Computational Chemistry
Vol.05 No.01(2017), Article ID:73618,14 pages
10.4236/cc.2017.51005
NBO Population Analysis and Electronic Calculation of Four Azopyridine Ruthenium Complexes by DFT Method
N’Guessan Kouakou Nobel, Kafoumba Bamba*, Ouattara Wawohinlin Patrice, Nahossé Ziao
Laboratoire de Thermodynamique et de Physico-Chimie du Milieu, UFR-SFA, Université Nangui Abrogoua, Abidjan, Côte d’Ivoire

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: October 20, 2016; Accepted: January 15, 2017; Published: January 19, 2017
ABSTRACT
The molecular structure, the Natural Bond orbital (NBO) and the Time Dependent- DFT of both isomers cis or γ-Cl and trans or δ-Cl of RuCl2(L)2, where L stands respectively for 2-phenylazopyridine (Azpy), 2,4-dimethyl-6-[phenylazo]pyridine (Dazpy), 2-[(3,5-dimethylphenyl)azopyridine] (Mazpy) and 2-pyridylazonaphtol (Nazpy) were calculated with DFT method at B3LYP/LANL2DZ level. The prediction of the frontier orbitals (Highest Occupied Molecular Orbital or HOMO and Lowest Unoccupied Molecular Orbital or LUMO) shows that the most active complexes suitable for electronic reactions are admitted to be the trans isomers. Moreover, δ-RuCl2 (Azpy)2 is discovered to react more actively as photo-sensitizer since its energy gap is the minimum. Besides, electronic structures of all complexes through NBO calculation indicate that Ru-N bonds are made of delocalization of occupancies from lone pair orbital of N atoms to the ruthenium. Moreover, Ru was assumed to have almost the same charge regardless the structure of the azopyridine ligands in the complex indicating that the ligands provide only a steric effect that is responsible for the ruthenium’s selectivity. Concerning the transition state, NBO analysis also highlights that the transition LP(Ru)?π*(N1-N2) does correspond to t2g?π*(L). This transition is assumed to correspond to Metal to Ligand Charge Transfer (MLCT) that is responsible for the photo-sensitiveness of the metallic complex. Besides, TDDFT calculation of complexes showed that δ-RuCl2(Nazpy)2 displays the largest band during the absorption. For that reason, it is admitted to be the best photosensitizer due to a large system of conjugation provided by Nazpy ligand.
Keywords:
Natural Bond Orbital (NBO), HOMO, LUMO, Azopyridine Ligand, MLCT, LLCT

1. Introduction
Since azopyridine complexes of ruthenium have been of real interest for their capability not merely to limit the metal degree of oxidation to II or III rendering it more selective but also to throw off cancer disease [1] [2] [3] [4] , no theoretical investigation was performed to well account for their electron transfer. In our former papers, we showed up that all azopyridine ligands are bidentate e.g. they link to ruthenium by two nitrogen atoms forming then a five center stable complex [5] . We also showed that thanks to the asymmetry of ligands, five isomers are normally obtainable. But when complex is synthesized by combining RuCl3・3H2O with any azopyridine ligand, only both isomers cis and trans are obtained as displayed by Figure 1 [6] .
We assumed therefore that by this method of synthesis, the most producible complex is up today γ-Cl. Moreover, literature explains that azopyridine complexes of ruthenium can be used as sensitizer better than bipyridine complexes of ruthenium [2] . In this work, we object to compare the reactivity of complexes when the ligand changes and investigate the nature of Ru-N bonding by using natural bond orbital (NBO) analysis since NBO is admitted to highlight the electronic structure of a molecule [7] . Moreover, through time dependent DFT (TD-DFT) we intend to find out which complex must be electronically active to behave as photo-sensitizer. Our study regards four azopyridine ligands of ruthenium named RuCl2(Azpy)2, RuCl2(Dazpy)2, RuCl2(Mazpy)2 and RuCl2(Nazpy)2 whose ligands are displayed by Figure 2.

