Open Journal of Pathology
Vol.4 No.3(2014), Article
ID:47941,6
pages
DOI:10.4236/ojpathology.2014.43017
Hemoglobin K-Woolwich (Hb KW): Its Combination with Sickle Cell Trait
Ruchika Sharma1, Melissa Rhodes2, Elizabeth Varga1, Samir B. Kahwash3*
1Division of Hematology/Oncology, Nationwide Children’s Hospital, Columbus, Ohio, USA
2Department of Pediatrics, University of Mississippi Medical Center, Jackson, Mississippi, USA
3Department of Pathology and Laboratory Medicine, Nationwide Children’s Hospital, Columbus, Ohio, USA
Email: *Samir.Kahwash@nationwidechildrens.org
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 11 April 2014; revised 11 May 2014; accepted 30 May 2014
ABSTRACT
Hemoglobin K-Woolwich (Hb KW) is a rare hemoglobin variant with very few cases reported. It is most prevalent in West African countries, particularly Nigeria, Ghana, and the Ivory Coast. Some reports suggest Hb KW may be a clinically benign trait, whereas others indicate it may behave similarly to a β+ thalassemia. The combination of hemoglobin S and hemoglobin KW (Hb S/KW) is a rare double heterozygous disorder with little known clinical characteristics. We report the hematologic and clinical data on three patients with Hb S/KW to help describe the characteristics of this patient population. The first two cases represent first cousins, ages 3 and 2 years. They are clinically asymptomatic. They have normal hemoglobin and mean corpuscle volume (MCV) levels without reticulocytosis. The third case is of a 14-year-old male who is non-anemic with no microcytosis. He has been clinically well except for abdominal pain upon dehydration. On hemoglobin electrophoresis, these patients have Hb S levels slightly higher than typically observed with sickle cell trait and a delay of hemoglobin F to adult levels. There exists a need for more reports to better delineate the clinical course and management of these patients.
Keywords:Hemoglobin K-Woolwich, Hemoglobin Electrophoresis, Fast Hemoglobin Variant, Sickle-Cell Anemia

1. Introduction
Hemoglobin K-Woolwich (Hb KW) is a rare genetically inherited hemoglobin variant with very few cases reported. It was first described under this name in a West Indian family in 1963. It has since been reported in Jamaica and several African countries including Nigeria, Ghana, and the Ivory Coast. Although it has been described as a heterozygous trait and in association with Hb β+ thalassemia, Hb S and Hb C, little is known about the phenotype of Hb S/KW due to the rarity of this combination. There is a need in the medical literature to further describe this patient population and get a better understanding of the best management for this rare disorder. We describe below three cases in children being followed in our Comprehensive Hemoglobinopathy Center.
2. Case One
A 2-month-old female presented to the Hematology clinic due to abnormal newborn screen results. She was born full term via C-section with no maternal or fetal complications. Newborn screen showed Hb F and Hb S. Father is from Dominican Republic and has a history of sickle cell trait, as does the patient’s five year old sister. Mother is of mixed Central American and African ancestry and reported “abnormal hemoglobin” on her side of the family; although she was not tested herself.
On initial presentation, the child had a Hb of 12.5 g/dl, MCV of 84 fl, and there was no laboratory or clinical evidence of hemolysis. Sickle solubility test was positive and peripheral smear showed few target cells and anisocytosis. Parents were told the differential included sickle cell disease (Hb S/S) or Hb S/β0 thalassemia. Mother was offered evaluation including CBC and Hb electrophoresis, but did not follow-up with testing. The patient was started on penicillin prophylaxis and continued to do well with no evidence of pain, dactylitis, acute chest syndrome or splenomegaly. At 12 months of age, the patient had a Hb of 12.7 g/dl and an MCV of 68 fl. Sickle solubility test was positive and peripheral smear showed microcytic hypochromic cells with target cells and anisocytosis. Repeat Hemoglobin electrophoresis testing also done at 12 months of age showed Hemoglobins S (45%), a new fast variant hemoglobin (36%) and Hb F (16%). On the Isoelectric focusing gel, the fast variant ran between hemoglobin A and hemoglobin N positions. On the cellulose acetate gel, the fast variant ran well beyond the hemoglobin A position, close to the hemoglobin J position. On citrate agar, the variant was not distinctly seen as a separate band, but the hemoglobin F band was noted to be wider than normal. The patient is currently 3 years old and is asymptomatic.
