Journal of Materials Science and Chemical Engineering
Vol.05 No.04(2017), Article ID:75927,11 pages
10.4236/msce.2017.54005
Enhancement of Oxygen Evolution Activity of Ruddlesden-Popper-Type Strontium Ferrite by Stabilizing Fe4+
Toshihiro Takashima1*, Koki Ishikawa2, Hiroshi Irie1
1Clean Energy Research Center, University of Yamanashi, Yamanashi, Japan
2Special Doctoral Program for Green Energy Conversion Science and Technology, Interdisciplinary Graduate School of Medicine and Engineering, University of Yamanashi, Yamanashi, Japan

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: March 16, 2017; Accepted: April 27, 2017; Published: April 30, 2017
ABSTRACT
Development of active iron based water oxidation for designing an ideal artificial photosynthesis devices operating under benign neutral pH is highly demanded. We investigated the electrocatalytic activity of Ruddlesden-Pop- per-type strontium ferrite (Sr3Fe2O7) toward the oxygen evolution reaction (OER). Owing to the temperature-dependent efficiency of the charge disproportionation of Fe4+, the OER activity of Sr3Fe2O7 varied with the temperature, and the onset potential for the OER at a neutral pH underwent a negative shift of approximately 200 mV by increasing the temperature for the stabilization of Fe4+. When metal substitution was made to Sr3Fe2O7 for stabilizing Fe4+ at room temperature, the temperature dependence of the OER activity disappeared and the OER was driven at a small overpotential without increasing the temperature, indicating that the stabilization of Fe4+ is substantially important for achieving high OER activity.
Keywords:
Oxygen Evolution, Charge Disproportionation, Water-Splitting, Sr3Fe2O7

1. Introduction
The ever-growing global energy consumption has triggered considerable interest in addressing the challenge of storing renewable energy in a chemical form [1] [2] . One promising solution to this issue is to produce hydrogen (H2) by using solar energy to split water into H2 and oxygen (O2) [3] [4] [5] . As a four- electron transfer reaction, the O2 evolution reaction (OER, 2H2O → O2 + 4H+ + 4e−), which is a half-reaction of water splitting, suffers from sluggish kinetics owing to a large overpotential and has been considered to be the efficiency- limiting step of water splitting. Therefore, extensive research has been devoted to developing O2 evolution catalysts [6] - [15] . Iridium oxide (IrO2) and ruthenium oxide (RuO2) are effective OER catalysts for solar water splitting as they exhibit high turnover frequencies under neutral pH conditions [6] [7] [8] ; however, their high cost and scarcity render their use impractical for large-scale applica- tions. Thus, it is important to develop active OER catalysts using earth-abundant elements.
In the past few decades, many earth-abundant metal oxides have been investigated [9] [10] [11] as potential OER catalysts to replace IrO2 and RuO2. Among them, iron (Fe) oxide is attractive because Fe is the most abundant first- row transition metal on Earth and nontoxic. Several Fe-based OER catalysts have been reported to show excellent activity [12] [13] [14] [15] . For example, Ba0.5Sr0.5Co0.8Fe0.2O3δ and CaCu3Fe4O12 have high activity comparable to that of IrO2 and RuO2 in an alkaline solution [12] [13] . Despite these achievements; however, substantial improvements in the design and preparation of catalysts are still needed because few Fe-based OER catalysts function effectively under neutral pH conditions [16] [17] [18] . Therefore, for the successful application of Fe- based catalysts as components for solar fuel production systems, improvement of their OER activity under neutral pH conditions is essential.