Figure 1. Synthesis of azopyridine ruthenium complexes. In this paper, azopyridine ligands were formed by combinations of both R and R1. Thus, when phenyl ring replaces R, and R1 is H, then we have Azpy ligand. Nazpy ligand corresponds to R replaced by naphtol ring and R1 by H. when R1 is CH3 substituent and R is phenyl ring, then we have Dazpy ligand. Whereas Mazpy, it is formed of R replaced by dimethylphenyl group and R1 by hydrogen atom. In all structures, chlorine atoms are in trans position.

Figure 2. Azopyridine ligands involved in ruthenium complexes formations. Numbers indicate carbon atoms.
The regarding isomers are all admitted to be C2-symmetrical except δ-RuCl2(Nazpy)2 that is Ci-symmetrical. Anyhow, they all present a C2 axis that makes both azopyridine ligands identical within each complex [8] .
2. Method
All geometry optimizations were performed with DFT method using Becke’s hybrid three parameters exchange functional of non local correlation functional of Lee Yang and Parr (B3LYP). They were carried out using an effective core Lanl2dz basis set owing to the relativist effect of core electrons from ruthenium atom [9] . The resulting geometries were verified as minima by frequency calculation. NBO whose program is embedded in Gaussian 09 package used for calculations [10] [11] was developed at B3LYP/Lanl2dz level on optimized molecules. The natural bond orbital analysis emphasizes the role of intermolecular orbital interaction or charge transfer in the complex. It is performed by considering all possible interaction between filled donor and empty acceptor NBOs and estimating their energetic importance by second-order perturbation theory. The stabilization energy E2 [12] associated with electron delocalization between electron donor NBO(i) and electron acceptor NBO(j) is evaluated according to equation below.
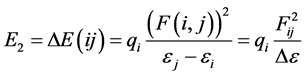 (1)
(1)
where qi stands for the orbital occupancy, εi and εj are diagonal elements (orbital energies) and Fij is the off-diagonal NBO Fock matrix element. The Natural Localized Molecular Orbital NLMO that displays the delocalization from a Lewis orbital to a non Lewis orbital was also predicted at B3LYP/Lanl2dz level. It thus provides additional evidence of the intermolecular delocalization effects [13] . Practically, each semi-loca- lized NLMO Ωi is expressed as a linear combination of the parent Lewis-type NBO σi (with coefficient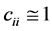 ) and residual weak contributions (
) and residual weak contributions ( ) from non-Lewis NBOs
) from non-Lewis NBOs 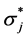 as indicated in Equation (2).
as indicated in Equation (2).
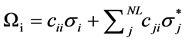 (2)
(2)
Clearly, NLMO completes the NBO summary and the perturbation theory energy. Regarding electronic prediction, it was carried out using TDDFT method with combined basis set. Thus Ru was lonely calculated with ECP Lanl2dz while the remaining atoms were performed with polarized split valence 6-31G(d) basis set.
3. Results and Discussions
3.1. Frontier Molecular Orbital Energies
The frontier molecular orbitals comprising the highest occupied molecular orbital (HOMO), the lowest unoccupied molecular orbital (LUMO) with the energy gap between HOMO and LUMO (ΔE) of the complexes are calculated and displayed in Table 1. Frontier orbital highlights the reactive property of complexes. The HOMO is the first orbital provider of the electron and the LUMO is the first orbital acceptor of the electron, and the gap between HOMO and LUMO characterizes the molecular chemical stability. The energy gap between the HOMO and the LUMO molecular orbitals is a critical parameter in determining molecular electrical transport properties though it is a measure of electron conductivity [14] . The chemical activity of the molecule is also observed from Eigen values of LUMO and HOMO and from the energy gap value calculated from them. Thus, ΔE is the result of a significant degree of intermolecular charge transfer (ICT) from the electron donor to the efficient electron acceptor group. Besides, from HOMO and LUMO which are admitted to be respectively the ionization potential and the affinity energy, the chemical hardness was determined according to Koopman’s theory given by:
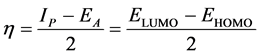 (3)
(3)
where Ip = −EHOMO is ionization potential (Kcal・mol−1), EA = −ELUMO is electronic affinity (Kcal・mol−1).
