Open Journal of Gastroenterology
Vol.4 No.2(2014), Article ID:42891,8 pages DOI:10.4236/ojgas.2014.42013
Polymorphisms in methylenetetrahydrofolate reductase gene: Their impact on liver steatosis and fibrosis of chronic hepatitis c patients
![]()
1Gastroenterology Department, Faculty of Medicine, University of Mersin, Mersin, Turkey
2Department of Medical Biology and Genetic, Faculty of Medicine, University of Mersin, Mersin, Turkey
3Department of Pathology, Faculty of Medicine, University of Mersin, Mersin, Turkey
4Department of Biostatistics, Faculty of Medicine, University of Mersin, Mersin, Turkey
Email: *enginaltintas@mersin.edu.tr
Copyright © 2014 Engin Altintas et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Engin Altintas et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received 29 November 2013; revised 31 December 2013; accepted 8 January 2014
KEYWORDS
Fibrosis; Hepatitis C; Gene Polymorphism; Methylenetetrahydrofolate Reductase; Steatosis
ABSTRACT
Aim & Background: The mechanism of steatosis in Hepatitis C virus infection is multifactorial; therefore, it is complex and unclear. The aim of this study was to investigate the effects of methylentetrahydrofolate reductase (MTHFR) gene polymorphisms on the course of chronic hepatitis C virus infection and the development of steatosis due to hepatitis C virus. Methods: This study included 109 patients with chronic hepatitis C virus infection. Necroinflammatory activity, degrees of fibrosis and steatosis and MTHFR gene polymorphisms were investigated. Polymerase chain reaction-restriction fragment length polymorphism was used to determine MTHFR C677T and A1298C polymorphisms. Results: Fibrosis was correlated with age (r = 0.336, p = 0.002), platelet (r = −0.448, p < 0.0001), ALT (r = 0.241, p = 0.026), AST (r = 0.361) and GGT (r = 0.224, p = 0.039). Steatosis was only correlated with fibrosis. MTHFR C677T and A1298C polymorphisms did not have a significant effect on the degree of steatosis (p = 0.857, p = 0.202 respectively). There was a relation between MTHFR C677T and the degree of fibrosis but not A1298C (p = 0.014, p = 0.187 respectively). Conclusion: We found that MTHFR C677T polymorphism contributed to the development of fibrosis in patients with chronic hepatitis C virus infection.
1. INTRODUCTION
Hepatitis C virus (HCV) is a major cause of chronic liver disease worldwide. According to the most recent WHO estimate, the prevalence of HCV is approximately 2.2%, affecting approximately 123 million people in the world [1]. Hepatic steatosis refers to excessive fat accumulation in the liver, which also is very common in the general population. Hepatic steatosis is the most common cause of elevated aminotransferases and probably the most common cause of chronic liver disease worldwide [2]. Over the last decade, it has become apparent that liver steatosis in the setting of HCV infection is a distinct condition with specific clinical and prognostic features [3-8].
An increased prevalence of steatosis in patients with HCV is well established [3-9]. Most studies have reported approximately 50% prevalence of steatosis among patients undergoing a liver biopsy because of HCV [10-21].
HCV may induce liver steatosis by interfering with lipid metabolism in hepatocytes, and, indirectly, by influencing the level of insulin resistance. As a consequence, the metabolic steatosis, although largely due to disorders independent of the viral infection, may at least partially be ameliorated by antiviral therapy. It has to be noted, however, that as many as 30% of patients with fatty liver who do not drink alcohol and are infected with genotypes other than 3a have normal BMI and HOMA score, suggesting that other causes of fatty liver exist in hepatitis C [22].
A relatively new field is represented by the metabolic impact of cytokines secreted by adipose tissue, i.e. the so-called adipokines. As an example, an association has been reported for serum levels of adiponectin and HCVrelated steatosis [23].
Apart from viral and metabolic factors, some specific host genetic polymorphisms may also play a role in the pathogenesis of steatosis. Hyperhomocysteinemia, by inducing endoplasmic reticulum (ER) stress, causes deregulation of the endogenous sterol response pathway via SREBP, leading to increased hepatic biosynthesis and uptake of cholesterol and triglycerides [24]. This in turn leads to steatosis. It has also been suggested that hyperhomocysteinemia may increase oxidative stress by inhibiting the expression of several antioxidant enzymes, thus sensitizing hepatocytes to the cytotoxic effect of pro-oxidant agents [25,26]. Interestingly, a close association has been reported between the severity of steatosis and homocysteine serum levels in chronic hepatitis C patients [27].
Three enzymes utilize homocysteine (Hcy) as a substrate: methionine synthase (MS) and betaine-homocysteine methyltransferase (BHMT), which convert homocysteine back to methionine, and cystathionine ß-synthase (CBS), the first enzyme in the transsulfuration pathway [28]. The distribution of Hcy among them depends on metabolic conditions: when methionine is relatively deficient, remethylation of Hcy is favored, whereas in situations of methionine excess, the transsulfuration pathway prevails [28,29]. S-Adenosylmethionine (AdoMet), the first metabolite of methionine, modulates the flow of Hcy through these metabolic pathways: increased levels of AdoMet activate CBS and inhibit the activity of MS and BHMT [28,30]. Impairment of Hcy remethylation or transsulfuration leads to hyperhomocystinemia. Such situations may develop as a consequence of genetic defects in the enzymes MS, CBS, or methylenetetrahydrofolate reductase (MTHFR) (the enzyme that synthesizes the MS cosubstrate 5-methyltetrahydrofolate) [29,31].
Accordingly, the aims of the present study were: 1) to assess their relationship with the MTHFR C677T and A1298C polymorphisms; and 2) to establish whether such metabolic and genetic alterations are related to the development and severity of steatosis and to the progression of liver fibrosis in CHC patients.