Recently, numerous studies have been conducted to investigate the mechanism of the OER on Fe oxides by using various spectroelectrochemical techniques [18] [19] [20] [21] [22] . On the basis of the results of in situ UV-vis measurement, we have reported that Fe4+ is the intermediate species of the OER on a hematite (α-Fe2O3) electrode [18] [19] . Hamann et al. observed a potential- dependent infrared absorption peak, which they attributed to a high-valent Fe4+-oxo species, and proposed that the rate-determining step of the OER is Fe3+-OH → Fe4+ = O + e− + H+ [20] . Chen et al. conducted in situ Mössbauer measurements and observed signals indicating that the formation of Fe4+ proceeds on NiFe hydroxide during the electrocatalysis of the OER [21] . Concerning a descriptor for OER activity, Suntivich et al. reported that near-unity occupancy of the eg orbitals of transition-metal ions at the B-site of perovskites (formula ABO3) is essential to obtain high OER activity [12] . Notably, the Fe4+ ion that is formed on metal oxides has the high-spin d4 configuration of t2g3eg1 [23] [24] , and its formation satisfies the conditions required for high OER activity. On the basis of these reports, we hypothesize that the accessibility to Fe4+ is a possible descriptor for the OER activity of Fe-based catalysts [25] . Ruddlesden-Popper-type stron- tium ferrite (Sr3Fe2O7) contains Fe4+ which is unstable and consumed by charge disproportionation (CD) (2Fe4+→ Fe3+ + Fe5+) [26] . Notably, the CD of Fe4+ in Sr3Fe2O7 can be suppressed by regulating the temperature and its chemical composition [26] [27] [28] .
Thus, to examine the validity of the hypothesis that the accessibility to Fe4+ is a descriptor for the OER activity of Fe-based catalysts, we have investigated the OER activities of Sr3Fe2O7 and its La- or Ti-substituted compounds at different temperatures using electrochemical measurements. By increasing the tem- perature or substituting the foreign elements to suppress the CD of Fe4+, the enhancement of the OER activity was observed.
2. Experimental Section
2.1. Preparation of Electrodes
Sr3Fe2O7 powder was synthesized by a solid-state reaction [26] . Stoichiometric amounts of α-Fe2O3 (Kojundo Chemical Lab., 99.9%) and strontium carbonate (SrCO3, Kojundo Chemical Lab., 99.9%) were ground in a ball mill and calcinated in air at 900˚C for 9 h. The resulting powder was pressed into pellets and sintered in air at 1300˚C for 24 h. When Sr2.6La0.4Fe2O7 and Sr3FeTiO7 were prepared, stoichiometric amounts of lanthanum oxide (La2O3, Kanto Chemical, 98.0%) and titanium dioxide (TiO2, Kanto Chemical, 98.0%) were added to the starting materials as reported in the literature [27] [28] . All chemical reagents were used without further purification.
Electrodes were prepared using a spray deposition method as reported previously [25] . Briefly, 300 mg of the synthesized powder sample was suspend- ed in 200 mL of ethanol. The suspension was sprayed onto a clean conducting glass substrate (FTO-coated glass, resistance: 20 Ω/square; SPD Laboratory Inc.) at 170˚C using an automatic spray gun (Lumina, ST-6; Fuso Seiki Co., Ltd.). After coating, the resultant transparent black film was calcinated at 500 oC in air for 2 h.
2.2. Characterization
The crystal structures of the electrocatalysts were analyzed by X-ray diffraction (XRD) using a PW-1700 X-ray diffractometer (PANalytical) with monochromatic Cu Kα radiation. XRD patterns were recorded from 15˚ to 80˚ in 2θ with a step size of 0.02˚ and a scan rate of 0.25˚/min. Scanning electron microscopy (SEM) inspection was performed using a scanning electron microscope (JSM- 6500F, JEOL).
2.3. Electrochemical Measurements
Polarization curves were obtained with a commercial potentiost at and potential programmer (HZ-5000, Hokuto Denko). A platinum wire was used as a counter electrode. All potentials were measured against a silver/silver chloride reference electrode (Ag/AgCl/KCl(sat.)) and converted to the standard hydrogen electrode (SHE) reference scale using the equation U(versus SHE) = U(versus Ag/AgCl/ KCl (sat.)) + 0.197. The electrolyte solution used for all experiments was 0.1 M sodium sulfate (Na2SO4) aqueous solution, which was prepared from highly pure Milli-Q water (18 MΩ∙cm) and Na2SO4 (Kanto Kagaku, 99.0%). The pH was adjusted to 7 using 0.1 M sulfuric acid (H2SO4, Kanto Kagaku, 96.0%) and 0.1 M sodium hydroxide (NaOH, Kanto Kagaku, 97.0%). No agent for pH buffering was added to the electrolyte solution to avoid effects from the adsorption of multivalent anions. Prior to the measurement, the electrolyte was maintained at a certain temperature and bubbled with argon gas for at least 15 min. Polarization curves were measured by sweeping the electrode potential from the rest potential to 1.5 V at a scan rate of 10 mV/s and the concentration of O2 dissolved in the electrolyte was monitored during the potential sweep using a needle-type O2 microsensor (Microx TX3-trace, PreSens). The current density was normalized to the geometric surface area of the electrode.