The hardness emphasizes the reactivity of the complex. Therefore, the larger the gap between HOMO and LUMO is, the harder the molecule is and the worse its reactivity is. In contrary, the chemical softness that is defined as the inverse of hardness highlights the capacity of an atom or group of atoms to receive electrons. Therefore, the soft molecule needs small energy to liberate an electron from HOMO since the gap ΔE is narrow. Besides, the chemical potential was calculated to account for the capability for

Table 1. Frontier orbital energies characterizing the reactivity of azopyridine ruthenium complexes in kcal・mol−1 calculated at B3LYP/LANL2DZ level.
aDE = LUMO-HOMO.
electron to leave from the molecule through Equation (3) as:
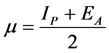 (4)
(4)
In Table 1, we can well see with each complex that the most reactive isomer represents δ-Cl for which energy gap is small. This can certainly be explained by the fact that both ligands are in Trans position where the repulsion brought about by the steric effect between them is minimized. Moreover, and specifically, δ-RuCl2(Azpy)2 is the most reactive complex with 41.5 Kcal・mol−1. In consequence, it is assumed to be the soft molecule though it displays the low value of hardness [15] . However, the reactive RuCl3・3H2O presents the highest energy gap with 57.5 Kcal・mol−1. Therefore, it is admitted to be the hardest molecule.
3.2. NBO Analysis
The natural bond orbital was performed on complexes using the pseudo-potential LANL2DZ basis set whose particularity is to freeze the core electrons [16] within the ruthenium atom. Therefore, Table 2 displays the electronic structure of ruthenium for each of the complexes. The calculations were made on the complexes, the reactive RuCl3・3H2O and also on ruthenium atom.
According to NBO analysis, Table 2 shows up that the structure of the valence orbital of Ru is characterized by 4d5s. Regarding the core electrons, Table 3 shows that LANL2DZ provides approximately 36 electrons regardless the structure of the ligand. Whereas the Rydberg orbitals (5p5d6p), they account for the more extended bits of the bases functions used including polarization functions electrons. Their occupancies within the complexes are low and constant with 0.06 electrons except Ru atom that does not display any value. We can assume that the Rydberg electrons are certainly related to the presence of ligands. Almost the same remark concerns the valence electrons where only Ru atom displays 8 electrons while all complexes show almost 7.40 electrons. Regarding the total electrons in ruthenium atom that is 44 when it is isolated, RuCl2(L)2 complexes display almost a constant value of 43.44 electrons while RuCl3・3H2O displays 43.1 electrons. Consequently, we can admit that these results tend to highlight the electron donor’s strength of azopyridine ligands to Ru. In reality, the nominal
Table 2. Electronic structure of ruthenium undergoing the pseudo-potential effect to minimize the relativistic effect.
Table 3. Allotment of ruthenium electrons between core, valence and Rydberg orbitals.
charge of Ru in RuCl2(L)2 is +2. So, the natural atomic charge that corresponds to the difference between the nuclear charge of Ru (44) and its total electron population in Table 3 within each complex is very low (lower than +2) confirming a significant transfer of electron density from the ligands to Ru [17] . However, regarding RuCl3・3H2O, the nominal charge of Ru is +3 due to the presence of three chloride atoms while the natural charge is +0.9. This discrepancy of charge is attributed to the presence of water molecules that are known to be strong electrons donors.
