Open Journal of Biophysics
Vol.04 No.04(2014), Article ID:51126,6 pages
10.4236/ojbiphy.2014.44016
Human Prion Protein Conformational Changes Susceptibility: A Molecular Dynamics Simulation Study
Laura Alejandra Mandujano-Rosas1, Daniel Osorio-González1*, Pedro Guillermo Reyes-Romero2, Jorge Mulia-Rodríguez1
1Laboratory of Molecular Biophysics, Faculty of Sciences, Autonomous University of State of Mexico, Toluca, Mexico
2Laboratory of Advaced Physics, Faculty of Sciences, Autonomous University of State of Mexico, Toluca, Mexico
Email: *dog@uaemex.mx
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 29 August 2014; revised 30 September 2014; accepted 29 October 2014
ABSTRACT
Prion proteins are related to the development of incurable and invariably fatal neurodegenera- tive diseases in humans and animals. The pathogenicity involves the conversion of the host-en- coded-alpha rich isoform of prion protein, PrPC, into a misfolded beta-strand rich conformer, PrPSc. Although it has already been described that many punctual mutations alter the stability of PrPC, making it more prone to adopt an abnormal misfolded structure, the majority of cases reported among general population are sporadic in wild-type organisms. Thus, in this work we studied the dynamics and stability profiles of wild-type human prion protein by Molecular Dynamics (MD) simulation at different solvent temperatures. This analysis brought out certain residues and segments of the prion protein as critical to conformational changes; these results are consistent with experimental reports showing that protein mutants in those positions are related to the development of disease.
Keywords:
Prion Protein, Misfolding Susceptibility, Molecular Dynamics Simulation

1. Introduction
The human prion protein (hPrP) is a membrane N-linked glycosylphosphatidylinositol-anchored glycoprotein that has 253 amino acids when it is newly synthesized. It is expressed in various tissues, but is particularly present in nervous system. Its primary structure includes an N-terminal 22-residue signal peptide, which mediates translocation into the endoplasmic reticulum [1] , and is cleaved during importation. Soon after importation event, core glycans are attached to N181 and N197, and also a glycosylphosphatidylinositol anchor is trans- ferred to S230 [2] [3] . Core glycans are subjected to further processing in the Golgi compartment [4] . hPrP also has oxidation-susceptible methionines at positions 129, 134, 154, 166, 205, 206, 231 [5] . Two cysteine residues at positions 179 and 214 are linked by a single disulfide bond [6] . The last C-terminal 22 amino acid residues comprise another signal sequence, which is also absent in the 209-residue mature form of the protein [1] .
Sequence PQGGGGWGQ is among important structural regions of hPrP, beginning at residue number 51, and followed by four repetitions of the octapeptide PHGG(G/S)WGQ which encompasses residues 60 to 91; such octapeptides are cofactor binding sites. It also has a palindromic sequence AGAAAAGAA that spans residues 113 to 120, as part of a downstream tightly packed hydrophobic core. Residues at positions 127 and 129 are polymorphic, as it could be either glycine or valine in the first case, and methionine or valine in the second one [7] .
In accordance with the protein-only hypothesis, a misfolding of human prion protein is the cause of prion diseases. Experimental evidence supports that this is the sole etiologic agent of a group of neurodegenerative diseases related to the conversion of the native isoform, an alpha-helix rich cellular protein (PrPC), into a beta- strand rich misfolded conformer, named “Scrapie” prion protein (PrPSc) [8] . The PrPSc is thought to be infectious through nucleation of complete or partial PrPSc forming rod-like aggregates. Template assistance and nucleated polymerization models have been proposed to explain the mechanisms of conformational conversion and aggregation, but they are still poorly understood [5] . Thus, this work is aimed to provide insight into them by using a molecular dynamics approximation to the problems.
PrPC has a flexibly disordered N-terminal tail and a structured C-terminal globular domain from amino acid 125 to 231. The C-terminal globular domain includes three alpha-helixes known as H1, H2 and H3 at 144 - 154, 173 - 194 and 200 - 228 positions, and a short antiparallel beta-strand region at 127 - 130 and 161 - 164 residues (S1 and S2). It also has three zones of structural irregularity at 167 - 171, 187 - 194 and 219 - 228 [9] . H1 and H2 are linked by a loop formed by 165 - 171 residues, better known as X-loop. The protein hydrophobic core comprises interactions between H2 and H3 and between H3 and the loop preceding H1 (S1-H1 loop), as well as contacts between the beta-strand with the rest of the globular domain [5] .