3. Results and Discussion
3.1. Characterization of the Prepared Electrodes
Figure 1 shows the XRD patterns of Sr3Fe2O7 and its substituted materials Sr2.6La0.4Fe2O7 and Sr3FeTiO7. All materials exhibited XRD patterns indexed to the tetragonal space group I4/mmm as reported in the literature [26] [27] [28] , and no peaks assignable to other crystal phases were detected. The peak position of the prepared Sr3Fe2O7 (Figure 1(a)) matched with a reference data (Figure 1(b), ICSD no. 163173). In contrast, the intensity of the diffraction peaks corresponding to (00h) planes was particularly intense for our prepared Sr3Fe2O7. This can be understood by the preferred orientation of the Sr3Fe2O7 particles which have plate-like shapes (Figure 2(a)) owing to its two-dimensional (2-D)

Figure 1. XRD patterns of synthesized crystalline powder ((a) Sr3Fe2O7; (b) Sr2.6La0.4Fe2O7 and (c) Sr3FeTiO7) and (d) reference data (Sr3Fe2O7, ICSD no. 163173). Peaks marked with (▲) in trace (a) correspond to (00h) diffraction peaks. (e) Shift of diffraction peaks to a lower angle upon substituting metal ions.

Figure 2. SEM images of the prepared electrodes ((a) Sr3Fe2O7; (b) Sr2.6La0.4Fe2O7 and (c) Sr3FeTiO7); (d) Cross-section image of a Sr3Fe2O7 electrode. The inset of (d) shows an optical image of the Sr3Fe2O7 electrode.
layered crystal structure composed of stacked rock salt and perovskite layers with the sequence of SrO-(SrFeO3)2. For Sr2.6La0.4Fe2O7 and Sr3FeTiO7, shifts of the diffraction peaks to lower angles were observed (Figure 1(e)), confirming that cationic substitution had taken place. The peak shift was larger for Sr3FeTiO7 as the expansion of the crystal lattice has been reported to be more prominent for Sr3FeTiO7 than Sr2.6La0.4Fe2O7 [27] [28] .
Figure 2 shows SEM images of the prepared film electrodes. The Sr3Fe2O7 particles had a diameter ranging from 1 μm to 8 μm (Figure 2(a)). In contrast, the particle sizes of Sr2.6La0.4Fe2O7 and Sr3FeTiO7 were approximately from 0.3 μm to 1.2 μm (Figure 2(b) and Figure 2(c)). All the samples were uniformly deposited on the electrodes. From the cross-section image in Figure 2(d), the thickness of the deposited film was found to be approximately 500 nm.
3.2. Electrocatalytic Activity of Sr3Fe2O7 Electrocatalysts
Figure 3 shows polarization curves of a Sr3Fe2O7 film electrode measured at pH 7. Irrespective of the temperature, we observed simultaneous increases in the anodic current and O2 concentration while neither of them was observed at this potential by using a bare FTO electrode (data not shown). In contrast to the typical polarization curves for OER, Sr3Fe2O7 showed a slight decrease in the anodic current upon sweeping the electrode potential. This decrease is because a part of Sr3Fe2O7 transformed to Sr3Fe2(OH)12 during the measurements by intercalation of water molecules between its two rock-salt-type SrO layers [29] . However, because this transformation causes no anodic current and O2 formation, we can consider that the observed results indicate that the OER was electrocatalyzed on Sr3Fe2O7.