Table 4 displays the atomic charges for each atom involved in the formation of Ru- Ligand bonds. It shows that the charge of Ru in the complex is almost the same regardless both the nature of the ligand and the configuration of the complex’s isomer [2] [18] . Thus, we can admit that azopyridine ligands behave seemingly and the hindrance that they provide should have no electronic influence on the charge of Ru. However, it must certainly induce a steric effect necessary for selective reactions [6] and for the symmetry. For instance, while the three complexes δ-RuCl2(Azpy)2, δ-RuCl2(Dazpy)2 and δ-RuCl2(Mazpy)2 are C2-symmetrical with different chloride atomes, δ-RuCl2 (Nazpy)2 presents a Ci symmetry indicating an inversion center [19] . Herein, all ligands are consequently identical by pairs and thus, symmetric atoms display the same charge. Regarding the charges of nitrogen atoms both on ligands (Table 5) and on ruthenium complexes (Table 4), the lowest charges are located on both N1 and Npy. Therefore, since the azopyridine ligands are known to be bidentate structures [5] , we can believe that the electrons density on N1 should be delocalized either on Npy or on N2 that leads to the formation of Ru-Npy and Ru-N2 bonds. This process will give then rise to a five-ring stable shape of complex [5] [20] . Table 6, displays the interaction between N1, N2 and Npy atoms through NLMO delocalization of N1 electrons within the four azopyridine ligands.
Through Table 6, we can see that the four ligands delocalize the occupancies of the lone pair LP(N1) in the same natural atomic orbitals NAOs. Principally, LP(N1) delocalizes greatly onto the C1-Npy antibond. This fact confirms the identical electronic behavior of the azopyridine ligands.
Table 4. The NBO atomic charges of atoms involved in formation of Ru-Ligand bonds within azopyridine complexes of ruthenium calculated at B3LYP /Lanl2dz level.
Table 5. The NBO atomic charges of nitrogen atoms of azopyridine ligands involved in ruthenium complex formation at B3LYP/Lanl2dz level.
Table 6. Linear combination of NBO LP(N1) in each ligand with the parent Lewis LP(N1) and non Lewis orbitals in which its electrons can delocalize through NLMO analysis. The predictions were performed at B3LYP/Lanl2dz level.
The NBO Occupancy within the Ru Complexes
All azopyridine complexes studied herein are admitted to show a C2 axis [19] . So, both azopyridine ligands (ref. Figure 1) are identical and nitrogen atoms involved in the formation of Ru-ligand bonds are assumed to be in the same environment by pair. Regarding both chlorine atoms, they covalently bind to Ru atom since its nominal charge is +II. Here, the occupancy of NBO was performed at B3LYP/Lanl2dz level. So, Table 7 and Table 8 list the calculated occupancies of natural orbitals and natural hybrids of several atoms involved in the formation of Ru-N bonds in RuCl2(L)2 comprising electron delocalization. L stands for 2-phenylazopyridine (Azpy), 2-phenylazo-4,6-dime- thylpyridine (Dazpy), 2,6-dimethylphenylazo-2-pyridine (Mazpy) and 2-pyridylazo- naphtol (Nazpy). In these aforementioned Tables, only the AOs that occupancies are far lower than the ideal occupancy (2.0e) and the MOs involved in electronic transition are presented. It is assumed that the low occupancies of an AO disclose the orbital to be involved in MO formation [21] .
With azopyridine ligands and according to the reactive RuCl3・3H2O, ruthenium is involved in an octahedral structure with six single bonds. Regarding RuCl2(L)2, Cl atoms that are responsible for the nominal charge +II of Ru are involved in an ionic
Table 7. Occupancy of natural orbitals (NBOs) and hybrids on Cl. N and Ru atoms involved in formation of Ru-Ligand in γ- and δ-RuCl2(L)2 at B3LYP/LANL2DZ level.
aLP represents Lone Pair Orbital; bHybrid concerns the first atom of the bond; cLinear combination of NAOs of the atom concerned in the NBO hybrid; dPercentage contribution of each NAO in the NBO hybrid.