3. Case Two
A 1-month-old female presented to the Hematology clinic due to abnormal newborn screen results. She was born at 39 weeks via C-section and did not have any complications. Newborn screen had detected Hb F and Hb S. Mother is African American and has a history of Hb S trait. Father is of mixed Central American and African ancestry and reported having an “abnormal hemoglobin gene called Hb J Toronto”. Two older siblings had also been told that they had “Hb J Toronto trait”. The family reported that the father is a full sibling to the mother of Case One (Figure 1).
At initial presentation the patient’s Hb was 10.6 g/dl and MCV was 94 fl. Her sickle solubility test was negative and peripheral smear showed target cells with anisopoikilocytosis. Patient was started on penicillin prophylaxis and was followed. She continued to do well without evidence of pain, dactylitis, acute chest syndrome or splenomegaly. At her 6-month routine check-up visit, her Hb was 11.6 g/dl, MCV was 75 fl, and she continued to have no evidence of hemolysis with normal reticulocyte count, normal bilirubin and normal lactate dehydrogenase (LDH).
To clarify the diagnosis, hemoglobinopathy genotyping for the father and the child were sent to a specialized reference laboratory. On CBC, father had a normal hemoglobin of 15.0 g/dl with an MCV of 80 fl. Hemoglobin analysis by BioRad Variant II HPLC revealed Hb A 57%, a variant Hb (that elutes ahead of Hb A at 1.3 minutes) 38.6%, Hb A2 3.3%, and Hb F 0.5%. Genomic DNA was extracted from peripheral blood leukocytes. The β-globin genes were amplified by PCR and direct nucleotide sequencing was done. Testing revealed that the father was heterozygous for Hb KW, β codon 132 AAA > CAA or Lys132Gln.
Hemoglobin analysis on the patient by BioRad Variant II HPLC revealed Hb S 41.1%, a variant Hb that elutes just behind Hb F at 1.3 minutes 38.6%, Hb F 15.4%, and Hb A2 3.1%. Sequencing of genomic DNA revealed that the patient was a compound heterozygote for two globin gene mutations: Hb S and Hb KW (β codon 132 AAA > CAA or Lys132Gln). In light of these results, it was determined that the cousin (Case One), who has a similar variant, was also a Hb S/KW compound heterozygote. At the time of this report, the patient is 2 years

Figure 1. Family pedigree for cases one and two. Circles: female relatives; Squares: male relatives; Hb K-Woolwich: Hemoglobin K-Woolwich trait; HBB: beta globin gene; Lys: lysine; Gln: glutamine; NBS: newborn screen; Hb electro: hemoglobin electrophoresis; SAB: spontaneous abortion; Hb S: sickle cell trait; Hb A: hemoglobin A.
of age and is asymptomatic.
Study of This Patient’s Siblings
CBC on the patient’s 11-year-old sister showed normal hemoglobin of 12.4 g/dl with an MCV of 78.3fl. Hemoglobin electrophoresis showed Hb A 55.5%, a variant consistent with Hb KW of 38.5%, Hb A2 of 3.0% and Hb F of 3.0%. The peripheral blood smear showed normocytic, normochromic red blood cells with rare target forms. The CBC on the patient’s 8-year-old sister was similar, with normal hemoglobin of 13.1 g/dl and MCV of 78.6 fl. Hemoglobin electrophoresis showed Hb A 55.0%, a variant Hb consistent with Hb KW 39.0%, Hb A2 3.5% and Hb F 2.0%. Like the older sister, her peripheral blood smear showed normocytic, normochromic red blood cells with rare target forms.
