Open Journal of Immunology
Vol.04 No.03(2014), Article ID:49494,20 pages
10.4236/oji.2014.43013
Temporal Transcriptional Regulation of IL-10-Induced Anti-Inflammatory
Genes
in LPS-Triggered Macrophages
Amanda
F.
Dillow
*,
Leah
N.
Cardwell
*,
Tyler
J.
Smith
*,
Brad
D.
Groppe
,
Brian
A.
Peterson
,
Maxwell
A.
Sickman
,
Brian
K.
Weaver
#
Department of Biology, Missouri State University, Springfield, MO, USA
Email: #brianweaver@missouristate.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 30 June 2014; revised 30 July 2014; accepted 30 August 2014
ABSTRACT
Interleukin-10 (IL-10) mediates an anti-inflammatory response that is executed through the expression of IL-10-induced genes. Certain IL-10-induced genes, exemplified by TNIP3, are induced by IL-10 only in conjunction with a pro-inflammatory signal. We sought to characterize the mechanism whereby IL-10 and Toll-like receptor signals synergized to induce expression of genes like TNIP
3 in
macrophages. Stimulation with IL-10 and lipopolysaccharide (LPS) synergistically induced an increase in the transcription rate of TNIP3, while having no effect on its mRNA stability. This transcriptional mechanism proved to be generalizable to 14 other genes that also were synergistically induced by IL-10 and LPS in monocytes/macrophages. Although all of the genes had this synergistic transcriptional regulation in common, they could be divided into three subsets based on their differential requirements for de novo protein synthesis and kinetics of expression: namely, primary response genes, early secondary response genes, and late secondary response genes. This coordinated and temporal pattern of transcriptional regulation in response to IL-10 and LPS was conserved in both human and mouse monocytes/macrophages, and it was associated with differential dependencies on PI3K and JNK signaling pathways. These results underscore the complex nature of the IL-10-induced transcriptional response that occurs specifically in LPS-trig- gered macrophages.
Keywords:
IL-10, TLR, Macrophage, Inflammation, Transcription

1. Introduction
The innate immune system can rapidly respond to danger signals presented by invading pathogens. Macrophages play an important role in this response by initiating and promoting inflammation [1] - [4] . Innate pathogen re- cognition is mediated by interaction between a diverse range of pathogen-associated molecular patterns (PAMPs) and a collection of specific pattern recognition receptors (PRRs) expressed by macrophages and other host cells [5] - [8] . Lipopolysaccharide (LPS) of gram-negative bacteria serves as a prototypical PAMP that is recognized by Toll-like Receptor 4 (TLR4), a member of the TLR family of membrane-bound PRRs. As a group, the TLRs’ mediate responses to PAMPs derived from bacteria, fungi, protozoa, and viruses [9] - [12] .
Upon PAMP binding, TLRs form dimeric structures and activate conserved signal transduction pathways through their homologous cytoplasmic Toll/IL-1R (TIR) domains [13] - [15] . The receptor TIR domains engage with adapter proteins of the MyD88 family, which recruit and activate protein kinases and ubiquitin ligases of the IRAK and TRAF families that, in turn, lead to activation of the latent transcription factors NF-κB, AP-1, and IRF3/7. The activated transcription factors then drive the expression of numerous pro-inflammatory genes, including the cytokines: TNF, IL-1, IL-6, IL-12, and IFN-α/β. Successful elimination of invading pathogens depends on the qualitative and quantitative effects produced by these newly expressed inflammatory mediators.
Although a robust inflammatory response is critical for the host defense, it must also be negatively regulated in order to prevent excessive host tissue damage. The anti-inflammatory cytokine interleukin-10 (IL-10) plays an important part in this negative regulation. IL-10 can be produced in a constitutive or inducible manner by several types of immune cells, including TLR-triggered macrophages [16] - [18] . Once secreted, IL-10 can act on macrophages to suppress TLR responses and pro-inflammatory gene expression [19] - [22] . IL-10 works in a selective manner to inhibit specific pro-inflammatory genes via mechanisms that include transcriptional repression, mRNA destabilization, and translational inhibition [19] [20] [23] - [27] . The cytokine’s importance is evident by the development of autoimmune/inflammatory syndromes, particularly enterocolitis, in mice and humans with defects in IL-10 responses [28] - [33] . Furthermore, mice engineered to specifically lack IL-10-respones in macrophages display similar inflammatory phenotypes, revealing these cells as important targets of the cytokine in vivo [34] - [36] .
IL-10 acts on macrophages by binding to the cell-surface IL-10 receptor, which is a heterodimer of two receptor chains, IL-10R1 and IL-10R2, that form a tetrameric structure upon binding to cytokine [16] [17] [37] . IL-10 receptor ligation induces the activation of two protein tyrosine kinases, JAK1 and TYK2, which phosphorylate specific tyrosine residues within the cytoplasmic tail of IL-10R1. Tyrosine-phosphorylated IL-10R1 then recruits the latent transcription factor STAT3, and subsequent JAK-dependent tyrosine phosphorylation of STAT3 leads to its activation and induction of target gene expression [38] [39] . In order to better understand the IL-10-mediated anti-inflammatory response, researchers have endeavored to identify and characterize the IL- 10/STAT3-induced genes [40] - [47] . More than one hundred IL-10-induced genes have been identified, and a number of these, including SOCS3, Bcl-3, NF-IL3, DUSP1, TTP, and TNIP3, have been shown to play specific roles in the anti-inflammatory response [17] [19] [20] [22] [43] [48] [49] .
