhani, R., Pascual, M. and Bonventre, J.V. (1996) Acute Renal Failure. New England Journal of Medicine, 334, 1448-1460.
http://dx.doi.org/10.1056/NEJM199605303342207 - 4. Rihal, C.S., Textor, S.C., Grill, D.E., Berger, P.B., Ting, H.H., et al. (2002) Incidence and Prognostic Importance of Acute Renal Failure after Percutaneous Coronary Intervention. Circulation, 105, 2259-2264.
http://dx.doi.org/10.1161/01.CIR.0000016043.87291.33 - 5. Lam, M.F., Li, F.K., Choy, B.Y., Tang, S., Lo, W.K., et al. (2000) The Impact of the Establishment of a Multiorgan Transplantation Program on Cold Ischemia Time and Delayed Graft Function in Renal Transplantation. Transplantation Proceedings, 32, 1611-1612.
http://dx.doi.org/10.1016/S0041-1345(00)01451-2 - 6. Pagtalunan, M.E., Olson, J.L., Tilney, N.L. and Meyer, T.W. (1999) Late Consequences of Acute Ischemic Injury to a Solitary Kidney. Journal of the American Society of Nephrology, 10, 366-373.
- 7. Wald, R., Quinn, R.R., Luo, J., Li, P., Scales, D.C., et al. (2009) Chronic Dialysis and Death among Survivors of Acute Kidney Injury Requiring Dialysis. JAMA, 302, 1179-1185.
http://dx.doi.org/10.1001/jama.2009.1322 - 8. Ishani, A., Xue, J.L., Himmelfarb, J., Eggers, P.W., Kimmel, P.L., et al. (2009) Acute Kidney Injury Increases Risk of ESRD among Elderly. Journal of the American Society of Nephrology, 20, 223-228.
http://dx.doi.org/10.1681/ASN.2007080837 - 9. Schainuck, L.I., Striker, G.E., Cutler, R.E. and Benditt, E.P. (1970) Structural-Functional Correlations in Renal Disease. II. The Correlations. Human Pathology, 1, 631-641.
http://dx.doi.org/10.1016/S0046-8177(70)80061-2 - 10. Bonventre, J.V. and Yang, L. (2011) Cellular Pathophysiology of Ischemic Acute Kidney Injury. Journal of Clinical Investigation, 121, 4210-4221.
http://dx.doi.org/10.1172/JCI45161 - 11. Jin, L., Beswick, R.A., Yamamoto, T., Palmer, T., Taylor, T.A., et al. (2006) Increased Reactive Oxygen Species Contributes to Kidney Injury in Mineralocorticoid Hypertensive Rats. Journal of Physiology and Pharmacology, 57, 343-357.
- 12. Palm, F. (2006) Intrarenal Oxygen in Diabetes and a Possible Link to Diabetic Nephropathy. Clinical and Experimental Pharmacology and Physiology, 33, 997-1001.
http://dx.doi.org/10.1111/j.1440-1681.2006.04473.x - 13. Wiggins, K.J., Tiauw, V., Zhang, Y., Gilbert, R.E., Langham, R.G., et al. (2008) Perindopril Attenuates Tubular Hypoxia and Inflammation in an Experimental Model of Diabetic Nephropathy in Transgenic Ren-2 Rats. Nephrology (Carlton), 13, 721-729.
http://dx.doi.org/10.1111/j.1440-1797.2008.01008.x - 14. Kim, J., Seok, Y.M., Jung, K.J. and Park, K.M. (2009) Reactive Oxygen Species/Oxidative Stress Contributes to Progression of Kidney Fibrosis Following Transient Ischemic Injury in Mice. American Journal of Physiology-Renal Physiology, 297, F461-F470.
http://dx.doi.org/10.1152/ajprenal.90735.2008 - 15. Saenz-Morales, D., Escribese, M.M., Stamatakis, K., Garcia-Martos, M., Alegre, L., et al. (2006) Requirements for Proximal Tubule Epithelial Cell Detachment in Response to Ischemia: Role of Oxidative Stress. Experimental Cell Research, 312, 3711-3727.
http://dx.doi.org/10.1016/j.yexcr.2006.05.024 - 16. Kong, T., Xu, D., Yu, W., Takakura, A., Boucher, I., et al. (2009) Gα12 Inhibits α2β1 Integrin-Mediated Madin-Darby Canine Kidney Cell Attachment and Migration on Collagen-I and Blocks Tubulogenesis. Molecular Biology of the Cell, 20, 4596-4610.
http://dx.doi.org/10.1091/mbc.E09-03-0220 - 17. Meyer, T.N. (2002) Zonula Occludens-1 Is a Scaffolding Protein for Signaling Molecules. Gα12 Directly Binds to the Src Homology 3 Domain and Regulates Paracellular Permeability in Epithelial Cells. Journal of Biological Chemistry, 277, 24855-24858.
http://dx.doi.org/10.1074/jbc.C200240200 - 18. Sabath, E., Negoro, H., Beaudry, S., Paniagua, M., Angelow, S., et al. (2008) Gα12 Regulates Protein Interactions within the MDCK Cell Tight Junction and Inhibits Tight-Junction Assembly. Journal of Cell Science, 121, 814-824.
http://dx.doi.org/10.1242/jcs.014878 - 19. Yanamadala, V., Negoro, H., Gunaratnam, L., Kong, T. and Denker, B.M. (2007) Gα12 Stimulates Apoptosis in Epithelial Cells through JNK1-Mediated Bcl-2 Degradation and Up-Regulation of IκBα. Journal of Biological Chemistry, 282, 24352-24363.
http://dx.doi.org/10.1074/jbc.M702804200 - 20. Meyer, T.N., Hunt, J., Schwesinger, C. and Denker, B.M. (2003) Gα12 Regulates Epithelial Cell Junctions through Src tyrosine Kinases. American Journal of Physiology, 285, C1281-C1293.
http://dx.doi.org/10.1152/ajpcell.00548.2002 - 21. Sabath, E., Hunt, J. and Denker, B.M. (2005) Gα12 Activates c-Src and Increases Paracellular Permeability in Cultured Podocytes. Journal of the American Society of Nephrology, 16, 440A.