4. Case Three
A 14-year-old African American male presented to our Hematology clinic. He carried a diagnosis of Hb S/KW compound heterozygote, diagnosed on newborn screening at an outside institution. He was followed in the sickle cell clinic there for the first 9 years of his life. Mother reports that the patient had an episode of dactylitis at 9 months of age. He was on Amoxicillin prophylaxis until 5 years of age. He never had acute chest syndrome, priapism, invasive bacterial infection, or needed opioid medications at home for treatment of pain. In the past 5 years, he had an average of 1 - 2 ER visits every year for abdominal pain after sickness or dehydration; these visits never required overnight hospitalization or the need to go home with pain medication. He also had a past history of asthma and Attention Deficit Disorder (ADD). At presentation to the Hematology clinic his hemoglobin level was 13.6 g/dl, with an MCV of 80 fl. His peripheral blood smear showed normochromic, normocytic red blood cells with rare target and tear-drop shaped forms (Figure 2). There was no laboratory evidence of increased red cell turnover, including normal reticulocyte count, normal bilirubin and normal LDH. Sickle solubility test was positive and peripheral smear showed normocytic normochromic cells. Hemoglobin electrophoresis revealed Hb S 48.0%, Hb “other” 50% and Hb A2 2.0%. Isoelectric focusing showed hemoglobin bands in the S
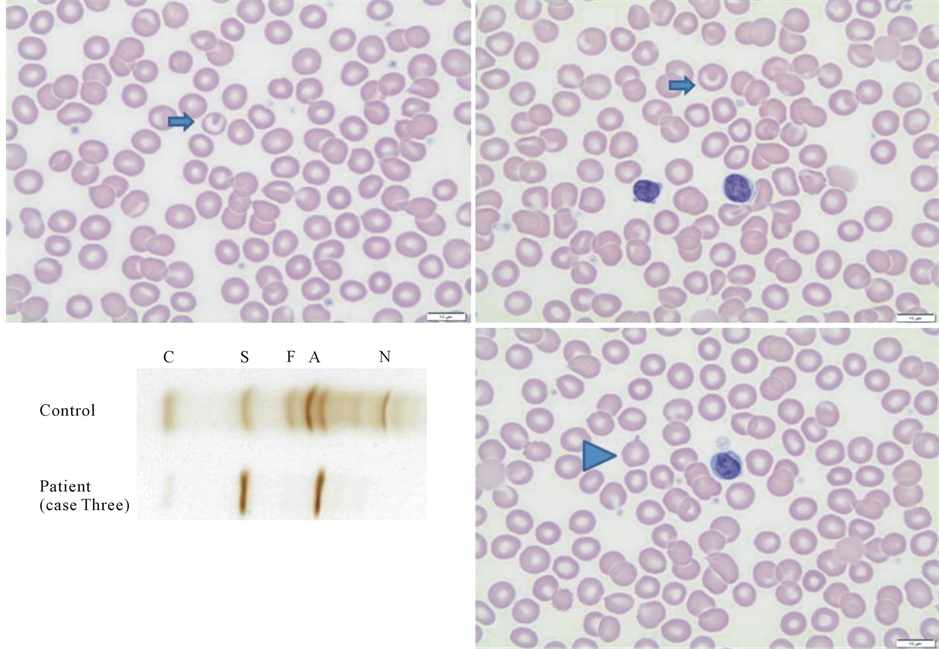
Figure 2. Peripheral blood smear of patient in case three. Shows rare target (arrows) and rare tear-drop shaped RBC’s (arrowhead). Isoelectric focusing plate shows a band in the S position and another band slightly anodal to the A position.
portion, along with a “fast variant” consistent with the diagnosis of Hb S/KW compound heterozygote (Figure 2).
5. Discussion
Hemoglobin K-Woolwich (Hb KW) is one of the K group of hemoglobins. These are Beta chain variants that share migrating to the same “fast” position on hemoglobin electrophoresis; due to a gain of one negative charge, resulting from one amino acid substitution. The first report of an Hb K variant was made in Berbers, an indigenous ethnic group of North Africa, in Algeria by Cabannes and Buhr in 1955 [1] . It was described as a fast variant of Hemoglobin A that moved further towards the anode on hemoglobin electrophoresis at alkaline pH. Several Hb K variants were subsequently described [2] . Hb KW was first described in a West Indian family by researchers in Woolwich, England [3] . Two members of the same family (a man of West Indian descent and his maternal aunt) had Hb KW associated with Hb S. Hemoglobin electrophoresis for one patient showed 40% Hb KW and no Hb A. Both these patients were not anemic but had a history of occasional joint pain. A second instance of a combination of Hb S/KW was reported from Nigeria [4] . In this report, the underlying defect of Hb KW was described as a substitution of a Lysine residue (positively charged) by Glutamine (neutral net charge).