Although it is clear that STAT3-dependent genes are responsible for executing the anti-inflammatory response, a model based solely on activated STAT3 inducing all of the target genes does not sufficiently explain the characteristics of the IL-10-induced transcriptome in macrophages. Specifically, while certain genes can be induced by IL-10 stimulation alone, others are only up-regulated by IL-10 when macrophages are co-stimulated with a TLR agonist such as LPS [41] - [44] . The genes in the latter category have been referred to as IL-10 super-induced genes, since IL-10 increases their expression only in a synergistic manner with TLR stimulation [42] . As a result, genes induced by IL-10 can be described in two transcriptional modules: IL-10-induced genes versus IL-10 super-induced genes. This situation serves as an example of signal integration wherein macrophages adjust their transcriptional output to multiple environmental cues: exposure to either a TLR agonist or IL-10 alone versus IL
-10 in
the concurrent presence of a TLR agonist. The existence of these distinct transcriptional modules may allow quantitatively or qualitatively different anti-inflammatory responses to take place in cells under different situations. For example, induction of specific genes by IL-10 alone may provide for the inactivation of resting macrophages prior to their exposure to PAMPs or other inflammatory mediators. On the other hand, the ability of IL-10 to super-induce certain genes only in the presence of concurrent PAMP stimulation may provide for the resolution of inflammatory responses by activated cells. From a mechanistic perspective, a distinction should exist between the signaling or transcriptional pathways controlling the two gene modules.
While direct targeting of promoters by STAT3 alone could explain the expression of the IL-10-induced gene module, it is less clear as to how the IL-10 super-induced genes become responsive to IL-10 only in TLR-trig- gered cells. Therefore, we sought to characterize the mechanisms controlling the expression of a previously identified IL-10 super-induced gene—TNIP3 (also known as ABIN-3) [43] . Herein, we show that the synergistic induction of TNIP3 by IL
-10 in
LPS-triggered macrophages occurs through an increase in the gene’s transcription rate, and not due to an increase in its mRNA stability. Through extending the investigation to additional genes, we found that the entire IL-10 super-induced gene module responds likewise at the level of transcriptional synergy. Despite their universal control at the transcriptional level, the IL-10 super-induced genes could be differentiated into subsets of primary response and secondary response genes based on their dependencies on de novo protein synthesis. The secondary response genes were found to be further divisible into early and late subsets based on their differential expression kinetics. These results demonstrate that macrophages respond to IL-10 and LPS together with the synergistic induction of a set of anti-inflammatory genes in a multipart and temporally-regulated transcriptional cascade.
2. Materials and Methods
2.1. Reagents
Recombinant human and mouse IL-10 were purchased from eBioscience. Rat anti-mouse IL-10R1 neutralizing monoclonal antibody (clone 1B
1.3a
) was purchased from BD Biosciences. LPS (from Escherichia coli O127:B8), actinomycin D, and cycloheximide were purchased from Sigma. The Universal 18S PCR Primer Set was purchased from Ambion/Life Technologies. The RPL
13A
and PPIA PCR primer pairs were purchased as part of the Human and Mouse Housekeeping Gene Primer Sets from RealTimePrimers.com. All other PCR primers were designed as described below and obtained from Integrated DNA Technologies (IDT). The pharmacological inhibitors for JAK1 (JAK Inhibitor I), PI3K (LY294002), ERK-1/2 (ERK Inhibitor II, FR180204), JNK-
1/2/3
(JNK Inhibitor II, SP600125), and p38 MAPK (SB203580) were purchased from EMD Chemicals/Calbiochem.
2.2. Cell Culture
Cell cultures were maintained in a 37˚C incubator with a humidified 5% CO2 atmosphere. The human monocyte/macrophage cell lines, THP-1 and U937, were cultured at a concentration of 0.5 − 2 × 106 cells/ml in RPMI 1640 culture medium (Hyclone), supplemented with 10% fetal bovine serum (Hyclone),
2 mM
L-glutamine (Hyclone),
1 mM
sodium pyruvate (Hyclone), 50 U/ml penicillin (Hyclone), 50 μg/ml streptomycin (Hyclone), 0.0375% sodium bicarbonate (Hyclone), and 50 μM 2-mercaptoethanol (Sigma). Human peripheral blood mononuclear cells (PBMC) were obtained from
Astarte
Biologics
and cultured in complete RPMI 1640 medium as described above. HEK 293T cells were cultured in DMEM culture medium (Hyclone), supplemented with 10% fetal bovine serum,
2 mM
L-glutamine,
1 mM
sodium pyruvate, 50 U/ml penicillin, and 50 μg/ml streptomycin. Mouse bone marrow-derived macrophages (BMM) were prepared and cultured as described previously [43] [50] . In brief, femurs from 8-week-old C57BL/6J mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and bone marrow was eluted and plated at a density of 5 × 106 cells per
100 mm
Petri dish (Fisher) in DMEM, supplemented with 10% fetal bovine serum, 5% donor horse serum (Atlanta Biologicals), 20% L cell-condi- tioned media,
2 mM
L-glutamine,
1 mM
sodium pyruvate, 50 U/ml penicillin, 50 μg/ml streptomycin, and 1 ng/ml M-CSF (eBioscience). After 7 days, macrophages (6 − 8 × 106 cells per dish) were re-plated into tissue culture plates and fed with resting media (lacking 20% L cell-conditioned media) to allow the cells to become quiescent.
2.3. RNA Isolation
THP-1 and U937 cells were plated at a concentration of 2 − 3 × 106 cells/ml in
35 mm
or
60 mm
tissue culture plates (Corning) and incubated for 24 h prior to treatments. PBMC were plated at 4 × 106 cells/ml in
35 mm
plates and incubated for 48 h prior to treatments. BMM were plated at a density of 3 × 106 cells per
35 mm
plate and incubated in resting media for 24 h prior to treatments. Total RNA was purified using either the RNA STAT-60 reagent (Tel-Test, Inc.) followed by DNase treatment (Promega) or the RNeasy Mini Kit (Qiagen) and on-column DNase treatment (Qiagen) according to the manufacturer’s protocols. RNA yield and integrity were assessed by UV spectrophotometry and agarose gel electrophoresis.