2. PATIENTS AND METHODES
2.1. Patient Selection
Data were collected from the hospital records kept between 6 March, 2006 and 6 March, 2010. The following conditions were excluded: concurrent active hepatitis B virus (positive for hepatitis B surface antigen) or human immunodeficiency virus infection, autoimmune hepatitis, drug-induced hepatitis, steatohepatitis, primary biliary cirrhosis, primary sclerosing cholangitis, haemochromatosis, Wilson’s disease and α1-antitrypsin deficiency by appropriate history and by biochemical and serological tests. Patients with type 2 diabetes mellitus (T2DM) were excluded. The patients were selected through who were diagnosed with chronic hepatitis C virus (HCV) infection, underwent laboratory investigations for complete blood count, liver chemistry, fasting blood sugar, total cholesterol, triglyceride and insulin levels within one month before they had liver biopsy. All patients were genotype 1. A complete clinical evaluation was performed on each patient. Baseline characteristics collected at the time of liver biopsy included the age, height, weight and body mass index (BMI). The BMI was calculated as weight in kg/(height in meters)2. Information regarding the average current daily alcohol intake (g/day) in the past 6 months and past alcohol intake (g/day) before the last 6 months was noted. No patient had clinical evidence of hepatic decompensation (jaundice, hepatic encephalopathy, ascites, hepatorenal syndrome, or variceal bleeding) at the time of biopsy. There were eligible 109 patients. Control group was consisted 172 blood donors. All patients gave written and oral informed consent and approval was obtained from the ethical committee of Mersin University Medical Faculty Hospital (06.03.2006-8).
2.2. DNA Extraction and Analysis
A blood sample was drawn from each individual. Venous blood samples were collected in tubes containing ethylenediaminetetraacetic acid. DNA was extracted from whole blood by salting out procedure [32].
2.3. Molecular Analysis of MTHFR C677T and A1298C Polymorphisms
Polymerase Chain Reaction and Restriction Fragment Length Polymorphism (PCR-RFLP) assays were used to determine MTHFR C677T and A1298C polymorphisms. The primer pairs used were forward 5’-TGAAGGAGAAGGTGTCTGCGGGA-3’, reverse 5’-AGGACGGTGCGGTGAGAGTG-3’ for MTHFR C677T polymorphism [33,34], and forward 5’-TGAAGGAGAAGGTGTCTGCGGGA-3’ reverse 5’-AGGACGGTGCGGTGAGAGTG-3’ for MTHFR A1298C polymorphism [35]. PCR was performed in a 25 μl volume with 100 ng DNA, 100 μM dNTPs, 20 pmol of each primer, 1.5 mM MgCl2, 1 × PCR buffer with (NH4)2SO4 (Fermentas, Sigma Chemical Co., St. Louis, MA, USA) and 1 U Taq DNA polymerase (Fermentas, Sig-ma Chemical Co., St. Louis, MA, USA). Amplification was performed on an automated Thermal Cycler
(Techne Flexigene, Cambridge, UK). PCR conditions were 2 minutes for initial denaturation at 95˚C; 35 cycles at 95˚C for 45 seconds for denaturation, 1 minute at 60˚C for annealing (30 seconds at 64˚C for MTHFR A1298C polymorphism) and 1.5 minutes at 72˚C for extension, followed by 7 minutes at 72˚C for final extension. PCR products were digested with specific restriction enzymes. Digestion of the PCR product was carried out using 10 U HinfI (Fermentas, Sigma Chemical Co., St. Louis, MA, USA) for MTHFR C677T polymorphism and 10 U SatI (Fermentas, Sigma Chemical Co., St. Louis, MA, USA) for MTHFR A1298C polymorphism, overnight at 37˚C. The HinfI restricted products of MTHFR C677T; genotypes CC, CT and TT had band sizes of 198bp, 198 bp/175 bp/23 bp and 175 bp/23 bp, respectively. The SatI restricted products of MTHFR A1298C; genotypes AA, AC and CC had band sizes of 138 bp, 138 bp/119 bp/19 bp and 119 bp/19 bp, respectively. The digest products were resolved at 120 V for 30 - 40 minutes on a 3.5% agarose gel containing 0.5 μg/ml ethidium bromide.
2.4. Liver Biopsy
Necroinflammatory activity was scored based on Knodell’s Histological Activity Index (HAI); 0 - 4 corresponded to portal inflammation, 0 - 4 to lobular degeneration and necrosis and 0 - 10 to periportal necrosis [36]. The disease stage was determined based on Scheuer’s classification; 0 corresponded to absence of fibrosis, 1 to enlarged fibrotic portal tracts, 2 to periportal or portoportal septa but regular architecture, 3 to fibrosis of irregular architecture without overt cirrhosis and 4 to possible or confirmed cirrhosis [37]. Fibrosis is determined as absent, mild or severe if fibrosis scores are ‘‘0’’, “1 - 2’’ or ‘‘3 - 4’’ respectively. Liver steatosis was graded as in the following: 0 corresponded to steatosis in less than 5% of hepatocytes, 1 to steatosis in 5% - 34% of hepatocytes, 2 to steatosis in 35% - 69% of hepatocytes and 3 to steatosis in more than 69% of hepatocytes.
2.5. Statistical Analyses
Chi-square test was to determine frequencies, Student’s t test or Kruskal-Wallis to analyse demographic data and laboratory results when appropriate, Spearman’s rank correlation to determine the correlation between the variables, ANNOVA to determine the relation between the variables, and a logistics regression analysis which to find the main factor of the liver steatosis and fibrosis. Data were analysed with SPSS 10.0 (SPSS for Windows, Chicago, IL). P < 0.05 was considered significant.
3. RESULTS
The study included 42 males with a median age of 48 years and 67 females with a median age of 53 years (Table 1). Out of 109 patients included in the study, 88.99% had fibrosis and 66.05% had steatosis. Fibrosis was correlated with age (r = 0.336, p = 0.002), platelet (r = −0.448, p < 0.0001), ALT (r = 0.241, p = 0.026), AST (r = 0.361) and GGT (r = 0.224, p = 0.039). Steatosis was only correlated with fibrosis. Further statistical analyses showed a relation between steatosis and age platelet and GGT (p = 0.019, p = 0.004 and p = 0.028 respectively); but no relation between steatosis and ALT and AST. The logistics regression analysis was done that to examine the real relationship between gene polymorphisms and the liver steatosis and fibrosis. A relationship between gene polymorphisms and the liver steatosis and fibrosis is not found; p = 0.636 between MTHFR C677T polymorphism and fibrosis, p = 0.157 between MTHFR C677T polymorphism and steatosis, p = 0.489 between MTHFR A1298C polymorphism and fibrosis, and p = 0.943 between MTHFR A1298C polymorphism and steatosis.