Hb KW was also found in 24 members of 2 tribes which lived in different regions of Ghana with various genotypes including Hb A/KW, Hb S/KW and Hb C/KW [5] . In the Ivory Coast, of 42 subjects having Hb KW, 41 belonged to the Akan group but one subject was of Voltaic origin [6] .
Lang et al. considered Hb KW to be equivalent to a β+ thalassemia on the basis of results from a family in which α thalassemia and Hb KW interacted. In this family, elevated levels of Hb A2 were observed in heterozygotes and reduced synthesis of βKW chains, compared to βA chains observed in one case [7] . In 1980, Cabannes et al. described a family from Akan population in which the proband was homozygous for KW and several relatives were heterozygous [8] . The proband as well as the heterozygotes were clinically and hematologically normal, except for malarial crises. Cabannes et al. did not measure globin synthesis in this family.
Six years later, Zago et al. described the globin synthesis and hematologic characteristics of a Brazilian black family with six Hb KW heterozygotes [9] . The proband was a 2-year-old boy who presented with microcytic and hypochromic anemia and ancylostomiasis (Hookworm). Hemoglobin electrophoresis was suggestive of Hb KW in the child, his father and four siblings. Using high-performance liquid chromatography, an abnormal peptide was identified; amino acid analysis confirmed the substitution of lysine by glutamine at the 132nd position, consistent with Hb KW. Globin chain synthesis was measured for the proband and his father. The βKW and βA/α ratios were 1.00 and 1.13 while the βKW/βA ratios were 1.12 and 0.90 respectively, consistent with balanced globin synthesis, and a low probability for α thalassemia trait. The only hematologic abnormalities on CBC were an MCH of 20 pg in the proband, who concomitantly had iron levels on the low end of normal. It is also notable that a sibling of the proband had an MCV of 71 fl and overt of iron deficiency.
Tolo et al. described 130 cases of Hb KW in the Ivory Coast, diagnosed between 1968 and 1992 [10] . Three cases were homozygous, 110 cases were heterozygous, 1 case had Hb KW with β+ thalassemia, 2 cases had Hb C/KW and 9 had Hb S/KW. Approximately 30% of all subjects in this cohort had a moderate anemia with microcytic, hypochromic red blood cells, but the authors noted that the prevalence of iron deficiency in this population is approximately 30% - 40%. Based on the later fact on iron deficiency may better explain the anemia than the hemoglobinopathy in this study. The median hemoglobin in this cohort was 12.7 g/dl with a range of 11.2 to 14.2 g/dl, and the median MCV was 80.9 fl with a range of 64 - 100 fl. The authors did not stratify complete blood count data by genotype. Of the patients with Hb S/KW, the median Hb S concentration was 54.0%, Hb KW 42.7%, and Hb A2 3.3%. The authors of this paper conclude that in their view, Hb KW is a clinically asymptomatic trait.
The three patients we present here are compound heterozygotes for Hb S and Hb KW. The 14-year-old male is non-anemic with no microcytosis. He has been clinically well except for abdominal pain when dehydrated. The other two patients are young children and are clinically doing well with no evidence of dactylitis, acute chest syndrome or splenic sequestration. All three patients have normal hemoglobin levels without reticulocytosis, and generally unremarkable red blood cell indices similar to Hb S trait (Table 1). However, these patients have slightly higher Hb S levels on hemoglobin electrophoresis than sickle cell trait patients. This indicates that, compared to patients with sickle cell trait, patients with Hb S/KW compound heterozygote are more likely to have a slightly increased risk for sickling episodes, and should be considered to be at risk for pain crises, acute chest syndrome and other sickle related pathology.
There is limited data available to determine the best management for patients with Hb S/KW. Although much of the literature suggests Hb S/KW may behave similarly to sickle cell trait, others suggest similarities to a Hb S/Hb β+ thalassemia phenotype. We opted to follow these patients closely for infection risk and recommended standard penicillin prophylaxis as well as immunizations against encapsulated organisms. Given the normal hemoglobin level and lack of hemolysis, risk of stroke is likely very low. We are therefore not monitoring our patients with trans-cranial Doppler studies. With the small number of patients with Hb KW in the literature, there is a need for more reports to shed light on the clinical course and best management of these patients.