Notably, when the temperature of the electrolyte was increased from 30˚C to 70˚C, the anodic current showed a negative shift of the onset potential of approximately 200 mV. Since the onset potential for O2 formation was similarly
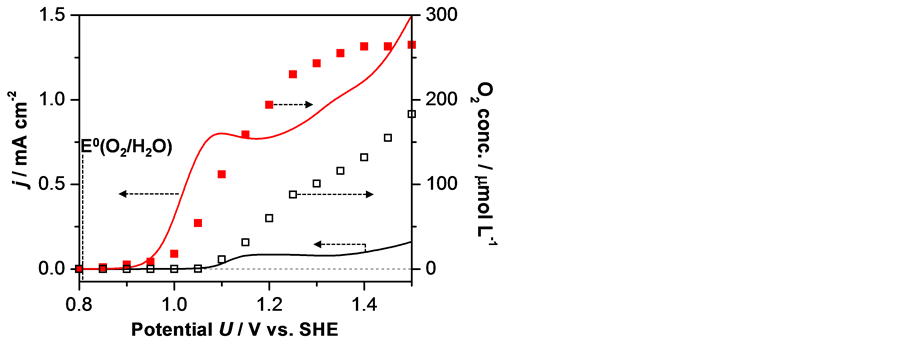
Figure 3. Potential dependences of current density (line) and dissolved O2 concentration (squares) for Sr3Fe2O7 electrodes in 0.1 M Na2SO4 aqueous solution (pH 7) during a potential sweep at 10 mV/s at 70˚C (red line and closed squares) and 30˚C (black line and open squares). E˚ represents the standard potential of the OER at pH 7.
shifted, these results indicate that the OER activity of Sr3Fe2O7 is improved by increasing the temperature. As demonstrated in a solid oxide electrolysis cell (SOEC), the OER is thermodynamically more favorable at a high temperature and the standard potential of the OER becomes more negative with increasing temperature owing to a decrease in the Gibbs free energy required for the OER [30] . However, the potential shift due to the change in the Gibbs free energy is estimated to be at most only 40 mV at 70˚C because of the small temperature difference between 30˚C and 70˚C. Thus, the observed improvement of the OER activity should originate from a temperature-dependent property of Sr3Fe2O7.
As described in the introduction, Fe4+ is considered to play an important role in the OER on Fe-based catalysts; however, Fe4+ is unstable against CD and rapidly disappears in usual [26] [28] [29] [31] [32] [33] According to the literature, the efficiency of CD is closely related to the electronic bandwidth of σ* bonding composed of Fe-3d and O-2pσ* orbitals, and CD occurs when the bandwidth of σ* bonding is narrow [31] [32] . For Fe-based perovskite com- pounds, the bandwidth broadens at high temperatures because with increasing temperature, the Fe-O-Fe bond angle increases and the electronic interaction between Fe and O strengthens [31] [32] . Kuzushita et al. investigated the temperature dependence of the Fe4+ stability by performing Mössbauer measure- ments and found that there is a critical temperature for the CD of Sr3Fe2O7 at 70˚C ± 10˚C, indicating that CD is suppressed above 70˚C [26] . Therefore, the observed enhancement of OER activity at 70˚C is considered to be due to the suppression of the CD of Fe4+.
To examine the validity of the interpretation that the stabilization of Fe4+ leads to the enhancement of the OER activity of Sr3Fe2O7, we also investigated the effect of Fe4+ stability on the OER activity using the metal-substituted materials. Figure 4(a) shows the polarization curves of Sr2.6La0.4Fe2O7 measured at 30˚C and 70˚C. Unlike Sr3Fe2O7, the anodic current initiated to increase from essentially the same potential for both temperatures, which is consistent with the
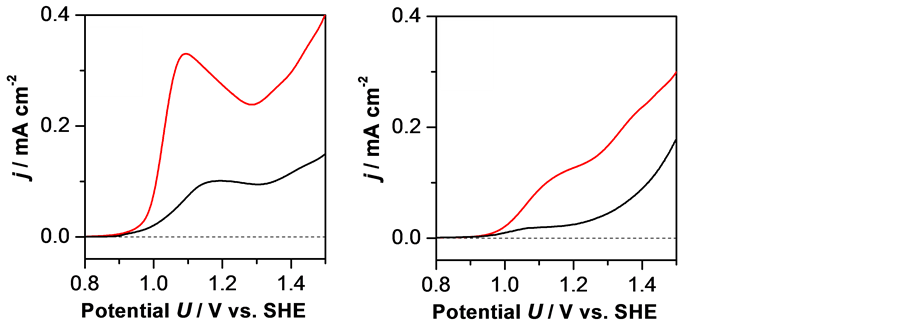 (a) (b)
(a) (b)
Figure 4. Polarization curves of (a) Sr2.6La0.4Fe2O7 and (b) Sr3FeTiO7 electrodes measured at 70˚C (red line) and 30˚C (black line).