3.1. Distribution of MTHFR C677T Gene Polymorphism Genotypes
Out of 109 patients, 61(56%) had C/C genotype, 36 (33%) had C/T genotype and 12 (11%) had T/T genotype of MTHFR C677T polymorphism. There was no statistically significant difference between the patient group and the control group for MTHFR C677T gene mutation (p = 0.336) (table 2). The distribution of MTHFR C677T polymorphism with or without steatosis was homogenous and there was no relation with steatosis. (p = 0.857) (Figure 1). There was a relationship with the severity of fibrosis and MTHFR C677T (p = 0.014) (Figure 2).
3.2. Distribution of MTHFR A1298C Gene Polymorphism Genotypes
One patients were not found to have MTHFR A1298C polymorphism. Out of the remaining 108 patients, 58 (53.7%) had A/A genotype, 30 (27.8%) had A/C genotype and 20 (18.5%) had C/C genotype. The frequency of MTHFR A1298C gene mutation was high in the patient group as twice as that of the control group. [20 (18.5%) versus 16 (9.8). But, according to MTHFR A1298C gene mutation, there was no statistically significant difference between the two groups (p = 0.994) (table 2).
There was not a significant difference between the distribution of MTHFR A1298C polymorphism and degree of fibrosis and degree of steatosis (p = 0.187 and p = 0.202; respectively) (Figures 3 and 4).
4. DISCUSSION
In this study, we found that MTHFR C677T and A1298C polymorphisms did not contribute to liver steatosis and
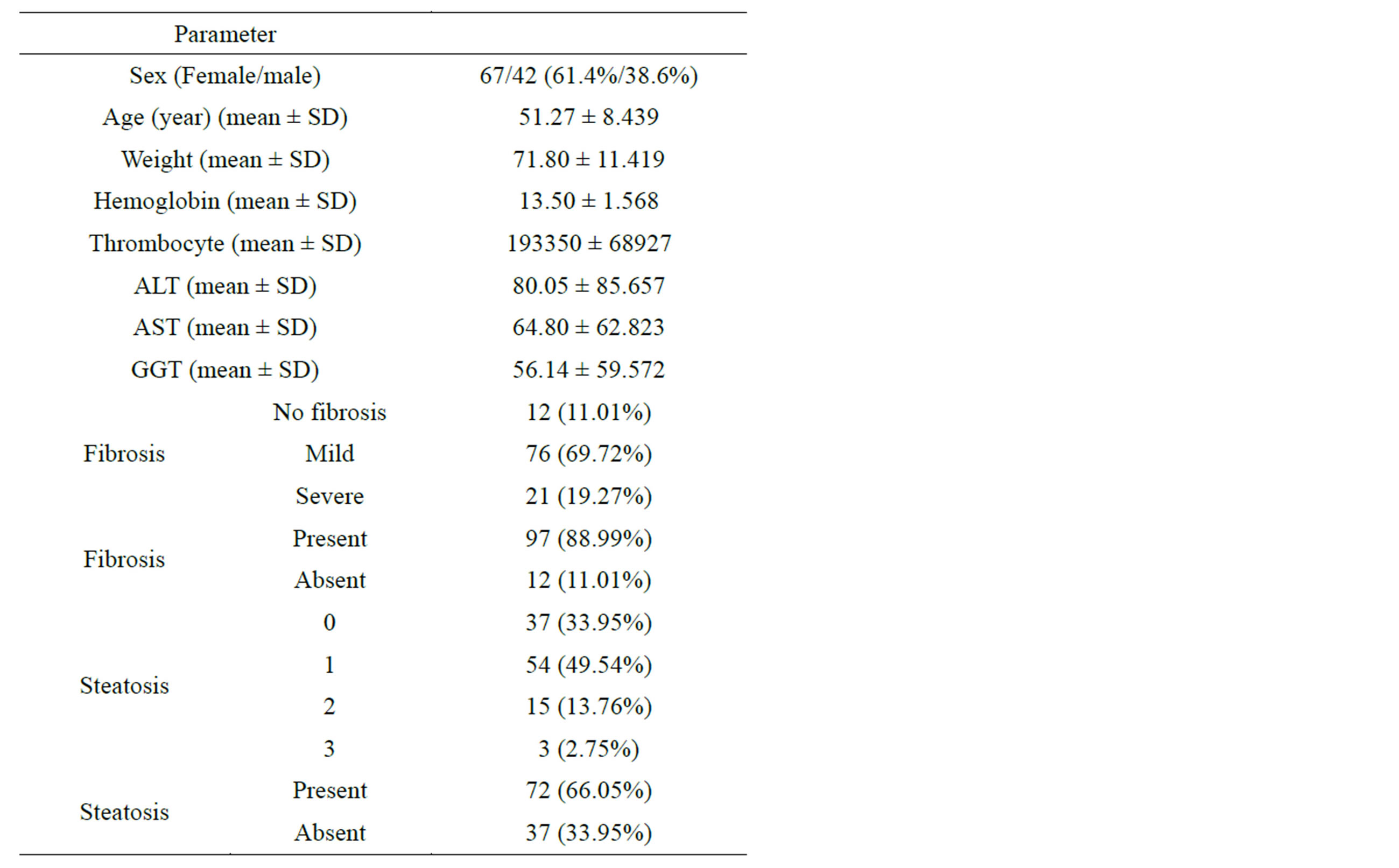
Table 1. The characteristics and demographic data of patients were shown (n = 109).