Table 1. A summary table of hematologic data in the three patients presented.
Hb: hemoglobin; RBC: red blood count; MCV: mean corpuscle volume; MCH: mean corpuscle hemoglobin; RDW: red blood cells distri-bution width; S: hemoglobin S; KH: hemoglobin K-Woolwich; F: hemoglobin F.
6. Conclusion
Our cases demonstrate the importance of obtaining a thorough family history and, in some cases, performing genetic testing for clarification of unusual hemoglobinopathies. Newborn screening results for Cases One and Two were initially thought to be consistent with Hb S/S or Hb S/β0 thalassemia, leading to incorrect prognostic and genetic counseling of the families. Practitioners should keep in mind that minor bands of hemoglobin variants (such as Hb KW) may be missed at newborn screen, at a time when Hb F levels are high. Follow-up testing after hemoglobin F levels decrease are helpful in the workup of challenging cases. Our cases also demonstrate that persons with compound heterozygosity for Hb KW and Hb S are usually asymptomatic clinically with no anemia or significant hematologic abnormalities.
Acknowledgements
The authors thank Maria Nunez, Laurie Dillemuth, and Flora Yazigi for their help in preparing this manuscript.
References
- Cabannes, R. and Buhr, L. (1955) [A New Human Hemoglobin Fraction with an Electrophoretic Migration More Rapid ThanHemoglobin A and AlowerThan Hemoglobin H; Preliminary Note]. Pediatrie, 10, 888-889.
- Livingstone, F., Miller, M., Neel, J., Robinson, A. and Zuelzer, W. (1956) Two Fast Hemoglobin Components in Liberian Blood Samples. Blood, 11, 902-906.
- O’Gorman, P., Allsopp, K.M., Lehmann, H. and Sukumaran, P.K. (1963) Sickle-Cell Haemoglobin K Disease. British Medical Journal, 2, 1381-1382. http://dx.doi.org/10.1136/bmj.2.5369.1381
- Allan, N., Beale, D., Irvine, D. and Lehmann, H. (1965) Three Hemoglobins K: Woolwich, an Abnormal, Cameroon and Ibadan, Two unusual Variants of Human Hemoglobin A. Nature, 208, 658-661. http://dx.doi.org/10.1038/208658a0
- Ringelhann, B., Konotey-Ahulu, F.L., Talapatra, N.C., Nkrumah, N.K., Wiltshire, B.G. and Lehmann, H. (1971) Haemoglobin K Woolwich (α2β2132 Lysine → Glutamine) in Ghana. Acta Haematologica, 45, 250-258. http://dx.doi.org/10.1159/000208632
- Cabannes, R., Renaud, R., Mauran, A., Pennors, H., Charlesworth, D., Price, B.G. and Lehmann, H. (1972) [Two Fast Hemoglobins in Ivory-Coast: Hb K Woolwich and a New Hemoglobin Hb J Abidjan (alpha-51 Gly-Asp)]. Nouvelle revue francaise d’hematologie, 12, 289-300.
- Lang, A., Lehmann, H. and King-Lewis, P.A. (1974) Hb K Woolwich the Cause of a Thalassaemia. Nature, 249, 467-469. http://dx.doi.org/10.1038/249467a0
- Cabannes, R., Amegnizin, P., Sangare, A., Arne, D., Casey, R. and Lehmann, H. (1980) Haemoglobin K Woolwich: A Study of the Family of a Homozygote. Journal of Medical Genetics, 17, 183-186. http://dx.doi.org/10.1136/jmg.17.3.183
- Zago, M., Costa, F., Greene, L. and Bottura, C. (1986) Balanced Globin Synthesis by Hb K Woolwich Heterozygotes. British Journal of Haematology, 64, 207-210. http://dx.doi.org/10.1111/j.1365-2141.1986.tb07590.x
- Tolo, A., Toure, H., Elenga, J., Allangba, O., Sanogo, I., Meite, M. and Sangare, A. (1995) [Epidemiologic, Clinical and Hematologic Profile of K Woolwich Hemoglobinopathies in Ivory Coast]. Medicine Tropicale, 55, 143-145.
NOTES

*Corresponding author.