More than 30 different punctual mutations in the open reading frame of the human prion gene (PRNP) have been studied as they are linked to human prion inherited diseases, such as Gerstmann-Sträussler-Scheinker syndrome, fatal familial insomnia and familial Creutzfeldt-Jakob disease. All these diseases have variations in clinical, physiological and pathological characteristics, which are probably explained by the differences in the mechanism of destabilization of PrPC, conversion into PrPSc, aggregation, and by the specific brain regions where all these processes occur. It is particularly remarkable that many missense point mutations are clustered within H2 and H3, and the loop between them. Mutations leading to disease have also been reported, although much less represented, at N-terminal domain, particularly at the octapeptide region and the disordered zone before S1 [10] - [14] .
Mutations may alter either the thermodynamic stability of PrPC, change surface properties that in turn modify interactions, and vary the response to cellular pathways control processes, or a combination of these mechanisms. However, solution structures of hPrP mutants are conformationally similar to the wild-type protein, and only molecular simulation techniques have been capable of elucidate differences between them [15] - [18] .
Table 1 summarizes different mutations associated with the development of human prion inherited diseases.
2. Material and Methods
Extensive all-atom molecular dynamics simulations at 37˚C and different pH values were made to study the structure of wild-type human prion protein, first in the vacuum, and then using water as solvent. We used spatial coordinates of human prion protein structured region (amino acid residues 125 - 228) as in 1QLZ code of Protein Data Bank.
In order to relax the protein in vacuum, MD was applied at minimum energy conditions and under the influence of GROMOS96 force field at GROMACS 4.0.7 suite of programs [19] . Consecutively, periodic frontier conditions were incorporated by using a rhombic dodecahedral water box which volume was 283 nm3. This process was repeated using 7778 water molecules as solvent with Simple Point Charge (SPC) model.

Table 1. This table shows the most important structural and punctual mutations reported to have influence over the con- formational stability/pathological capacity of human prion protein.
The system protein-water was neutralized by addition of sodium and chloride ions, and the potential energy was defined as the sum of two types of non-bonded interactions (electrostatic and van der Waals) and three types of bonded interactions (chemical, angles and twisting).
The equilibration process was started with a set of velocities taken from a Maxwell-Boltzmann distribution until it reached 305 K in about 350 ps using the Berendsen thermostat for both systems (protein and solvent). The production process was implemented with the same thermostat and the Parrinello-Rahman barostat; the total simulation length was 80 ns. The performance of computer system was 5.0 ns per day.
The measure of the deviation between the position of particle 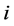 and some reference position was calculated with the Root Mean Square Fluctuation (RMSF).
and some reference position was calculated with the Root Mean Square Fluctuation (RMSF).
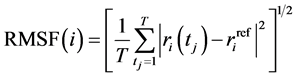 (1)
(1)
where  is the time over one wants to calculate the average,
is the time over one wants to calculate the average, 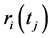 is the position of the particle
is the position of the particle 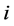 on the time
on the time 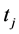 and
and 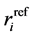 is the reference position of the particle
is the reference position of the particle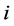 . In order to obtain the relative vibrational motion of different regions of the protein we use the B-factor
. In order to obtain the relative vibrational motion of different regions of the protein we use the B-factor
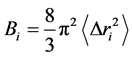 (2)
(2)
where  is the mean square displacement and is related with the self-diffusion coefficient
is the mean square displacement and is related with the self-diffusion coefficient 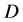 via the Einstein relation
via the Einstein relation
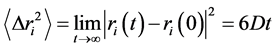 (3)
(3)
where  is the initial position of the particle
is the initial position of the particle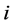 .
.
Also we used the  -component pressure tensor
-component pressure tensor 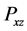 to estimate the shear viscosity
to estimate the shear viscosity  which evaluates the rates of possible changes in the protein conformation, both variables are related by
which evaluates the rates of possible changes in the protein conformation, both variables are related by
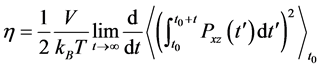 (4)
(4)
Here, 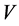 is the volume,
is the volume, 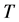 the temperature,
the temperature, 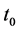 the initial time and
the initial time and 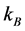 the Boltzmann constant [21] [22] .
the Boltzmann constant [21] [22] .
3. Results and Discussion
The solvated protein was stabilized with an initial temperature of 310 K, and then the temperature of the solvent was modified in order to stimulate the protein and identify the regions with higher vibrational states. Figure 1 shows that such regions correspond to residues 144 - 149, 178 - 181, 193 - 197 and 221 - 223. The RMSF was calculated with (1).