Table 8. Second-order interaction energy (E2. kcal/mol) between donor and acceptor orbitals in RuCl2(L)2 complexes at B3LYP/ LANL2DZ level.
aStared label (*) indicates anti-bonding, LP (A) is a valence lone pair orbital on atom A.
bonding. Therefore, σ(Ru-Cl) is a strong bond. Whereas Ru-N bonds, they are actually formed of electron transfer from lone pair LP(N) to Ru atom. Table 7 displays the structures of LP(N) orbital expected to bind to Ru atomic orbital owing to their low occupancies. We can see that LP(Npy) and LP(N2) have almost identical populations indicating their equal ability to form Ru-N bond and confirming therefore the bidentate state of the ligands. These interactions are highlighted through Table 8 by the second order perturbation theory where higher energies reflect the stability of the donor-ac- ceptor interactions. Here, Table 8 displays in fact the electron delocalization (ED) from filled donor orbital to empty acceptor orbital.
Besides, both transitions LP(N2)?LP*(Ru) and LP(Npy)?LP*(Ru) show that only Npy and N2 are involved in bonding with the same LP*(Ru) orbital.
Moreover, Table 7 shows LP(N1) with 1.94e as occupancy. This carries out its non involvement in Ru-N bondings. However, it delocalizes its electrons in the C1-Npy bonding as confirmed by Table 6 and Table 8.
Whereas LP(Ru)?p*(N1-N2), it indicates the electron delocalization regarding the metal to ligand charge transfer (MLCT) transition . This result is consistent with literature where Azopyridine ligands are admitted to be π-acceptor and σ-donor [11] . This transition is also consistent with the ability for δ-Cl complexes to better behave as photo-sensitizer though the structure of their donor LP(Ru) is exclusively made of combination of dxz and dyz as indicated in Table 7.
. This result is consistent with literature where Azopyridine ligands are admitted to be π-acceptor and σ-donor [11] . This transition is also consistent with the ability for δ-Cl complexes to better behave as photo-sensitizer though the structure of their donor LP(Ru) is exclusively made of combination of dxz and dyz as indicated in Table 7.
3.3. TDDFT Analysis
TD-DFT is performed to understand the electronic absorption and find out the ability for the complex to behave as sensitizer [22] . Since an efficient photosensitizer shows a strong absorption band in the visible region (400 - 800 nm) [23] , Figure 3 shows up the electronic spectra of azopyridine ruthenium complexes. We can see here that δ-Cl isomers present the highest and the widest wavelengths. Therefore, Table 9 lists the calculated wavelengths of the complexes.
Table 9 displays the main wavelengths (λmax) calculated for each complex, their excited energy, the frontier orbital’s composition and the main transitions regarding the

Figure 3. Simulated absorption spectra of both isomers γ-Cl and δ-Cl of azopyridine complexes including reactive RuCl3・3H2O recorded from 400 to 800 nm.
Table 9. Absorption properties of complexes comprising the maximum wavelength, oscillation frequency f and the main transitions involved by the energy.