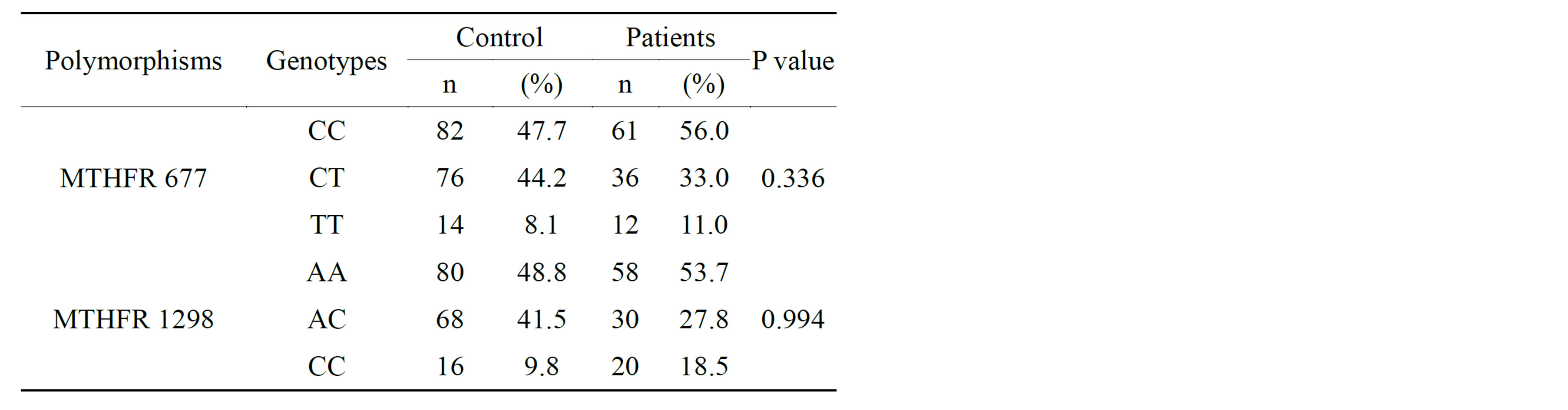
Table 2. Comparison of MTHFR C677T and MTHFR A1298C polymorphism frequencies in control subjects and patients with HCV.

Figure 1. The distrubution of MTHFR C677T polymorphism with or without steatosis was homogenous and there was no relation with steatosis (p = 0.857). In the logistics regression analysis, there was not relationship between steatosis and MTHFR C677T (p = 0.157).
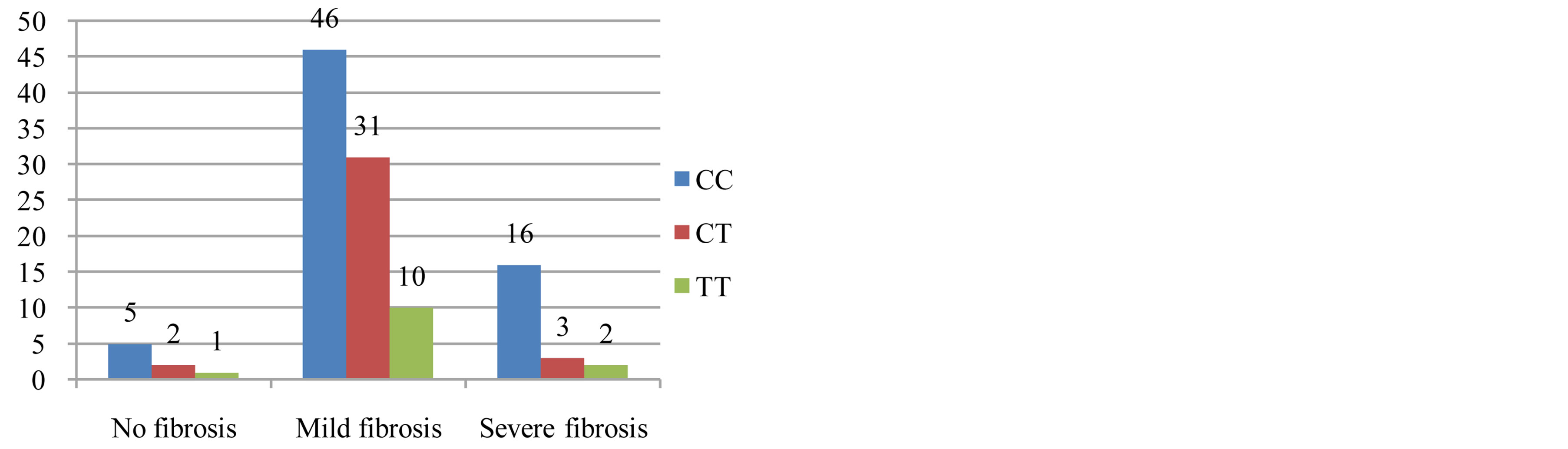
Figure 2. A relationship was found between the severity of fibrosis and MTHFR C677T and this relationship was favored to wild type (p = 0.014). But in the logistics regression analysis, there was not relationship between fibrosis and MTHFR C677T (p = 0.636).

Figure 3. The distribution of MTHFR A1298C and that of steatosis (absent or existent) were homogenous and there was no relationship (p = 0.202). In the logistics regression analysis, there was not relationship between steatosis and MTHFR A1298C (p = 0.943).
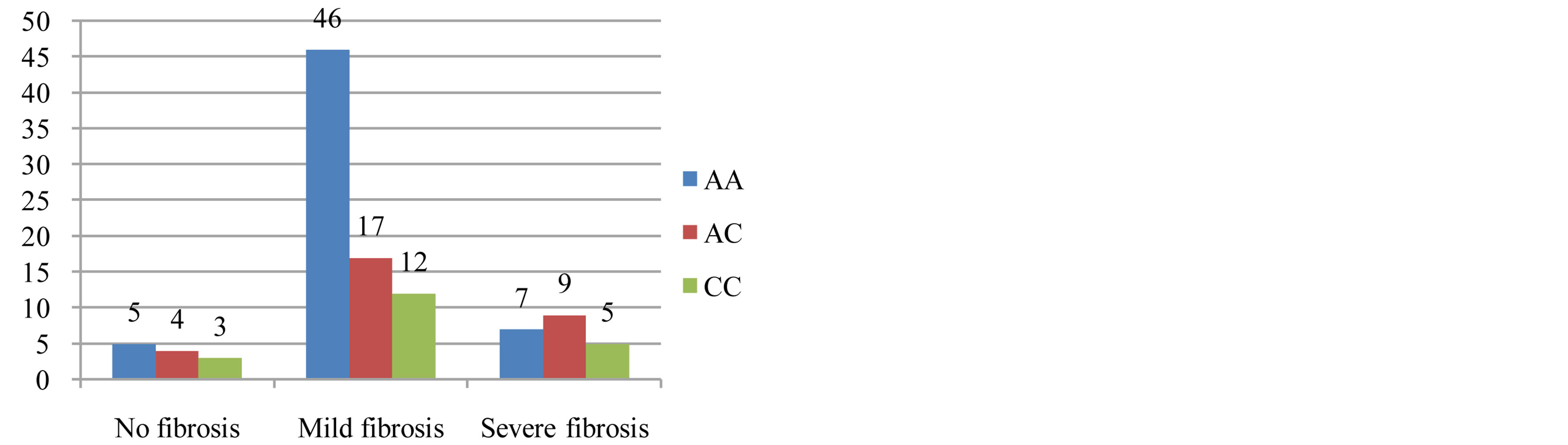
Figure 4. There was no relationship between the severity of the fibrosis and MTHFR A1298C (p = 0.187). In the logistics regression analysis, there was not relationship between fibrosis and MTHFR A1298C (p = 0.489).