- 22. Yu, W., Beaudry, S., Negoro, H., Boucher, I., Tran, M., et al. (2012) H2O2 Activates G Protein α12 to Disrupt the Junctional Complex and Enhance Ischemia Reperfusion Injury. Proceedings of the National Academy of Sciences of the United States of America, 109, 6680-6685.
http://dx.doi.org/10.1073/pnas.1116800109 - 23. Roche, J.K., Keepers, T.R., Gross, L.K., Seaner, R.M. and Obrig, T.G. (2007) CXCL1/KC and CXCL2/MIP-2 Are Critical Effectors and Potential Targets for Therapy of Escherichia coli O157:H7-Associated Renal Inflammation. The American Journal of Pathology, 170, 526-537.
http://dx.doi.org/10.2353/ajpath.2007.060366 - 24. Meyer, T.N., Schwesinger, C., Ye, J., Denker, B.M. and Nigam, S.K. (2001) Reassembly of the Tight Junction after Oxidative Stress Depends on Tyrosine Kinase Activity. The Journal of Biological Chemistry, 276, 22048-22055.
http://dx.doi.org/10.1074/jbc.M011477200 - 25. Kong, T., Xu, D., Tran, M. and Denker, B.M. (2010) Regulation of Integrin Expression by Gα12: An Additional Potential Mechanism Modulating Cell Attachment. Cell Adhesion & Migration, 4, 372-376.
http://dx.doi.org/10.4161/cam.4.3.11639 - 26. Yu, W., Kong, T., Beaudry, S., Tran, M., Negoro, H., et al. (2010) Polycystin-1 Protein Level Determines Activity of the Gα12/JNK Apoptosis Pathway. The Journal of Biological Chemistry, 285, 10243-10251.
http://dx.doi.org/10.1074/jbc.M109.070821 - 27. Simmons, E.M., Himmelfarb, J., Sezer, M.T., Chertow, G.M., Mehta, R.L., et al. (2004) Plasma Cytokine Levels Predict Mortality in Patients with Acute Renal Failure. Kidney International, 65, 1357-1365.
http://dx.doi.org/10.1111/j.1523-1755.2004.00512.x - 28. Smithson, A., Sarrias, M.R., Barcelo, J., Suarez, B., Horcajada, J.P., et al. (2005) Expression of Interleukin-8 Receptors (CXCR1 and CXCR2) in Premenopausal Women with Recurrent Urinary Tract Infections. Clinical and Vaccine Immunology, 12, 1358-1363.
http://dx.doi.org/10.1128/cdli.12.12.1358-1363.2005 - 29. Ember, J.A., Sanderson, S.D., Hugli, T.E. and Morgan, E.L. (1994) Induction of Interleukin-8 Synthesis from Monocytes by Human C5α Anaphylatoxin. The American Journal of Pathology, 144, 393-403.
- 30. Nagamatsu, Y., Nishida, M., Onohara, N., Fukutomi, M., Maruyama, Y., et al. (2006) Heterotrimeric G Protein Gα13-Induced Induction of Cytokine mRNAs through Two Distinct Pathways in Cardiac Fibroblasts. Journal of Pharmacological Sciences, 101, 144-150.
http://dx.doi.org/10.1254/jphs.FP0051036 - 31. Ahlstrom, A., Hynninen, M., Tallgren, M., Kuusela, P., Valtonen, M., et al. (2004) Predictive Value of Interleukins 6, 8 and 10, and Low HLA-DR Expression in Acute Renal Failure. Clinical Nephrology, 61, 103-110.
http://dx.doi.org/10.5414/CNP61103 - 32. Mahe, Y., Mukaida, N., Kuno, K., Akiyama, M., Ikeda, N., et al. (1991) Hepatitis B Virus X Protein Transactivates Human Interleukin-8 Gene through Acting on Nuclear Factor κB and CCAAT/Enhancer-Binding Protein-Like Cis-Elements. The Journal of Biological Chemistry, 266, 13759-13763.
- 33. Ki, S.H., Choi, M.J., Lee, C.H. and Kim, S.G. (2007) Gα12 Specifically Regulates COX-2 Induction by Sphingosine 1-Phosphate. Role for JNK-Dependent Ubiquitination and Degradation of IκBα. The Journal of Biological Chemistry, 282, 1938-1947.
http://dx.doi.org/10.1074/jbc.M606080200 - 34. Jian, S.L., Hsieh, H.Y., Liao, C.T., Yen, T.C., Nien, S.W., et al. (2013) Gα12 Drives Invasion of Oral Squamous Cell Carcinoma through Up-Regulation of Proinflammatory Cytokines. PLoS ONE, 8, e66133.
http://dx.doi.org/10.1371/journal.pone.0066133
NOTES

*Corresponding author.