2.4. Quantitative RT-PCR (RT-qPCR) Assay
First-strand cDNA was made from 0.5 μg of DNase-treated RNA using the Verso cDNA
Kit
(Thermo Scientific) according to the manufacturer’s protocol. First, RNA samples were heated to 70˚C for 5 min and then quick- cooled to remove secondary structure. The RNA was then subjected to reverse transcription at 50˚C for 60 min using a mix of random primers and oligo-dT primers, followed by enzyme inactivation at 95˚C for 2 min. Completed reactions were diluted with nuclease-free water to a volume of 250 μl (~2 ng/μl) and 10 μl was used as template for PCR reactions. Relative gene expression was measured using SYBR Green-based real-time/quan- titative PCR (qPCR). Numerical values were assigned to unknowns using standard curves made from serially diluted cDNA. Relative expression values for genes of interest were determined after normalization to expression levels of multiple housekeeping/reference genes to ensure the fidelity of the results. PCR reactions were performed in duplicate with a final reaction volume of 25 μl consisting of 12.5 μl GoTaq qPCR Master Mix (Promega), 10.0 μl cDNA template, PCR primers (final concentration of 200 nM each), and nuclease-free water. PCR was performed using an Mx3005P QPCR system (Agilent Technologies). PCR amplification was as follows: 95˚C (10 min), followed by 40 cycles of 95˚C (30 s), 60˚C (60 s), and a final dissociation curve segment to monitor the specificity of amplification.
PCR primer pairs were designed using the NCBI Primer-BLAST primer design program and oligonucleotides were obtained from Integrated DNA Technologies (IDT). Primer pairs were designed and tested based on the following parameters: primer length of 18 - 26 nt, primer melting temperature between 58˚C - 62˚C, amplicon length between 75 - 250 bp, hybridization to different exons spanning one or more introns of the target cDNA, PCR efficiency of >85% over a linear dynamic range of 6 log10, and correlation coefficient (r2) > 0.95. Primer pairs for detecting mature mRNAs were designed to hybridize to different exons spanning one or more introns of the target cDNA, whereas primer pairs for detecting primary transcripts were designed to hybridize to an exon and adjoining intron of the target cDNA. Primer sequences are provided in Table 1.
2.5. Chromatin Immunoprecipitation (ChIP) Assay
BMM were plated at a density of 12 × 106 cells per
100 mm
tissue culture plate and incubated in resting media for 24 h prior to treatments. Chromatin immunoprecipitation (ChIP) was performed using the Pierce Agarose ChIP Kit (Thermo Scientific) according to the manufacturer’s protocol. Cells were fixed in 1% formaldehyde for 10 min, followed by cell lysis, and isolated nuclei were incubated with 3 units of Micrococcal Nuclease in digestion buffer for 15 min at 37˚C to yield sheared chromatin in the range of 200 - 1000 bp. Digested chromatin was pre-cleared with Protein A/G agarose before being split and subjected to immunoprecipitation overnight at 4˚C using 1 μg of rabbit polyclonal IgG against RNA Pol II (Santa Cruz; sc-899) or normal rabbit IgG (Santa Cruz; sc-2027). Immune complexes were bound to Protein A/G agarose by incubating for 1 h at 4˚C. After washing, immune complexes were eluted and cross-links were reversed by incubation with Proteinase K at 65˚C. The DNA was recovered in 50 μl of elution buffer using the provided DNA Clean-Up columns. Eluted DNA was analyzed by qPCR using primer pairs targeting the proximal promoters for the mouse TNIP3 gene (−122 to +93; forward
5’
-AGCTAGGTTAGCCCCAGGCAT and reverse
5’
-GATGTGGGGCAGATTGGCCTT) and mouse GAPDH gene (−266 to −189; forward
5’
-TCCCCCTATCAGTTCGGAGCC and reverse
5’
-TCCT- CCCTCTCTTTGGACCCG). Relative level of promoter DNA present was determined using standard curves made from serially diluted genomic DNA and compared to 10% input chromatin template.
2.6. Plasmid Constructs and Reporter
Gene
Assay
A 2.5 kb DNA fragment of the human TNIP3 gene promoter (nucleotide position −2420 to +40 relative to the transcriptional start site) was isolated by PCR using THP-1 genomic DNA as template. The forward primer (
5’
-GGttaattaaGAATGCCTCCAATAACTTATAG) included a PacI restriction site and the reverse primer (
5’
-GggatccAAATGAACAGAAGTGACTGTG) included a BamHI restriction site for subcloning purposes. PCR amplification and product subcloning were carried out using the Strataclone Ultra Blunt Kit (Agilent Technologies). The PCR product was excised from the pSC-B cloning vector with EcoRI (Promega), gel purified with the QIAquick Gel Extraction Kit (Qiagen), and then digested with PacI (Thermo Scientific) and BamHI
(Thermo Scientific). The linear PacI-BamHI promoter fragment was ligated to the FUGW self-inactivating retroviral vector that had been digested with PacI and BamHI to remove the ubiquitin promoter [51] . In the final plasmid vector, referred to as pFGW-hTNIP3pr, the human TNIP3 promoter fragment is in position to drive the expression of a GFP reporter gene.
The pFGW-hTNIP3pr vector was packaged in HEK 293T cells by co-transfection with the delta 8.9 and VSVg plasmids using Lipofectamine 2000 (Invitrogen). Retroviral supernatants were collected at 48 h and 72 h post-transfection. THP-1 cells were plated at a concentration of 1.5 × 106 cells/ml in
35 mm
plates and transduced with the retroviral supernatants in the presence of 8 μg/ml polybrene (Sigma). Afterwards, the cells were split and allowed to rest for 24 h prior to treatments. Expression levels of GFP were measured by flow cytometry using a BD Accuri C6 flow cytometer (BD Biosciences). Mock-transduced cells and cells transduced with the parent FUGW ubiquitin promoter vector were used to assess transduction efficiency and to determine the GFP-positive and negative gates. Transduction efficiency was approximately 15% of the total cells. For each experimental treatment on the pFGW-hTNIP3pr vector transduced cells, 20,000 total events were collected and cells in the GFP-positive gate are presented as histogram plots.