liver fibrosis in patients with chronic HCV infection. Out of 109 patients, 66.05% had steatosis and 88.99% had fibrosis. Fibrosis was correlated with age, platelet, ALT, AST, GGT and MTHFR C677T; steatosis was correlated with fibrosis. All above findings are consistent with the literature [10-21,27].
The hyperhomocysteinemia-induced steatosis model may explain why only some, but not all, HCV-infected patients develop steatosis, and why only a minority of patients, e.g. those with higher homocysteine levels, accumulate a greater amount of fat in the liver. Hyperhomocysteinemia may result from a MTHFR C677T polymorphism [27]. In that study, it was shown that the polymorphism of the MTHFR gene at position 677, which has a prevalence of 12% - 15% for the TT genotype in the general population, was associated with both hyperhomocysteinemia and a greater degree of steatosis in chronic hepatitis C patients. It was estimated that the relative risk of developing more severe steatosis was six-fold higher for patients with the CT genotype and 20-fold higher for those with the TT genotype [27]. However, in our study we found any relationship between steatosis with neither MTHFR C677T nor MTHFR A1298C (p = 0.857 and p = 0.202, respectively).
Impaired liver function leads to altered methionine and homocysteine metabolism. The observations of Tevijana et al. [38] have suggested impaired liver function could be a novel determinant in the development of hyperhomocysteinemia and a role for elevated homocysteine levels in the development of liver fibrosis. In a recent study, it was investigated to verify the role of recipient MTHFR polymorphism in favouring graft fibrosis progression in patients with recurrent HCV after orthotopic liver transplantation [39]. Authors have suggested that the MTHFR C677T polymorphism may play a role in influencing liver fibrosis progression in patients with recurrent hepatitis C, in conjunction with donor age, but not via steatosis promotion [39]. In a study from China, it has showed that hyperhomocysteinemia may be an independent risk factor for liver cirrhosis [40]. The genetic mutation of MTHFR C667T has been possibly an important mechanism of hyperhomocysteinemia in liver cirrhosis [40]. Authors have suggested that the level of plasma homocysteine may be an early indicator for liver cirrhosis [40]. In another study of Ventura et al. [41] MTHFR C677T mutation and disease stage has showed to be the most important predictive factors of hyperhomocysteinaemia in liver cirrhosis [41]. Hyperhomocysteinaemia is highly prevalent in liver cirrhosis but not in other chronic liver diseases. They have reported that hyperhomocysteinaemia may contribute to fibrogenesis and vascular complication of liver cirrhosis [41].
The most remarkable result of this study is the distribution of MTHFR gene polymorphisms’ (especially MTHFR A1298C) genotypes. 61(56%) had C/C genotype, 36 (33%) had C/T genotype and 12 (11%) had T/T genotype of MTHFR C677T polymorphism. Out of the remaining 108 patients, 58 (53.7%) had A/A genotype, 30 (27.8%) had A/C genotype and 20 (18.5%) had C/C genotype. The distribution of MTHFR C677T gene polymorphism genotypes and alleles determined in this study was comparable with that reported in the literature, while the MTHFR A1298C homozygote frequency (18.5%) detected in this study was two times higher than that reported in the literature [38]. This may be a characteristic of patients with HCV infection. Being homozygote for MTHFR A1298C polymorphism might increase tendency to HCV infection. Otherwise, to bear wild type MTHFR C677T (CC) was related to the fibrosis.
Hyperhomocysteinemia is frequent in the Caucasian population (more than 15%) and its role in vascular pathology has been clearly established [42]. In hepatology, experimental data in transgenic mice deficient in homocysteine metabolism enzymes have shown the presence of severe liver steatosis with occasional steatohepatitis [43]. In human beings, many studies have found a correlation between homocysteine and steatosis or even NASH [44,45]. Some authors have suggested a discriminating threshold to differentiate simple steatosis from NASH. In chronic hepatitis C, preliminary data have shown that hyperhomocysteinemia is an independent risk factor for steatosis or even fibrosis [27]. The physiopathological mechanism has now begun to be better understood. On one hand, there is a strong correlation between homocysteine and insulin resistance whatever its etiology [46- 48]. On the other hand, homocysteine has a direct effect on the liver, resulting in over expression of SREBP-1 and favouring steatosis [49]. It stimulates proinflammatory cytokine secretion such as NF kappa B increasing the risk of NASH [50,51]. Finally, homocysteine could increase the risk of fibrosis by stimulating TIMP 1 [51]. Moreover hepatitis C virus induces hypomethylation of STAT 1 and could decrease the antiviral activity of interferon [52]. Results from in vitro studies have shown that the normalisation of STAT 1 methylation by bringing betaine and S Adenosyl Methionine (which belongs to homocysteine cycle) restores the antiviral activity of interferon. These data should be confirmed to evaluate the importance of homocysteine dosage in the diagnosis of NASH. Finally, treatment of hyperhomocysteinemia could have favourable consequences in steatopathies and HCV infection. Betaine has been shown to be the safest, least expensive and most effective in attenuating ethanol-induced liver injury [53]. Betaine, by virtue of aiding in the remethylation of homocysteine, removes both toxic metabolites (homocysteine and S-adenosylhomocysteine), restores S-adenosylmethionine level, and reverses steatosis, apoptosis and damaged proteins accumulation [54].