Figure 1. Root mean square fluctuations per residue show the most vulnerable regions to vibrations. Labeled temperatures correspond to solvent. The residues 150, 184 and 210 do not show substantial changes with respect to the solvent temperature.
In order to identify the relative vibrational motion of different parts of the protein, we calculated de B-factor for the same solvent temperatures, as can be observed in Figure 2.
It is well known that calculation of viscosity with (4) converges very slowly [21] ; however we were interested in analyzing its evolution in early times because its variations stimulate conformational changes on the protein. Figure 3 shows that viscosity has partially uniform oscillations around 1.5 cP when the solvent has the lowest temperature, in other words, viscosity varies more uniformly at the lowest temperature than at other temperatures, maintaining values predominantly below 2 cP.

Figure 2. The B-factor reveals the regions with more thermal effects on the protein. These regions are the same that can be seen in Figure 1. Labeled temperatures correspond to solvent.

Figure 3. It shows the shear viscosity evolution in early times of production process simulation at three different solvent temperatures; it follows that the increasing-decreasing monotonic behavior when the solvent has a temperature lower than 310 K, is not a factor that interfere with the regions of increased flexibility in the protein.
4. Conclusion
The regions of high susceptibility to conformational changes are close to mutation sites experimentally tested and reported. According to Figure 1, the regions with higher vibrational states correspond to residues 144 - 149, 178 - 181, 193 - 197 and 221 - 223. The simulation showed important oscillations in nanosecond time scale order. These oscillations are not due to viscosity fluctuations, but due to temperature changes in solvent. The oscillations observed in three different properties indicate that the van der Waals term of the force field employed is very rigid, so we need to prove a more flexible potential with more realistic repulsion and attraction contributions, although it increases the computational time required for simulations.
Acknowledgements
The authors wish to thank Universidad Autónoma del Estado de México for financial support through project ID 3697/2014/CID.
References
- Rane, N.S., Chakrabarti, O., Feigenbaum, L. and Hegde, R. (2010) Signal Sequence Insufficiency Contributes to Neurodegeneration Caused by Transmembrane Prion Protein. The Journal of Cell Biology, 188, 515-526. http://dx.doi.org/10.1083/jcb.200911115
- Haraguchi, T., Fisher, S., Olofsson, S., Endo, T., Groth, D., Tarentino, A., Borchelt, D.R., Teplow, D., Hood, L., Burlingame, A., Lycke, E., Kobata, A. and Prusiner, S.B. (1989) Asparagine-Linked Glycosylation of the Scrapie and Cellular Prion Proteins. Archives of Biochemistry and Biophysics, 274, 1-13. http://dx.doi.org/10.1016/0003-9861(89)90409-8
- Stahl, N., Borchelt, D.R., Hsiao, K. and Prusiner, S.B. (1987) Scrapie Prion Protein Contains a Phosphatidylinositol Glycolipid. Cell, 51, 229-240. http://dx.doi.org/10.1016/0092-8674(87)90150-4
- Stimson, E., Hope, J., Chong, A. and Burlingame, A.L. (1999) Site-Specific Characterization of the N-Linked Glycans of Murine Prion Protein by High-Performance Liquid Chromatography/Electrospray Mass Spectrometry and Exoglycosidase Digestions. Biochemistry, 38, 4885-4895. http://dx.doi.org/10.1021/bi982330q
- Zhou, Z. and Xiao, G. (2013) Conformational Conversion of Prion Protein in Prion Diseases. Acta Biochimica et BiophysicaSinica, 45, 465-476. http://dx.doi.org/10.1093/abbs/gmt027
- Ranjan-Maiti, N. and Surewicz, W.K. (2001) The Role of Disulfide Bridge in the Folding and Stability of the Recombinant Human Prion Protein. The Journal of Biological Chemistry, 276, 2427-2431. http://dx.doi.org/10.1074/jbc.M007862200