3. Results
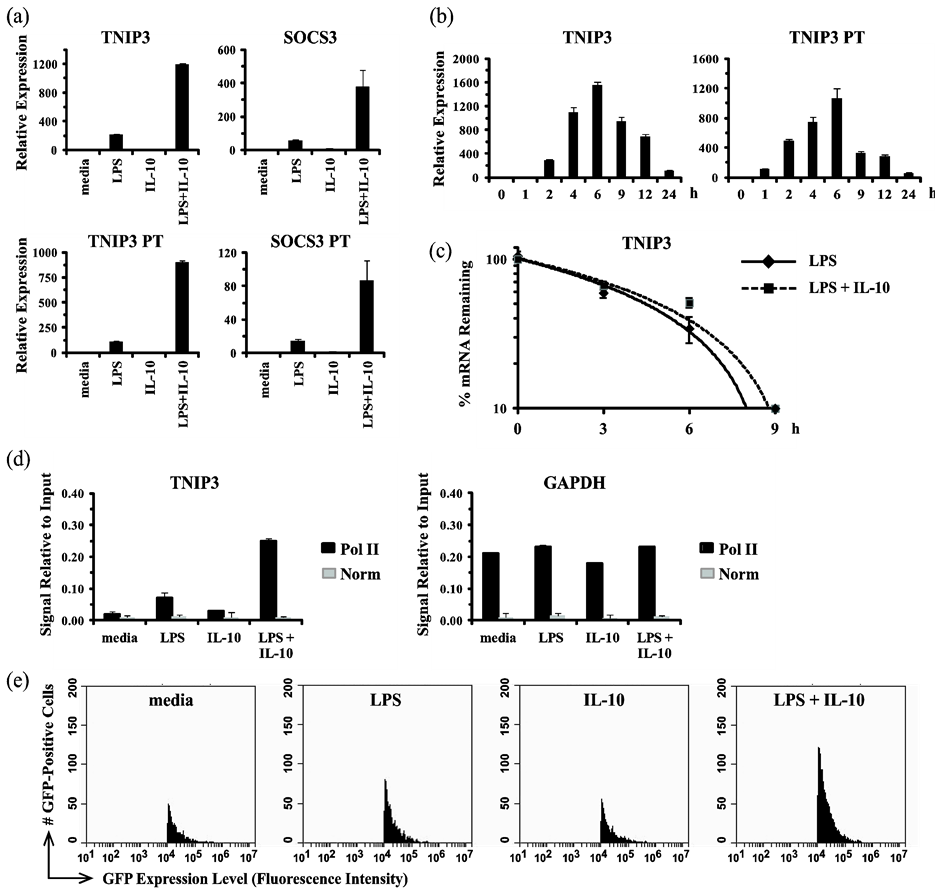
3.1. Synergistic Induction of TNIP3 by IL-10 and LPS Occurs through an Increase in Transcription Rate
To begin elucidating the mechanism whereby IL-10 super-induces TNIP3 expression in LPS-triggered macrophages, we examined whether IL-10 was acting on the gene at a transcriptional or post-transcriptional level. First, whether IL-10 acts to increase the transcription rate of TNIP3 was determined using a primary transcript (PT) RT-qPCR assay [24] [52] . This approach takes advantage of the fact that primary transcripts turnover rapidly as a result of RNA processing to remove introns and they are not subject to the same stabilization mechanisms observed for mature mRNAs; therefore, measurement of primary transcript levels provides a better marker of recent changes in transcription initiation rate. When the human monocytic cell line THP-1 was stimulated with LPS, IL-10, or LPS and IL
-10 in
combination, the TNIP3 primary transcript (TNIP
3 PT
) levels increased in a synergistic manner akin to what was observed for the mature transcript (TNIP3) levels measured by conventional RT-qPCR (Figure 1(a)). Notably, IL-10 stimulation led to this increase in TNIP3 transcription rate only in cells that were co-stimulated with LPS. For comparison and as a positive control for a gene known to be under direct transcriptional control by STAT3, we examined the expression of the IL-10-induced gene SOCS3. In agreement with its being controlled at the transcriptional level, the results showed that stimulation with LPS and IL-10 induced an increase in SOCS3 primary transcript levels in parallel with its mRNA induction (Figure 1(a)). The results for TNIP3 could be reproduced when using two additional sets of PCR primer pairs targeting different regions of the primary transcript, and in experiments using another human monocytic cell line U937 or primary human PBMC (data not shown). A time-course analysis of TNIP3 mRNA versus primary transcript expression showed that, relative to the mRNA, the primary transcript levels rose with slightly faster kinetics and declined more abruptly after a peak at 6 h (Figure 1(b)). Thus, in response to stimulation with IL-10 and LPS, TNIP3 transcription rate undergoes a synergistic increase that is measurable by 1 h, continues to climb over the next several hours, and then declines rapidly leading to a consequent decline in the mRNA levels. The relatively accelerated kinetic profile of the primary transcripts is consistent with them being the more rapidly induced and processed precursors to the mature mRNA. Second, to determine whether IL-10 acts to increase the stability of TNIP3 mRNA induced by LPS stimulation, the mRNA half-life was measured in cells in the absence or presence of IL-10. THP-1 cells were treated with either LPS or LPS and IL
-10 in
combination for 6 h, followed by addition of actinomycin D to stop transcription. TNIP3 mRNA levels were measured over a time-course post-actinomycin D addition to determine their decay rate (Figure 1(c)). The results showed that IL-10 stimulation did not synergistically increase the stability of TNIP3 mRNA in LPS-treated cells. Taken together, the results indicate that IL-10 synergizes with LPS to induce TNIP3 gene expression at a transcriptional level, and not at a post-transcriptional level.