5. CONCLUSIONS
It can be concluded that chronic liver disease contributes to hyperhomocysteinemia and that hyperhomocysteinemia causes more liver steatosis and liver fibrosis in patients with liver disease, suggestive of as vicious circle [54].
As a result of this study, MTHFR C677T and A1298C polymorphisms, both of which cause hyperhaemocysteinemia, did not contribute to the development of steatosis in patients with chronic hepatitis C virus infection. However, as MTHFR C677T (wild type), age and GGT increased and as platelet count dropped, the possibility of fibrosis increased. Moreover, being homozygote for MTHFR A1298C polymorphism might increase tendency to HCV infection. Future studies are required to confirm the results presented here.
REFERENCES
- Perz, J.F., Farrington, L.A., Pecoraro, C., Hutin, Y.J.F. and Armstrong, G.L. (2004) Estimated global prevalence of hepatitis C virus infection. 42nd Annual Meeting of the Infectious Diseases Society of America, Boston. http://dx.doi.org/10.1056/NEJMra011775
- Angulo, P. (2002) Nonalcoholic fatty liver disease. New England Journal of Medicine, 346, 1221-1231.
- Ramesh, S. and Sanyal, A.J. (2004) Hepatitis C and nonalcoholic fatty liver disease. Seminars in Liver Disease, 24, 399-413. http://dx.doi.org/10.1055/s-2004-860869
- Lonardo, A., Adinolfi, L.E., Loria, P., Carulli, N., Ruggiero, G. and Day, C.P. (2004) Steatosis and hepatitis C virus: Mechanisms and significance for hepatic and extrahepatic disease. Gastroenterology, 126, 586-597. http://dx.doi.org/10.1053/j.gastro.2003.11.020
- Charlton, M.R., Pockros, P.J. and Harrison, S.A. (2006) Impact of obesity on treatment of chronic hepatitis C. Hepatology, 43, 1177-1186. http://dx.doi.org/10.1002/hep.21239
- Asselah, T., Rubbia-Brandt, L., Marcellin, P. and Negro, F. (2006) Steatosis in chronic hepatitis C: Why does it really matter? Gut, 55, 123-130. http://dx.doi.org/10.1136/gut.2005.069757
- Lonardo, A., Loria, P., Adinolfi, L.E., Carulli, N. and Ruggiero, G. (2006) Hepatitis C and steatosis: A reappraisal. Journal of Viral Hepatitis, 13, 73-80. http://dx.doi.org/10.1111/j.1365-2893.2005.00669.x
- Leandro, G., Mangia, A., Hui, J., Fabris, P., RubbiaBrandt, L., Colloredo, G., Adinolfi, L.E., et al. (2006) HCV Meta-Analysis (on) Individual Patients’ Data Study Group. Relationship between steatosis, inflammation, and fibrosis in chronic hepatitis C: A meta-analysis of individual patient data. Gastroenterology, 130, 1636-1642. http://dx.doi.org/10.1053/j.gastro.2006.03.014
- Adinolfi, L.E., Utili, R. and Ruggiero, G. (1999) Body composition and hepatic steatosis as precursors of fibrosis in chronic hepatitis C patients. Hepatology, 30, 1530-1531. http://dx.doi.org/10.1002/hep.510300625
- Fiore, G., Fera, G., Napoli, N., Vella, F. and Schiraldi, O. (1996) Liver steatosis and chronic hepatitis C: A spurious association? European Journal of Gastroenterology & Hepatology, 125-129. http://dx.doi.org/10.1097/00042737-199602000-00006
- Czaja, A.J., Carpenter, H.A., Santrach, P.J. and Moore, S.B. (1998) Host and disease-specific factors affecting steatosis in chronic hepatitis C. Journal of Hepatology, 29, 198-206. http://dx.doi.org/10.1016/S0168-8278(98)80004-4
- Hourigan, L.F., Macdonald, G.A., Purdie, D., Whitehall, V.H., Shorthouse, C., Clouston, A. and Powell, E.E. (1999) Fibrosis in chronic hepatitis C correlates significantly with body mass index and steatosis. Hepatology, 29, 1215- 1219. http://dx.doi.org/10.1002/hep.510290401
- Clouston, A.D., Jonsson, J.R., Purdie, D.M., Macdonald, G.A., Pandeya, N., Shorthouse, C. and Powell, E.E. (2001) Steatosis and chronic hepatitis C: Analysis of fibrosis and stellate cell activation. Journal of Hepatology, 34, 314- 320. http://dx.doi.org/10.1016/S0168-8278(00)00096-9
- Hwang, S.J., Luo, J.C., Chu, C.W., Lai, C.R., Lu, C.L., Tsay, S.H., Wu, J.C., et al. (2001) Hepatic steatosis in chronic hepatitis C virus infection: Prevalence and clinical correlation. Journal of Gastroenterology and Hepatology, 16, 190-195. http://dx.doi.org/10.1046/j.1440-1746.2001.02407.x
- Adinolfi, L.E., Gambardella, M., Andreana, A., Tripodi, M.F., Utili, R. and Ruggiero, G. (2001) Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology, 33, 1358-1364. http://dx.doi.org/10.1053/jhep.2001.24432
- Petit, J.M., Benichou, M., Duvillard, L., Jooste, V., Bour, J.B., Minello, A., Verges, B., et al. (2003) Hepatitis C virus-associated hypobetalipoproteinemia is correlated with plasma viral load, steatosis, and liver fibrosis. The American Journal of Gastroenterology, 98, 1150-1154.