fact that the substitution of Sr with La suppresses CD at room temperature and that Fe4+ in Sr2.6La0.4Fe2O7 is stable at both 30˚C and 70˚C [27] . When the OER was conducted with Sr3FeTiO7, in which Fe4+ is stably contained at room temperature [28] , the onset potential was independent of temperature. Thus, the stabilization of Fe4+ is an effective means of enhancing the OER activity of Fe oxide. As shown in Figure 4(a) and Figure 4(b), a higher current density was observed at 70˚C.
Although the reason for this is unclear, it is assumed to be due to the greater convection of the electrolyte. Notably, the onset potentials observed with these substituted materials were almost the same as that observed for Sr3Fe2O7 at 70˚C, indicating that the efficient formation of Fe4+ on Sr3Fe2O7 derivatives enables the initiation of the OER around this potential. From a comparison of the polariza- tion curves (Figure 3, Figure 4(a) and Figure 4(b)), the current density of Sr3Fe2O7 at 70˚C was larger than those of metal substituted derivatives at the same temperature. This is likely to be due to higher concentration of Fe4+ in Sr3Fe2O7.
From the above results, the OER activity of Sr3Fe2O7 derivatives is considered to be determined by the efficiency of Fe4+ formation, and the suppression of CD was found to be effective for designing active OER catalysts. CD is known to take place not only with Fe4+ but also with other first-row transition-metal ions [31] [32] . Previously, one of the authors (T. T.) showed that the CD of Mn3+ is the primary origin of the pH-dependent OER activity of MnO2 and succeeded in enhancing OER activity under a neutral pH by suppressing CD [34] [35] [36] . Thus, by analogy with Fe4+ and Mn3+, the stabilization of other first-row tran- sition-metal ions by suppressing CD is likely to be a promising approach for the development of active OER catalysts made from abundant elements. Fe ions introduced in layer-structured metal (hydr) oxides have been reported to form Fe4+ without causing CD [37] [38] . The application of Fe-doped layered materials to the OER is currently underway in our laboratory.
4. Conclusion
In this study, the OER activity of Sr3Fe2O7 was investigated at 30˚C and 70˚C under neutral pH conditions. The onset potential for the OER of a Sr3Fe2O7 electrode was found to be dependent on the temperature and shifted by approximately 200 mV in the negative direction with increasing temperature. This enhancement of the OER activity is considered to be due to the fact that Fe4+ is stably formed by suppressing CD at 70˚C, and the stabilization of Fe4+ by metal substitution enabled efficient OER catalysis at room temperature. Unfortunately, Sr3Fe2O7 derivatives underwent the transformation in aqueous solution; however, these findings will provide insights for designing Fe oxide OER catalysts that can evolve O2 efficiently under neutral pH conditions.
Acknowledgements
This work was financially supported by the Program to Disseminate Tenure Tracking System by MEXT and by JKA with promotion funds from KEIRIN RACE (28-146).