visible region. Through this table we can see that γ-RuCl2(Nazpy)2 presents the highest wavelength with the important extinction coefficient (λmax = 748.1 nm and f = 0.106). We can assume that it should be the most sensitive complex. Moreover, all except one of them show that from the HOMO orbital until HOMO-4, molecular orbitals MOs are made principally of Ru orbital. Therefore, the regarding transitions are assumed to be metal to ligand charge transfer (MLCT) types. However, with γ-RuCl2(Nazpy)2, although maximum wavelength and extinction coefficient are also slightly important, HOMO is mainly made of ligand Nazpy orbital indicating that this transition is a ligand to ligand charge transfer (LLCT) type, which is not suitable for photochemical caracterisation since azopyridine ligands are reportedly insulator [6] . However, HOMO-1 is made of 63% Ru orbital and the second most important transition which wavelength is 538.2 nm with excited energy that is 2.3 ev is from H-6 to L. In consequence, we can assume that γ-RuCl2(Nazpy)2 is not sufficiently active as sensitizer. Besides, regarding LUMO and LUMO + 1 orbitals, they are exclusively made of Ligand orbitals in all complexes. Whereas the reactive RuCl3・3H2O, its HOMO and LUMO are both made of Ru orbitals and the maximum wavelength and oscillation strength f regarding transitions are low (λmax = 515.3 nm and f = 0.061). Since ΔE = 57.5 kcal・mol−1, we can see here the importance of ligands that improve the sensitivity of the ruthenium. Regarding excitation energy, we observe that δ-RuCl2(Azpy)2 presents the lowest value confirming its softness. Whereas the δ-RuCl2(Nazpy)2 it displays many metallic orbitals involved in the transition such as H-1, H-2 and H-4 with a large band of absorption. This strength can be attributed to the larger conjugate system that the ligand Nazpy provides [24] .
4. Conclusion
Four azopyridine complexes of ruthenium were predicted in this paper by NBO and TD calculations with DFT method. In order to recover the relativistic effect due to ruthenium atom, the pseudo-potential Lanl2dz basis set was used to perform calculation. Frontier molecular orbital energies calculation show first and foremost that δ- RuCl2(Azpy)2 is the most sensitive and soft complex expected to be used as sensitizer in photochemistry. Besides, the calculation shows that Ru atom in all complexes displays almost the same charge comprised between +0.53 and +0.59 that is significantly different from the nominal charge +2. This decrease in charge shows that azopyridine ligands are strong electrons donors. Nevertheless, the constant charge of ruthenium highlights that azopyridine ligands electronically behave similarly and the difference between them must be a steric effect for selective reactions. Furthermore, a natural bond orbital NBO analysis performed at B3LYP/Lanl2dz indicates that Ru-N bondings are made of delocalization of occupancies from Lone Pair atomic orbital of N2 and Npy to Ru. Moreover, as N1 does not link to ruthenium, it is assumed to delocalize its occupancies either in N2 or in Npy. This fact confirms the bidentate structure of azopyridine ligands. In addition, NBO shows that the transition regarding LP(Ru)?π*(N1-N2) corresponds to  confirming the ability for the azopyridine ruthenium complexes to be used as photo sensitizer. Therefore, the δ-Cl isomers regarding each azopyridine ligand are admitted to be the best reactive ruthenium complexes. Besides, TDDFT prediction performed confirms the softness of δ-RuCl2(Azpy)2. However, δ-RuCl2(Nazpy)2 is admitted to be the most sensitive with a large band of absorption and an involvement of many molecular orbitals in electron transfer. On behalf of that investigation, the coming work will consist on applying δ-RuCl2(Nazpy)2 as photo-sensitizer over a well known active semi-conductor compound as TiO2 anatase through dye-sensitized solar cell (DSSC) device.
confirming the ability for the azopyridine ruthenium complexes to be used as photo sensitizer. Therefore, the δ-Cl isomers regarding each azopyridine ligand are admitted to be the best reactive ruthenium complexes. Besides, TDDFT prediction performed confirms the softness of δ-RuCl2(Azpy)2. However, δ-RuCl2(Nazpy)2 is admitted to be the most sensitive with a large band of absorption and an involvement of many molecular orbitals in electron transfer. On behalf of that investigation, the coming work will consist on applying δ-RuCl2(Nazpy)2 as photo-sensitizer over a well known active semi-conductor compound as TiO2 anatase through dye-sensitized solar cell (DSSC) device.