To confirm that IL-10 increases TNIP3 transcription rate specifically in LPS/TLR-triggered cells, we performed chromatin immunoprecipitation (ChIP) and reporter gene assays to measure activation of the TNIP3 promoter. First, a ChIP assay was used to measure the recruitment of RNA polymerase II (RNA Pol II) to the TNIP3 promoter in response to LPS and/or IL-10 stimulation (Figure 1(d)). When macrophages were stimulated
![]()
Figure 1. IL-10 induces TNIP3 expression in LPS-stimulated monocytes/macrophages via a transcriptional mechanism. (a) THP-1 cells were mock treated (media) or treated with LPS (100 ng/ml), IL-10 (10 ng/ml), or LPS and IL
-10 in
combination for 4 h. Total RNA was isolated and analyzed by conventional or primary transcript (PT) RT-qPCR in duplicate. Relative expression values for genes of interest are presented as mean ± s.d. of duplicate reactions after normalization to the three reference genes: GAPDH, HPRT, and 18S rRNA. (b) THP-1 cells were treated with LPS (100 ng/ml) and IL-10 (10 ng/ml) in combination for the indicated times. Total RNA was isolated and analyzed by conventional or primary transcript (PT) RT-qPCR as described for panel (a). (c) THP-1 cells were mock treated or treated with LPS (100 ng/ml) in the absence (solid line) or presence of IL-10 (10 ng/ml) (dashed line) for 6 h, followed by addition of actinomycin D (5 μg/ml). Total RNA was isolated at the indicated times post-actinomycin D addition and analyzed by RT-qPCR in duplicate. Relative expression values (mean ± s.d. of duplicate reactions) for TNIP3 were determined after normalization to the reference gene 18S rRNA. (d) Mouse bone marrow-derived macrophages (BMM) were mock treated (media) or treated with LPS (100 ng/ml), IL-10 (10 ng/ml), or LPS and IL-10 for 2 h. Stimulation with LPS alone was done in the presence of a neutralizing anti-IL-10R1 mAb (0.5 μg/ml). Cells were harvested and subjected to ChIP assay using RNA Pol II or rabbit isotype control (
Norm
) antibodies. Purified DNA was analyzed for the TNIP3 and GAPDH proximal promoters using qPCR in duplicate. Normalized results (mean ± s.d. of duplicate reactions) are shown as signal relative to the total amount of input chromatin. (e) THP-1 cells were transduced with a reporter gene cassette in which a human TNIP3 promoter fragment controls expression of GFP. Cells were mock treated (media) or treated with LPS (100 ng/ml), IL-10 (10 ng/ml), or LPS and IL-10 for 8 h. GFP expression was measured by flow cytometry. For each sample, 20,000 events/cells were collected. Cells falling within the GFP-positive gate are shown as histogram plots. All results are representative of three independent experiments.
with LPS alone, there was a modest increase in RNA Pol II binding to the TNIP3 promoter, consistent with the slight increase in gene expression that can be observed in response to LPS (compare Figure 1(a) and Figure 1(d)). Notably, while stimulation with IL-10 alone had no effect, the cytokine was able to synergize with LPS to potentiate RNA Pol II recruitment (Figure 1(d)). In comparison, LPS and IL-10 had no effect on the binding of RNA Pol II to the GAPDH promoter. Second, a reporter gene assay was used to determine whether the TNIP3 promoter could drive expression of a heterologous gene akin to the endogenous TNIP3 gene. THP-1 cells were generated with a stably integrated reporter gene cassette consisting of a 2.5 kb human TNIP3 promoter fragment driving expression of GFP. When stimulated with LPS alone, GFP expression increased slightly over that of untreated (media) cells (Figure 1(e)). IL-10 stimulation alone did not affect GFP expression. However, when cells were stimulated with IL-10 and LPS in combination, GFP expression was increased above the level observed with LPS alone in a manner similar to the super-induction observed for endogenous TNIP3 gene expression. Collectively, the results support that IL-10 synergizes with LPS/TLR signaling to increase TNIP3 expression at a transcriptional level.
3.2. IL-10 Super-Induced Genes Display Differential Requirements for de novo Protein Synthesis
Previously, from experiments using mouse macrophages, TNIP3 induction in response to IL-10 and LPS was shown to be dependent on de novo protein synthesis, thus identifying it as a secondary response gene (i.e. it was not induced in the presence of the translation inhibitor cycloheximide) [43] . In contrast, SOCS3 exhibited the characteristics of a primary response gene whose induction was unaffected by cycloheximide. Those results suggest that a difference exists in the mechanism by which IL-10 super-induces the transcription of the two genes. In the present study, we addressed whether the same mechanistic division apparent between SOCS3 and TNIP
3 in
the mouse system also applies to the human system. Indeed, when THP-1 cells were cultured in the presence of cycloheximide, TNIP3 induction in response to LPS and IL-10 was markedly inhibited, while that of SOCS3 was not (Figure 2(a)).
In light of the mechanistic division between them, we hypothesized that SOCS3 and TNIP3 would be prototypical members of groups of primary response and secondary response genes induced synergistically by IL-10 and LPS. As our focal point had begun with the study of TNIP3 expression, we screened known IL-10-induced genes to identify those that are similarly super-induced by IL
-10 in