Cite this paper
Takashima, T., Ishikawa, K. and Irie, H. (2017) Enhancement of Oxygen Evolution Activity of Ruddlesden-Popper-Type Strontium Ferrite by Stabilizing Fe4+. Journal of Materials Science and Chemical Engineering, 5, 45- 55. https://doi.org/10.4236/msce.2017.54005
References
- 1. Gray, H.B. (2009) Powering the Planet with Solar Fuel. Nature Chemistry, 1, 7.
https://doi.org/10.1038/nchem.141 - 2. Lewis, N.S. and Nocera, D.G. (2006) Powering the Planet: Chemical Challenges in Solar Energy Utilization. Proceedings of the National Academy of Sciences, 103, 15729-15735.
https://doi.org/10.1073/pnas.0603395103 - 3. Fujishima, A. and Honda, K. (1972) Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature, 238, 37-38.
https://doi.org/10.1038/238037a0 - 4. Meyer, T. (2008) Catalysis: The Art of Splitting Water. Nature, 451, 778.
https://doi.org/10.1038/451778a - 5. Walter, M.G., Warren, E.L., McKone, J.R., Boettcher, S.W., Mi, Q., Santori, E.A. and Lewis, N.S. (2010) Solar Water Splitting Cells. Chemical Reviews, 110, 6446-6473.
https://doi.org/10.1038/451778a - 6. Lee, Y., Suntivich, J., May, K.J., Perry, E.E. and Shao-Horn, Y. (2012) Synthesis and Activities of Rutile IrO2 and RuO2 Nanoparticles for Oxygen Evolution in Acid and Alkaline Solutions. The Journal of Physical Chemistry Letters, 3, 399-404.
https://doi.org/10.1021/jz2016507 - 7. Chao, Y., Hernandez-Pagan, E.A., Vargas-Barbosa, N.M., Dysart, J.L. and Mallouk, T.E. (2011) A High Yield Synthesis of Ligand-Free Iridium Oxide Nanoparticles with High Electrocatalytic Activity. The Journal of Physical Chemistry Letters, 2, 402-406.
https://doi.org/10.1021/jz200051c - 8. Chandra, D., Takama, D., Masaki, T., Sato, T., Abe, T., Togashi, T., Kurihara, M., Saito, K., Yui, T. and Yagi, M. (2016) Highly Efficient Electrocatalysis and Mechanistic Investigation of Intermediate IrOx(OH)y Nanoparticle Films for Water Oxidation. ACS Catalysis, 6, 3946-3954.
https://doi.org/10.1021/acscatal.6b00621 - 9. Jin, K., Chu, A., Park, J., Jeong, D., Jerng, S.E., Sim, U., Jeong, H.-Y., Lee, C.W., Park, Y.-S., Yang, K.D., Pradhan, G.K., Kim, D., Sung, N.-E., Kim, S.H. and Nam, K.T. (2015) Partially Oxidized Sub-10 nm MnO Nanocrystals with High Activity for Water Oxidation Catalysis. Scientific Reports, 5, 10279.
https://doi.org/10.1038/srep10279 - 10. McCrory, C.C.L., Jung, S., Peters, J.C. and Jaramillo, T.F. (2013) Benchmarking Heterogeneous Electrocatalysts for the Oxygen Evolution Reaction. Journal of the American Chemical Society, 135, 16977-16987.
https://doi.org/10.1021/ja407115p - 11. Hunter, B.M., Gray, H.B. and Müller, A.M. (2016) Earth-Abundant Heterogeneous Water Oxidation Catalysts. Chemical Reviews, 116, 14120-14136.
https://doi.org/10.1021/acs.chemrev.6b00398 - 12. Suntivich, J., May, K.J., Gasteiger, H.A., Goodenough, J.B. and Shao-Horn, Y. (2011) A Perovskite Oxide Optimized for Oxygen Evolution Catalysis from Molecular Orbital Principles. Science, 334, 1383-1385.
https://doi.org/10.1126/science.1212858 - 13. Yagi, S., Yamada, I., Tsukasaki, H., Seno, A., Murakami, M., Fujii, H., Chen, H., Umezawa, N., Abe, H., Nishiyama, N. and Mori, S. (2015) Covalency-Reinforced Oxygen Evolution Reaction Catalyst. Nature Communications, 6, 8249.
https://doi.org/10.1038/ncomms9249 - 14. Smith, R.D.L., Prévot, M.S., Fagan, R.D., Zhang, Z., Sedach, P.A., Siu, M.K.J., Trudel, S. and Berlinguette, C.P. (2013) Photochemical Route for Accessing Amorphous Metal Oxide Materials for Water Oxidation Catalysis. Science, 340, 60-63.