Cite this paper
Nobel, N.K., Bamba, K., Patrice, O.W. and Ziao, N. (2017) NBO Population Analysis and Electronic Calculation of Four Azopyridine Ruthenium Complexes by DFT Method. Computational Chemistry, 5, 51-64. http://dx.doi.org/10.4236/cc.2017.51005
References
- 1. Kooijman, H., Hostze, C.G., Caspers, S.E., Haasnoot, J.G., Reedijk, J. and Spek, A.L. (2004) α-Dichlorobis(2-phenylazo-4,6-dimethyl-pyridine)ruthenium(II) Chloroform Solvate, Metal-Organic Papers. Acta Crystallographica Section E, E60, m247-m249.
https://doi.org/10.1107/S1600536804001618 - 2. Gowami, S., Chakravarty, A.R. and Chakrovorty, A. (1981) Chemistry of Ruthenium. 2. Synthesis, Structure, and Redox Properties of 2-(Arylazo) Pyridine Complexes. Inorganic Chemistry, 20, 2246-2250.
https://doi.org/10.1021/ic50221a061 - 3. Jorna, A.J., Boelrijk, A.E.M., Hoorn, H.J. and Reedijk, J. (1996) Heterogenization of a Ruthenium Catalyst on Silica and Its Application in Alcohol Oxidation and Stilbene Epoxidation. Reactive & Functional Polymers, 29, 101-114.
https://doi.org/10.1016/1381-5148(96)00005-3 - 4. Velders, A.H., Kooijman, H., Spek, A. L., Haasnoot, J.G., De Vos, D. and Reedijk, J. (2000) Strong Differences in the in Vitro Cytotoxicity of Three Isomeric Dichlorobis(2-phenyla-zopyridine)ruthenium(II) Complexes. Inorganic Chemistry, 39, 2966-2967.
https://doi.org/10.1021/ic000167t - 5. Affi, S.T., Bamba, K. and Ziao, N. (2015) Computational Characterization of Organometallic Ligands Coordinating Metal: Case of Azopyridine Ligands. Journal of Theoretical and Computational Chemistry, 14, Article ID: 1550006.
https://doi.org/10.1142/s0219633615500066 - 6. Bamba, K., Leger, J.-M., Garnier, E., Bachmann, C., Servat, K. and Kokoh, K.B. (2005) Selective Electro-Oxidation of D-Glucose by RuCl2(azpy)2 Complexes as Electrochemical Mediators. Electrochimica Acta, 50, 3341-3346.
https://doi.org/10.1016/j.electacta.2004.12.007 - 7. Chaitanya, K., Santhamma, C., Prasad, K.V. and Veeraiah, V. (2012) Molecular strucTure, Vibrational Spectroscopic (FT-IR, FT-Raman), First Order Hyperpolarizability, NBO Analysis, HOMO and LUMO Analysis, Thermodynamic Properties of 3,5-Dimethylben-zophenone by Ab Inito. Journal of Atomic and Molecular Sciences, 3, 1-22.
- 8. Ouattara, W.P., Bamba, K., Ziao, N. and N’guessan, K.N. (2016) Theoretical and Electrochemical Characterization of δ-RuCl2(Nazpy)2: Application to Oxidation of D-Glucose. American Journal of Analytical Chemistry, 7, 57-66.
https://doi.org/10.4236/ajac.2016.71006 - 9. Foresman, J.B. and Frisch, A. (1996) Exploring Chemistry with Electronic Structure Methods. 2nd Edition, Gaussian Inc., Pittsburgh.
- 10. Schuchardt, K. L., Didier, B.T., Elsethagen, T., Sun, L., Gurumoorthi, V., Chase, J., Li, J. and Windus, T.L. (2007) Basis Set Exchange: A Community Database for Computational Sciences. Journal of Chemical Information and Modeling, 47, 1045-1052.
https://doi.org/10.1021/ci600510j - 11. Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.A.J., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, O., Foresman, J.B., Ortiz, J.V., Cioslowski, J. and Fox, D.J. (2009) Gaussian 09, Revision E.01. Gaussian, Inc., Wallingford.