- Monto, A., Alonzo, J., Watson, J.J., Grunfeld, C. and Wright, T.L. (2002) Steatosis in chronic hepatitis C: Relative contributions of obesity, diabetes mellitus, and alcohol. Hepathology, 36, 729-736. http://dx.doi.org/10.1053/jhep.2002.35064
- Poynard, T., Ratziu, V., McHutchison, J., Manns, M., Goodman, Z., Zeuzem, S., Younossi, Z., et al. (2003) Effect of treatment with peginterferon or interferon alfa-2b and ribavirin on steatosis in patients infected with hepatitis C. Hepatology, 38, 75-85. http://dx.doi.org/10.1053/jhep.2003.50267
- Hu, K.Q., Kyulo, N.L., Esrailian, E., Thompson, K., Chase, R., Hillebrand, D.J. and Runyon, B.A. (2004) Overweight and obesity, hepatic steatosis, and progression of chronic hepatitis C: A retrospective study on a large cohort of patients in the United States. Journal of Hepatology, 40, 147-154. http://dx.doi.org/10.1016/S0168-8278(03)00479-3
- Rubbia-Brandt, L., Fabris, P., Paganin, S., Leandro, G., Male, P.J., Giostra, E., Carlotto, A., et al. (2004) Steatosis affects chronic hepatitis C progression in a genotype specific way. Gut, 53, 406-4012. http://dx.doi.org/10.1136/gut.2003.018770
- Patton, H.M., Patel, K., Behling, C., Bylund, D., Blatt, L.M., Vallee, M., Heaton, S., et al. (2004) The impact of steatosis on disease progression and early and sustained treatment response in chronic hepatitis C patients. Journal of Hepatology, 40, 484-490. http://dx.doi.org/10.1016/j.jhep.2003.11.004
- Muzzi, A., Leandro, G., Rubbia-Brandt, L., James, R., Keiser, O., Malinverni, R., Dufour, J.F., et al. (2005) Insulin resistance is associated with liver fibrosis in non-diabetic chronic hepatitis C patients. Journal of Hepatology, 42, 41-46. http://dx.doi.org/10.1016/j.jhep.2004.09.022
- Petit, J.M., Minello, A., Jooste, V., Bour, J.B., Galland, F., Duvillard, L., Verges, B., et al. (2005) Decreased plasma adiponectin concentrations are closely related to steatosis in hepatitis C virus-infected patients. Journal of Clinical Endocrinology and Metabolism, 90, 2240-2243. http://dx.doi.org/10.1210/jc.2004-1266
- Ascencio, C., Torres, N., Isoard-Acosta, F., Gomez-Perez, F.J., Hernandez-Pando, R. and Tovar, A.R. (2004) Soy protein affects serum insulin and hepatic SREBP-1 mRNA and reduces fatty liver in rats. Journal of Nutrition, 134, 522-529.
- Song, Z., Zhou, Z., Uriarte, S., Wang, L., Kang, Y.J., Chen, T., Barve, S., et al. (2004) S-adenosylhomocysteine sensitizes to TNF-alpha hepatotoxicity in mice and liver cells: A possible etiological factor in alcoholic liver disease. Hepatology, 40, 989-997.
- Wang, G. and Siow, Y.L. (2000) Homocysteine stimulates nuclear factor kappaB activity and monocyte chemoattractant protein-1 expression in vascular smooth-muscle cells: A possible role for protein kinase C. Biochemical Journal, 352, 817-826. http://dx.doi.org/10.1042/0264-6021:3520817
- Adinolfi, L.E., Ingrosso, D., Cesaro, G., Cimmino, A., D’Anto, M., Capasso, R., Zappia, V., et al. (2005) Hyperhomocysteinemia and the MTHFR C677T polymorphism promote steatosis and fibrosis in chronic hepatitis C patients. Hepatology, 41, 995-1003. http://dx.doi.org/10.1002/hep.20664
- Finkelstein, J.D. (1990) Methionine metabolism in mammals. The Journal of Nutritional Biochemistry, 1, 228- 236. http://dx.doi.org/10.1016/0955-2863(90)90070-2
- Refsum, H., Ueland, P.M., Nygard, O. and Vollset, S.E. (1998) Homocysteine and cardiovascular disease. Annual Review of Medicine, 49, 31-62. http://dx.doi.org/10.1146/annurev.med.49.1.31
- Mato, J.M., Alvarez, L., Ortiz, P. and Pajares, M.A. (1997) S-Adenosylmethionine synthesis: Molecular mechanisms and clinical implications. Pharmacology & Therapeutics, 73, 265-280. http://dx.doi.org/10.1016/S0163-7258(96)00197-0
- Mayer, E.L., Jacobsen, D.W. and Robinson, K. (1996) Homocysteine and coronary atherosclerosis. Journal of the American College of Cardiology, 27, 517-527. http://dx.doi.org/10.1016/0735-1097(95)00508-0
- Miller, S.A., Dykes, D.D. and Polesky, H.F. (1988) A simple salting out procedure for extracing DNA from human nucleated cells. Nucleic Acids Research, 16, 1215. http://dx.doi.org/10.1093/nar/16.3.1215
- Kim, Y.L. (2000) Methylenetetrahydrofolate reductase polymorphisms, folate, and cancer risk: A paradigm of genenutrient interactions in carcinogenesis. Nutrition Reviews, 58, 205-217. http://dx.doi.org/10.1111/j.1753-4887.2000.tb01863.x
- Daly, S.F., Molloy, A.M., Mills, J.L., Less, Y.J., Conley, M., Kirke, P.N., Weir, D.G. and Scott, J.M. (1999) The influence of 5,10 methylenetetrahydrofolate reductase genotypes on enzyme activity in placental tissue. British Journal of Obstetrics & Gynaecology, 106, 1214-1218. http://dx.doi.org/10.1111/j.1471-0528.1999.tb08151.x
- Weisberg, I., Tran, P., Christensen, B., Sibani, S. and Rozen, R. (1998) A second genetic polymorphism in methylenetetrahydrofolate reductase (MTHFR) associated with decreased enzyme activity. Molecular Genetics and Metabolism, 64, 169-172. http://dx.doi.org/10.1006/mgme.1998.2714
- Knodell, R.G., Ishak, K.G., Black, W.C., Chen, T.S., Craig, R., Kaplowitz, N., Kiernan, T.W. and Wollman, J. (1981) Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology, 1, 431-435. http://dx.doi.org/10.1002/hep.1840010511
- Scheuer, P.J. (1991) Classification of chronic viral hepatitis: A need for reassessment. Journal of Hepatology, 13, 372-374. http://dx.doi.org/10.1016/0168-8278(91)90084-O
- Tevijano, E.R.G., Berasain, C., Rodríguez, J.A., Corrales, F.J., Arias, R., Martín-Duce, A., Caballería, J., Mato, J.M. Matías and Avila, A. (2001) Hyperhomocysteinemia in liver cirrhosis mechanisms and role in vascular and hepatic fibrosis. Hypertension, 38, 1217-1221. http://dx.doi.org/10.1161/hy1101.099499
- Toniutto, P., Fabris, C., Falleti, E., Cussigh, A., Fontanini, E., Bitetto, D., Fornasiere, E., Minisini, R., De Feo, T., Marangoni, F. and Pirisi, M. (2007) Methylenetetrahydrofolate reductase C677T polymorphism and liver fibrosis progression in patients with recurrent hepatitis C. Liver International, 28, 257-263. http://dx.doi.org/10.1111/j.1478-3231.2007.01591.x
- Zhou, X.M., Lin, J.S., Sun, X.M., Tang, W.X., Zhang, W.Y., Yuan, S.Y. and Ai, L. (2005) The relationship between the plasma homocysteine level and the polymorphism of MTHFR gene C667T in liver cirrhosis. Zhonghua Gan Zang Bing Za Zhi, 13, 908-910.