https://doi.org/10.1126/science.1233638 - 15. Townsend, T.K., Sabio, E.M., Browning, N.D. and Osterloh, F.E. (2011) Photocatalytic Water Oxidation with Suspended Alpha-Fe2O3 Particles-Effects of Nanoscaling. Energy & Environmental Science, 4, 4270-4275.
https://doi.org/10.1039/c1ee02110a - 16. Gorlin, M., Gliech, M., De Araújo, J.F., Dresp, S., Bergmann, A. and Strasser, P. (2016) Dynamical Changes of a Ni-Fe Oxide Water Splitting Catalyst Investigated at Different pH. Catalysis Today, 262, 65-73.
- 17. Trzesniewski, B.J., Diaz-Morales, O., Vermass, D.A., Longo, A., Bras, W., Koper, M.T.M. and Smith, W.A. (2015) In Situ Observation of Active Oxygen Species in Fe-Containing Ni-Based Oxygen Evolution Catalysts: The Effect of pH on Electrochemical Activity. Journal of the American Chemical Society, 137, 15112-15121.
https://doi.org/10.1039/c1ee02110a - 18. Takashima, T., Ishikawa, K. and Irie, H. (2016) Detection of Intermediate Species in Oxygen Evolution on Hematite Electrodes Using Spectroelectrochemical Measurements. The Journal of Physical Chemistry C, 120, 24827-24834.
https://doi.org/10.1021/acs.jpcc.6b07978 - 19. Takashima, T., Ishikawa, K. and Irie, H. (2016) Efficient Oxygen Evolution on Hematite at Neutral pH Enabled by Proton-Coupled Electron Transfer. Chemical Communications, 52, 14015-14018.
https://doi.org/10.1039/C6CC08379J - 20. Zandi, O. and Hamann, T.W. (2016) Determination of Photoelectrochemical Water Oxidation Intermediates on Haematite Electrode Surfaces Using Operando Infrared Spectroscopy. Nature Chemistry, 8, 778-783.
https://doi.org/10.1038/nchem.2557 - 21. Chen, J.Y., Dang, L., Liang, H., Bi, W., Gerken, J.B., Jin, S., Alp, E.E. and Stahl, S.S. (2015) Operando Analysis of NiFe and Fe Oxyhydroxide Electrocatalysts for Water Oxidation: Detection of Fe4+ by Mossbauer Spectroscopy. Journal of the American Chemical Society, 137, 15090-15093.
https://doi.org/10.1038/nchem.2557 - 22. Barroso, M., Mesa, C.A., Pendlebury, S.R., Cowan, A.J., Hisatomi, T., Sivula, K., Gratzel, M., Klug, D.R. and Durrant, J.R. (2012) Dynamics of Photogenerated Holes in Surface Modified-Fe2O3 Photoanodesfor Solar Water Splitting. Proceedings of the National Academy of Sciences, 109, 15640-15645.
https://doi.org/10.1073/pnas.1118326109 - 23. Bocquet, A.E., Suga, S., Kimizuka, N., Takeda, Y. and Takano, M. (1992) Electronic Structure of SrFe4+O and Related Fe Perovskite Oxides. Physical Review B, 45, 1561-1570.
https://doi.org/10.1103/PhysRevB.45.1561 - 24. Yabuuchi, N. and Komaba, S. (2014) Recent Research Progress on Iron-and Manganese-Based Positive Electrode Materials for Rechargeable Sodium Batteries. Science and Technology of Advanced Materials, 15, Article ID: 043501.
https://doi.org/10.1088/1468-6996/15/4/043501 - 25. Takashima, T., Ishikawa, K. and Irie, H. (2014) Thermal Activiation of Sr3Fe2O7 Electrocatalysts for Water Oxidation at Neutral pH. ECS Transactions, 61, 35-41.
https://doi.org/10.1088/1468-6996/15/4/043501 - 26. Kuzushita, K., Morimoto, S., Nasu, S. and Nakamura, S. (2000) Charge Disproportionation and Antiferromagnetic Order of Sr3Fe2O7. Journal of the Physical Society of Japan, 69, 2767-2770.