- Coleman, B.M., Harrison, C.F., Guo, B., Masters, C.L., Barnham, K.J., Lawson, V.A. and Hill, H.F. (2014) Pathogenic Mutations within the Hydrophobic Domain of the Prion Protein Lead to the Formation of the Protease-Sensitive Prion Species with Increased Lethality. Journal of Virology, 88, 2690-2703. http://dx.doi.org/10.1128/JVI.02720-13
- Prusiner, S.B. (1982) Novel Proteinaceous Infectious Particles Cause Scrapie. Science, 216, 136-144. http://dx.doi.org/10.1126/science.6801762
- Zahn, R., Liu, A., Luhrs, T., Riek, R., von Schroetter, C., Lopez Garcia, F., Billeter, M., Calzolai, L., Wider, G. and Wuthrich, K. (2000) NMR Solution Structure of the Human Prion Protein. Proceedings of the National Academy of Sciences U.S.A., 97, 145-150. http://dx.doi.org/10.1073/pnas.97.1.145
- Rossetti, G., Cong, X., Caliandro, R., Legname, G. and Carloni, P. (2011) Common Structural Traits Across Pathogenic Mutants of the Human Prion Protein and Their Implications for Familial Prion Disorders. Journal of Molecular Biology, 411, 700-712. http://dx.doi.org/10.1016/j.jmb.2011.06.008
- Kiachopoulos, S., Bracher, A., Winklhofer, K.F. and Talzet, J. (2005) Pathogenic Mutations Located at the Hydrophobic Core of the Prion Protein Interfere with Folding and Attachment of the Glycosylphosphatidylinositol Anchor. The Journal of Biological Chemistry, 280, 9320-9329. http://dx.doi.org/10.1074/jbc.M412525200
- Cheng, W., van der Kamp, M.W. and Daggett, V. (2014) Structural and Dynamic Properties of the Human Prion Protein. Biophysical Journal, 106, 1152-1163. http://search.proquest.com/docview/1508983494?accountid=37347 http://dx.doi.org/10.1016/j.bpj.2013.12.053
- Ilc, G., Giachin, G., Jaremko, M., Jaremko, Ł., Benetti, F., Plavec, J., Zhukov, I. and Legname, G. (2010) NMR Structure of the Human Prion Protein with the Pathological Q212P Mutation Reveals Unique Structural Features. PLoS ONE, 5, e11715. http://dx.doi.org/10.1371/journal.pone.0011715
- Tatzelt, J., Ed. (2011) Topics in Current Chemistry 305: Prion Proteins. Springer Berlin-Heidelberg, Berlin.
- Taylor, D.R., Whitehouse, I.J. and Hooper, N.M. (2009) Glypican-1 Mediates Both Prion Protein Lipid Raft Association and Disease Isoform Formation. PLoS Pathogens, 5, e1000666. http://dx.doi.org/10.1371/journal.ppat.1000666
- Elmallah, M.I.Y., Borgmeyer, U., Betzel, C. and Redecke, L. (2013) Impact of Methionine Oxidation as an Initial Event on the Pathway of Human Prion Protein Conversion. Prion, 7, 404-411. http://dx.doi.org/10.4161/pri.26745
- Mani, K., Cheng, F., Havsmark, B., Jonsson, M., Belting, M. and Fransson, L.A. (2003) Prion, Amyloid Beta-Derived Cu(II) Ions, or Free Zn(II) Ions Support S-Nitroso-Dependent Autocleavage of Glypican-1 Heparan Sulfate. The Journal of Biological Chemistry, 278, 38956-38965. http://dx.doi.org/10.1074/jbc.M300394200
- Wu, D., Zhang, W., Luo, Q., Luo, K., Huang, L., Wang, W., Huang, T., Chen, R., Lin, Y., Pang, D. and Xiao, G. (2010) Copper (II) Promotes the Formation of Soluble Neurotoxic PrP Oligomers in Acidic Environment. The Journal of Cell Biochemistry, 111, 627-633. http://dx.doi.org/10.1002/jcb.22743
- Zhong, L. (2010) Exposure of Hydrophobic Core in Human Prion Protein Pathogenic Mutant H187R. Journal of Biomolecular Structure Dynamics, 28, 355-361. http://dx.doi.org/10.1080/07391102.2010.10507365
- Cheng, C.J. and Dagget, V. (2014) Different Misfolding Mechanisms Converge on Common Conformational Changes: Human Prion Protein Pathogenic Mutants Y218N and E196K. Prion, 8, 125-135. http://dx.doi.org/10.4161/pri.27807
- Hess, B., Kutzner, C., van der Spoel, D. and Lindahl, E. (2008) GROMACS 4: Algorithms for Highly Efficient, Load Balanced and Scalable Molecular Simulation. Journal of Chemical Theory and Computation, 4, 435-447. http://dx.doi.org/10.1021/ct700301q
- Hess, B. (2002) Determining the Shear Viscosity of Model Liquids from Molecular Dynamics. The Journal of Chemical Physics, 116, 209-217. http://dx.doi.org/10.1063/1.1421362
NOTES

*Corresponding author.