- Ventura, P., Rosa, M.C., Abbati, G., Marchini, S., Grandone, E., Vergura, P., Tremosini, S. and Zeneroli, M.L. (2005) Hyperhomocysteinaemia in chronic liver diseases: Role of disease stage, vitamin status and methylenetetrahydrofolate reductase genetics. Liver International, 25, 49-56. http://dx.doi.org/10.1111/j.1478-3231.2005.01042.x
- Sazci, A., Ergul, E., Kaya, G. and Kara, I. (2005) Genotype and allele frequencies of the polymorphic methylenetetrahydrofolate reductase gene in Turkey. Cell Biochemistry and Function, 23, 51-54. http://dx.doi.org/10.1002/cbf.1132
- Watanabe, M., Osada, J., Aratani, Y., Kluckman, K., Reddick, R., Malinow, M.R. and Maeda, N. (1995) Mice deficient in cystathionine beta-synthase: Animal models for mild and severe homocysteinemia. Proceedings of the National Academy of Sciences of the United States of America, 92, 1585-1589. http://dx.doi.org/10.1073/pnas.92.5.1585
- Gulsen, M., Yesilova, Z., Bagci, S., Uygun, A., Ozcan, A. and Ercin, C.N. (2005) Elevated plasma homocysteine concentrations as a predictor of steatohepatitis in patients with non-alcoholic fatty liver disease. Journal of Gastroenterology and Hepatology, 20, 1448-1455. http://dx.doi.org/10.1111/j.1440-1746.2005.03891.x
- Werstuck, G.H., Lentz, S.R., Dayal, S., Hossain, G.S., Sood, S.K. and Shi, Y.Y. (2001) Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. Journal of Clinical Investigation, 107, 1263-1273. http://dx.doi.org/10.1172/JCI11596
- Golbahar, J., Aminzadeh, M.A., Kassab, S.E. and Omrani, G.R. (2007) Hyperhomocysteinemia induces insulin resistance in male Sprague-Dawley rats. Diabetes Research and Clinical Practice, 76, 1-5. http://dx.doi.org/10.1016/j.diabres.2006.07.026
- Altinova, A.E., Toruner, F., Bukan, N., Yasar, D.G., Akturk, M., Cakir, N. and Arslan, M. (2007) Decreased plasma adiponectin is associated with insulin resistance and HDL cholesterol in overweight subjects. Endocrine Journal, 54, 221-226. http://dx.doi.org/10.1507/endocrj.K06-021
- Meigs, J.B., Jacques, P.F., Selhub, J., Singer, D.E., Nathan, D.M., Rifai, N., D’Agostino, R.B. and Wilson, P.W.F. (2001) Framingham offspring study. Fasting plasma homocysteine levels in the insulin resistance syndrome: The Framingham offspring study. Diabetes Care, 24, 1403-1410. http://dx.doi.org/10.2337/diacare.24.8.1403
- Adinolfi, L.E., Gambardella, M., Andreana, A., Tripodi, M.F., Utili, R. and Ruggiero, G. (2001) Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology, 33, 1358-1364. http://dx.doi.org/10.1053/jhep.2001.24432
- Hui, J.M., Kench, J., Farrell, G.C., Lin, R., Samarasinghe, D., Liddle, C., Byth, K. and George, J. (2002) Genotypespecific mechanisms for hepatic steatosis in chronic hepatitis C infection. Journal of Gastroenterology and Hepatology, 17, 873-881. http://dx.doi.org/10.1046/j.1440-1746.2002.02813.x
- Castera, L., Hezode, C., Roudot-Thoraval, F., Roudot-Thoraval, F., Lonjon, I., Zafrani, E.S., Pawlotsky, J.M. and Dhumeaux, D. (2004) Effect of antiviral treatment on evolution of liver steatosis in patients with chronic hepatitis C: Indirect evidence of a role of hepatitis C virus genotype 3 in steatosis. Gut, 53, 420-424. http://dx.doi.org/10.1136/gut.2002.009936
- Torres, L., García-Trevijano, E.R., Rodríguez, J.A., Bustos, M., Fernández, E., Eguinoa, E., Mato, J.M. and Avila, M.A. (1999) Induction of TIMP-1 expression in rat hepatic stellate cells and hepatocytes: A new role for homocysteine in liver fibrosis. Biochimica et Biophysica Acta, 1455, 12-22. http://dx.doi.org/10.1016/S0925-4439(99)00049-6
- Duong, F.H., Filipowicz, M., Tripodi, M., La Monica, N. and Heim, M.H. (2004) Hepatitis C virus inhibits interferon signaling through up-regulation of protein phosphatase 2A. Gastroenterology, 126, 263-277.
- Kharbanda, K.K. (2007) Role of transmethylation reactions in alcoholic liver disease. World Journal of Gastroenterology, 13, 4947-4954.
NOTES
*Corresponding author.