a cycloheximide-sensitive manner. We selected candidate genes from reports of IL-10-induced genes identified using different gene microarray platforms or PCR-based cloning approaches [40] -[45] . In total, 70 candidate genes were examined to determine if they could be super-induced by IL-10. Those genes meeting this condition were then differentiated based on whether their super-induction was sensitive to cycloheximide. To do this, THP-1 cells were stimulated with LPS, IL-10, or LPS and IL
-10 in
combination for 0, 2, 6, and 12 h in the absence or presence of cycloheximide. Total RNA was isolated and analyzed by RT-qPCR. The end-results of the candidate gene screening are presented in Figure 2 (cycloheximide-sensitivity) and Figure 3 (IL-10 super-induction). In addition to TNIP3 and SOCS3, we found 13 genes that could be super-induced by IL
-10 in
LPS-triggered THP-1 cells: BATF, CD163, CHI
3L
2, CXCL13, DUSP1, GJA1, IL1RN, IL7R, PTX3, RGS16, S
100A
8, S
100A
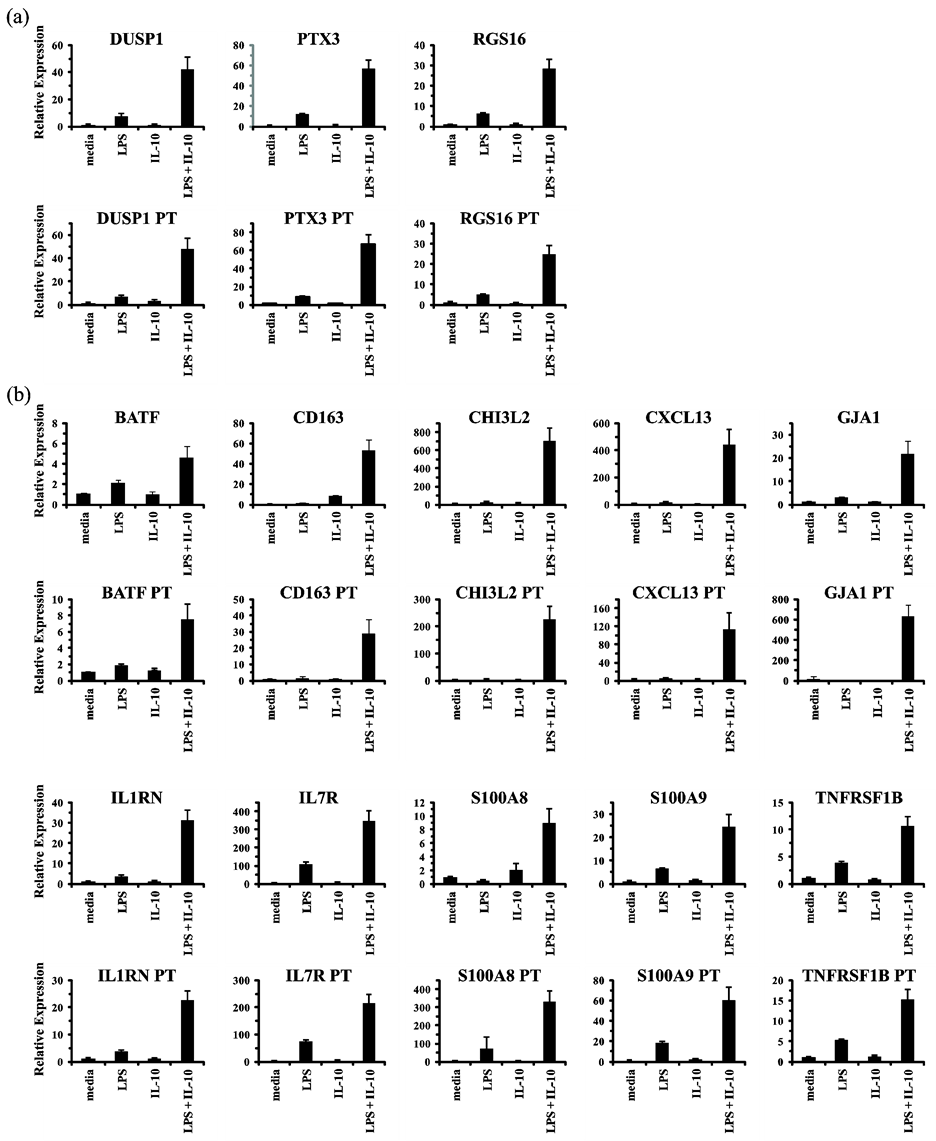
9, and TNFRSF1B. Of those 13 genes, 3 were found to be primary response genes (like SOCS3) whose induction was not inhibited by cycloheximide (Figure 2(b)), and the remaining 10 were found to be secondary response genes (like TNIP3) (Figure 2(c)). Therefore, the inducible regulation characteristic of TNIP3 is, in fact, representative of a group of IL-10 super-induced genes that are co-expressed in a delayed-response manner.
3.3. IL-10 Super-Induced Genes Are Universally Regulated through Enhancement of Gene Transcription
With the expanded group in hand, we addressed to what extent the genes were like TNIP
3 in
being regulated by IL-10 at the level of augmented transcription rate. THP-1 cells were stimulated with LPS, IL-10, or LPS and IL
-10 in
combination for 0, 2, 6, and 12 h. Total RNA was isolated and analyzed for primary transcript and mature transcript levels by RT-qPCR (Figure 3). For each of the newly identified IL-10 super-induced genes, regardless of whether a primary response gene (Figure 3(a)) or a secondary response gene (Figure 3(b)), IL-10 acted in concert with LPS signaling to potentiate an increase in primary transcript levels in a manner that mirrored the induction of their mature mRNA expression. These results indicate that IL-10 induces expression of TNIP3
![]()
Figure 2. Characterization of IL-10 super-induced primary response and secondary response genes. (a) THP-1 cells were mock treated (media) or treated with LPS (100 ng/ml) and IL-10 (10 ng/ml) for 6 h in either the absence or presence of cycloheximide (CHX; 10 μg/ml). Total RNA was isolated and analyzed by RT-qPCR in duplicate. Relative expression values are presented as mean ± s.d. of duplicate reactions after normalization to the three reference genes: GAPDH, HPRT, and RPL
13A
. (b) As described for panel (a), but showing the results for the additional genes identified as primary response genes. (c) As described for panel (a), but showing the results for the additional genes identified as secondary response genes. Additionally, RNA samples used for analyzing S
100A
8 and S
100A
9 were isolated after 12 h of stimulation instead of 6 h. All results are representative of three independent experiments.
and a cohort of genes in LPS-triggered cells universally by augmenting their transcription rate.
3.4. IL-10 Induces Anti-Inflammatory
Gene
Expression in LPS-Triggered Human Monocytes/Macrophages as a Cascade Involving Three Temporal Groups
The division of IL-10 super-induced genes into the two groups of primary response and secondary response genes indicates that a single mechanism does not underlie the synergy between IL-10 and LPS for all of the observed gene induction. Working towards elucidating these mechanisms, we next characterized the genes by analyzing their temporal expression patterns in IL-10 and LPS treated THP-1 cells. In accordance with being primary response genes whose induction is expected to depend solely on latent factors present in cells, DUSP1, PTX3, RGS16, and SOCS3 displayed the most rapid increases in expression, observable by 1 h after IL-10 and LPS stimulation (Figure 4(a)). Although expression of each primary response gene peaked and declined with slightly distinct kinetic profiles, they were generally induced rapidly (by 1 h), began to decline after 6 h, and had returned to near basal levels by 24 h post-stimulation. In comparison, the secondary response genes displayed more delayed induction kinetics, generally not being detectable until after 2 h of stimulation (Figure 4(b) and (c)). Interestingly, the group of secondary response genes appeared to further segregate into two subsets based on their temporal expression patterns. One subset, including BATF, IL1RN, IL7R, TNFRSF1B, and TNIP3,
![]()
Figure 3. Expression of IL-10 super-induced primary response and secondary response genes universally depends on synergistic enhancement of transcription rate. (a) THP-1 cells were mock treated (media) or treated with LPS (100 ng/ml), IL-10 (10 ng/ml), or LPS and IL-10 for 6 h. Total RNA was isolated and analyzed by conventional or primary transcript (PT) RT-qPCR in duplicate. Relative expression values are presented as mean ± s.d. of duplicate reactions after normalization to the three reference genes: GAPDH, HPRT, and 18S rRNA. The results for the primary response genes are shown in panel (a). (b) As described for panel (a), but showing the results for the secondary response genes. Additionally, RNA samples used for analyzing GJA1, S
100A
8, and S
100A
9 were isolated after 12 h of stimulation instead of 6 h. All results are representative of three independent experiments.