https://doi.org/10.1143/JPSJ.69.2767 - 27. Adler, P. (1997) Electronic State, Magnetism, and Electrical Transport Behavior of Sr3-xAxFe2O7 (x ≤ 0.4, A = Ba, La). Journal of Solid State Chemistry, 130, 129-139.
https://doi.org/10.1006/jssc.1997.7289 - 28. Adler, P. (1999) Charge Disproportionation in Iron (IV) Oxides: Electronic Properties and Magnetism in Sr3Fe2-xTixO7-y Annealed at High Oxygen Pressures. Journal of Materials Chemistry, 9, 471-477.
https://doi.org/10.1039/a806772d - 29. Matvejeff, M., Lehtimaki, M., Hirasa, A., Huang, Y.-H., Yamauchi, H. and Karppinen, M. (2005) New Water-Containing Phase Derived from theSr3Fe2O7 Phase of the Ruddlesden-Popper Structure. Chemistry of Materials, 17, 2775-2779.
https://doi.org/10.1021/cm050106z - 30. Ebbesen, S.D., Jensen, S.H., Hauch, A. and Mogensen, M.B. (2014) High Temperature Electrolysis in Alkaline Cells, Solid Proton Conducting Cells, and Solid Oxide Cells. Chemistry of Materials, 114, 10697-10734.
https://doi.org/10.1021/cr5000865 - 31. Goodenough, J.B. and Zhou, J.-S. (1998) Localized to Itinerant Electronic Transitions in Transition-Metal Oxides with the Perovskite Structure. Chemistry of Materials, 10, 2980-2993.
https://doi.org/10.1021/cm980276u - 32. Whangbo, M.-H., Koo, H.-J., Villesuzanne, A. and Pouchard, M. (2002) Effect of Metal-Oxygen Covalent Bonding on the Competition between Jahn-Teller Distortion and Charge Disproportionation in the Perovskites of High-Spin d4 Metal Ions LaMnO3 and CaFeO3. Inorganic Chemistry, 41, 1920-1929.
https://doi.org/10.1021/ic0110427 - 33. Jiang, L., Saldana-Greco, D., Schick, J.T. and Rappe, A.M. (2014) Enhanced Charge Ordering Transition in Doped CaFeO3 through Steric Templating. Physical Review B, 89, Article ID: 235106.
https://doi.org/10.1103/PhysRevB.89.235106 - 34. Takashima, T., Hashimoto, K. and Nakamura, R. (2012) Mechanisms of pH-De-pendent Activity for Water Oxidation to Molecular Oxygen by MnO2 Electrocatalysts. Journal of the American Chemical Society, 134, 1519-1527.
https://doi.org/10.1021/ja206511w - 35. Takashima, T., Hashimoto, K. and Nakamura, R. (2012) Inhibition of Charge Disproportionation of MnO2 Electrocatalysts for Efficient Water Oxidation under Neutral Conditions. Journal of the American Chemical Society, 134, 18153-18156.
https://doi.org/10.1021/ja306499n - 36. Takashima, T., Yamaguchi, A., Irie, H., Hashimoto, K. and Nakamura, R. (2014) In Situ UV-vis Absorption Spectra of Intermediate Species for Oxygen-Evolution Reaction on the Surface of MnO2 in Neutral and Alkaline Media. Electrochemistry, 82, 325-327.
https://doi.org/10.5796/electrochemistry.82.325 - 37. Axmann, P., Erdbrügger, C.F., Buss, D.H. and Glemser, O. (1996) Formation of FeIV and NiIV by Electrochemical and Chemical Oxidation of an Iron-Substituted Nickel (II) Hydroxide: The Direct Two-Electron Step NiII→NiIV + 2e-. Angewandte Chemie International Edition, 35, 1115-1118.
https://doi.org/10.1002/anie.199611151 - 38. Hamen, H.C.B. and Koch, C.B. (1994) Iron (IV) in Layered Cobalt-Iron Oxide Formed by Electrochemical Oxidation. Inorganic Chemistry, 33, 5363-5365.
https://doi.org/10.1021/ic00101a034