- 12. Weinhold, F. and Landis, C.R. (2001) Natural Bond Orbitals and Extensions of Localized Bonding Concepts. Chemistry Education Research and Practice, 2, 91-104.
- 13. Reed, A.E., Curtiss, L.A. and Weinhold, F. (1988) Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chemical Reviews, 88, 899-926.
https://doi.org/10.1021/cr00088a005 - 14. Sangeetha, C.C., Madivanane, R. and Pouchaname, V. (2014) The Vibrational Spectroscopic (FT-IR & FT Raman, NMR, UV) Study and HOMO & LUMO Analysis of Phthalazine by DFT and HF Studies. International Journal Of Engineering Research and General Science, 2, 222.
- 15. Bamba, K., Ouattara, W.P., N’Guessan, K.N. and Ziao, N. (2016) SARs Investigation of α-, β-, γ-, δ-, ε-RuCl2(Azpy)2 Complexes as Antitumor Drugs. Computational Chemistry, 4, 1-10.
https://doi.org/10.4236/cc.2016.41001 - 16. Cramer, C.J. (2004) Essentials of Computational Chemistry, Theories and Models, Effective Core Potentials. 2nd Edition, Wiley, Hoboken, 179.
- 17. Oziminski, W.P. and Narbutt, J. (2010) Theoretical Investigations on the Structure and Bonding in Neutral Trinitrate Complexes of Americium(III) and Europium(III) with 6,6’-Bis-(5,6-Diethyl-1,2,4-Triazin-Yl)-2,2 Bipyridine in Solvent Extraction Systems. Institute of Nuclear Chemistry and Technology, Warszawa, 35.
- 18. Shriver, D.F. and Atkins, PW. (1999) Inorganic Chemistry. 3rd Edition, Oxford University Press, Oxford, 292.
- 19. N’Guessan, K.N., Bamba, K., Ziao, N. and Ouattara, W.P. (2015) Molecular Structure, Vibrational Spectra and NMR Analyses on Two Azopyridine Ruthenium Complexes Using Density Functional Theory Calculations. Journal of Chemical and Pharmaceutical Research, 7, 246.
- 20. Velders, A.H., van der Schilden, K., Hoste, A.C.G., Reedijk, J., Kooijman, H. and Speck, A.L. (2004) Dichlorobis(2-Phenylazopyridine)Ruthenium(II) Complexes: Characterization, Spectroscopic and Structural Properties of Four Isomers. Dalton Transactions, 3, 448-455,
https://doi.org/10.1039/b313182c - 21. Ghiasi, R. and Ebrahimi, M.E. (2012) Natural Bond Orbital (NBO) Population Analysis of Iridabenzene (C5H5Ir)(PH3)3. Journal of Applied Chemical Research, 20, 7-13.
- 22. Fan, W.-J., Cai, J.-W., Yang, G.-J., Chi, J.-W., Zhou, D., Tan, D.-Z. and Zhang, R.-Q. (2016) Aggregation of Metal-Free Organic Sensitizers on TiO2(101) Surface for Use in Dye-Sensitized Solar Cells: A Computational Investment. Computational and Theoretical Chemistry, 1093, 1-8.
https://doi.org/10.1016/j.comptc.2016.08.006 - 23. Umer, M., Ibnelwaleed, A.H., Muhammad, D., Shakeel, A. and Khalil, H. (2015) Theoretical Study of Benzene/Thiophene Based Photosensitizers for Dye Sensitized Solar Cells (DSSCs). Dyes and Pigments, 118, 152-158.
- 24. Zhang, C.-R., Liu, Z.-J., Chen, Y.-H., et al. (2009) DFT and TDDFT Study on Organic Dye Sensitizers D5, DST and DSS for Solar Cells. Journal of Molecular Structure: THEOCHEM, 899, 86-93.
https://doi.org/10.1016/j.theochem.2008.12.015