![]()
Figure 4. Kinetic analysis of IL-10 super-induced gene expression in human monocytes/macrophages. (a) THP-1 cells were stimulated with LPS (100 ng/ml) and IL-10 (10 ng/ml) in combination for the indicated times. Total RNA was isolated and analyzed by RT-qPCR in duplicate. Relative expression values are presented as mean ± s.d. of duplicate reactions after normalization to the three reference genes: GAPDH, RPL
13A
, and 18S rRNA. The results for the primary response genes are shown in panel (a), while the results for the early and late secondary response genes are shown in panels (b) and (c), respectively. All results are representative of three independent experiments.
displayed induced expression initially between 2 - 4 h, peaked between 6 - 10 h, and were in decline by 24 h post- stimulation (Figure 4(b)). Whereas the second subset, including CD163, CHI
3L
2, CXCL13, GJA1, S
100A
8, and S
100A
9, generally showed a more delayed initial increase in expression between 4 - 6 h, and a later peak at 12 - 24 h without going into decline (Figure 4(c)). Hereafter, we will refer to these two respective subsets as “early” and “late” secondary response genes. These results expand on the concept that IL-10 induces a temporal cascade of gene expression specifically in TLR-triggered macrophages. We hypothesize that a commonality should exist in the mechanism controlling the super-induction of the primary response genes, and that this mechanism would be, in part, distinct from those controlling the expression of the secondary response genes. Furthermore, a difference is expected to exist between the control of the early and late secondary response genes resulting in their distinct kinetic profiles.
3.5. The Three-Tiered Modality of IL-10 Super-Induced Gene Transcription Is Conserved in Primary Mouse Macrophages
We addressed whether the temporal gene expression pattern observed using the human THP-1 cell line model was both characteristic of primary cells and conserved in the mouse system. Mouse bone marrow-derived macrophages (BMM) were treated with LPS alone (in the presence of an anti-IL-10 receptor neutralizing antibody to block autocrine effects of IL-10), IL-10 alone, or LPS and IL
-10 in
combination for 0, 2, 4, and 9 h. Total RNA was isolated and analyzed by RT-qPCR. Of the 15 genes characterized in the human cell system, 8 were found to be conserved with respect to being super-induced by IL
-10 in
LPS-triggered mouse macrophages (Figure 5(a)). The conserved genes included 2 of the primary response genes, namely DUSP1 and SOCS3, and 6 of the secondary response genes, namely BATF, IL1RN, TNIP3, CD163, GJA1, and S
100A
8. Next, we examined whether the genes displayed the three-wave temporal expression pattern in response to IL-10 and LPS as observed
![]()
Figure 5. Identification and kinetic analysis of IL-10 super-induced genes in primary mouse macrophages. (a) BMM were mock treated (media) or treated with LPS (100 ng/ml) in the presence of a neutralizing anti-IL-10R1 mAb (0.5 μg/ml), or IL-10 (10 ng/ml), or LPS and IL
-10 in
combination for 2 h (in the case of DUSP1, SOCS3), 4 h (in the case of BATF, IL1RN, TNIP3), or 9 h (in the case of CD163, GJA1, S
100A
8). Total RNA was isolated and analyzed by RT-qPCR in duplicate. Relative expression values are presented as mean ± s.d. of duplicate reactions after normalization to the three reference genes: HPRT, RPL
13A
, and PPIA. (b) BMM were treated with LPS (100 ng/ml) and IL-10 (10 ng/ml) in combination for the indicated times. Total RNA was isolated and analyzed by RT-qPCR as described for panel (a). All results are representative of three independent experiments.
in THP-1 cells. In BMM responding to IL-10 and LPS, the primary response genes, DUSP1 and SOCS3, were rapidly and strongly induced by 1 h, and then began to decline between 4 - 9 h (Figure 5(b)). Expression of each of the early secondary response genes, BATF, IL-1RN, and TNIP3, became detectable by 1 h, but peaked later than the primary response genes at a time between 2 - 4 h, and then returned to near basal levels by 16 - 24 h. The late secondary response genes, CD163, GJA1, and S
100A
8, displayed an even more delayed initial expression between 2 - 6 h, peaked between 6 - 12 h, and largely maintained their expression levels out to 24 h post-stimulation. Collectively, the results support the notion that IL-10 and TLR signals synergize for induced transcription of a set of anti-inflammatory genes expressed in coordinated and temporally distinct groups in both human and mouse macrophages.
3.6. Selective Requirements for PI3K and JNK in the Temporally Differential Expression of IL-10 Super-Induced
Genes
As an initial step into exploring the signaling mechanisms responsible for the observed temporal expression pattern of IL-10 super-induced genes, we tested the effects of inhibiting major protein kinases involved in the IL-10 and TLR signal transduction pathways [53] - [55] . In accordance with the obligate role JAK1 plays in IL
-10’
s activation of STAT3, cells stimulated with the combination of IL-10 and LPS in the presence of a JAK inhibitor displayed marked inhibition of each of the 8 conserved IL-10 super-induced genes (Figure 6). Inhibition of PI3K (phoshatidylinositol 3-kinase) led to a differential effect wherein the induction of primary response genes (DUSP1 and SOCS3) was not affected, while the induction of the secondary response genes, both early and late, was markedly inhibited. Inhibition of the JNK MAP kinase pathway resulted in strong suppression of late secondary response gene expression (CD163, GJA1, and S
100A
8), while having only a partial effect on the early secondary response genes: i.e. partially inhibiting IL1RN and TNIP3 but having no inhibitory effect on BATF. Like the early secondary response genes, the primary response genes were induced in a largely JNK-indepen- dent manner. Requirement for the other two MAP kinase pathways, p38 MAPK and ERK, did not appear to selectively coordinate or differentiate between the three temporal groups. While the inhibition of p38 MAPK strongly impaired DUSP1 induction, it only partially inhibited that of the other primary response gene SOCS3. Likewise, while two of the early secondary response genes (BATF and ILIRN) were partially suppressed by inhibiting p38, the expression of the third (TNIP3) was unaltered. The three late secondary response genes showed a strong dependency on p38, just as they had each shown for JNK. A dependency on ERK signaling also did not clearly differentiate the temporal groups. While inhibition of ERK resulted in strong inhibition of DUSP1 induction, it had no affect on SOCS3. Likewise, the early secondary response gene IL1RN showed a partial requirement for ERK signaling, whereas BATF and TNIP3 displayed no such requirement. The late secondary response genes showed the greatest dependency on MAP kinase signaling in general, displaying requirements for the JNK, p38, and ERK pathways. Collectively, the results indicate that a PI3K-dependent pathway is required for IL-10 and TLR signals to synergistically induce the secondary response genes, but not the primary response genes. And, a strong requirement for JNK signaling appears to distinguish the induction of the late secondary response genes from the early secondary response genes.
4. Discussion
In this study, we began to characterize the mechanism whereby the gene TNIP3 becomes inducible by IL-10 specifically in TLR-triggered macrophages. Since IL-10 is known to regulate gene expression through both transcriptional and post-transcriptional mechanisms, we first addressed at which of these levels the cytokine was promoting TNIP3expression. The results revealed that IL-10 induces TNIP3 through a transcriptional mechanism rather than one involving a post-transcriptional increase in mRNA stability. IL-10 potentiated TNIP3 transcription in TLR-triggered macrophages, but not in resting macrophages. Results from the reporter gene assay indicated that the cis-acting elements required for the TNIP3 gene to respond to IL-10 and LPS stimulation reside within 2.5 kb upstream of the transcriptional start site.
In comparing the mechanisms controlling the transcription of TNIP3 with another IL-10 super-induced gene, SOCS3, a difference was confirmed to exist: whereas SOCS3 is a primary response gene, TNIP3 is a secondary response gene whose induction by IL-10 is dependent on de novo protein synthesis. In an effort to assess the biological significance of IL-10 controlling TNIP3 expression in this way, we extended our comparative study to include more IL-10 super-induced genes. Of 70 candidate genes examined, 13 additional genes were identified that
![]()
Figure 6. Selective requirements for PI3K and JNK in IL-10 super-induced secondary response gene expression. THP-1 cells were mock treated (media) or stimulated with LPS (100 ng/ml) and IL-10 (10 ng/ml) in combination for 6 h. Each stimulation type was also performed in the presence of DMSO (vehicle) or a pharmacological inhibitor: JAK Inhibitor 1 (10 μM), PI3K inhibitor LY294002 (20 μM), JNK-
1/2/3
inhibitor SP600125 (25 μM), p38 MAPK inhibitor SB203580 (10 μM), or ERK-1/2 inhibitor FR180204 (50 μM). Total RNA was isolated and analyzed by RT-qPCR in duplicate. Relative expression values were determined as mean ± s.d. of duplicate reactions after normalization to the two reference genes: GAPDH and HPRT. Results are presented as percent inhibition relative to the maximum gene expression level induced by LPS and IL
-10 in
the presence of DMSO vehicle. Data bars are shaded and grouped in the order of primary response genes (PRG), early secondary response genes (SRG), and late SRGs. The results are representative of two independent experiments.
could be super-induced by IL
-10 in
LPS-triggered THP-1 cells. Several reasons might explain the relatively low number of positive genes identified from the candidate screen. First, the 70 candidates were selected from reports identifying IL-10-induced genes using different microarray platforms and varied cellular systems, including human monocytes, human dendritic cells, and mouse macrophages [40] - [45] . So, it is possible that a number of the reported genes are regulated in a cell type- or species-specific manner, and therefore are not IL-10-in- duced in the human monocytic cell line THP-1. Additionally, since THP-1 is an immortalized cell line, some of the genes and their regulatory mechanisms may be altered. Lastly, some of the fold inductions reported from the microarray experiments may simply be too low to be detected reproducibly by RT-qPCR. Nonetheless, 10 genes were identified that could be grouped with TNIP3 as IL-10 super-induced secondary response genes, and 3 additional genes were identified that could be grouped with SOCS3 as primary response genes. Mechanistically, IL-10 acted on all of the genes by increasing their transcription rates in LPS/TLR-triggered cells. Thus, the mechanisms underlying synergistic transcription in response to IL-10 and LPS are not limited to the TNIP3 gene, but instead appear to extend to a group of co-regulated genes. An examination of the kinetic expression profiles for the IL-10 super-induced genes revealed that the group can be further subdivided from two into three discrete temporal modules: chronologically, the primary response genes are the first to be activated, and they are followed by two sequential subsets of secondary response genes. Mechanistic differences would be expected to exist between the three temporal modules in terms of how the genes gain competency for transcriptional triggering in response to IL-10 and LPS.
Based on a growing body of research into the synergistic and temporal control of inducible gene expression, particularly in related systems of cellular responses to TLRs and cytokines, we can postulate on mechanisms at work in this system of positive crosstalk between IL-10 and LPS/TLR signaling [56] - [65] . First, with regard to the transcriptional synergy observed between IL-10 and TLR signaling, one potential mechanism explaining the induction of the primary response genes could be the direct and cooperative actions of TLR-activated NF-κB/ AP-1 along with IL-10-activated STAT3 binding simultaneously to enhancers within a gene’s regulatory region. There is evidence to support this notion in the case of SOCS3, which has had both types of enhancers characterized within its regulatory region [66] - [68] . During the course of our work, another group studying the synergistic induction of IL-1RN in response to IL-10 and LPS reported that the mechanism involves interdependent binding of both STAT3 and NF-κB to the IL-1RN promoter [69] . STAT3 binding was shown to facilitate an enhanced binding of NF-κB and a subsequent increase in histone acetylation at the IL-1RN promoter. It remains to be determined whether this mechanism also applies to TNIP3 and the other early secondary response genes, of which we identified IL-1RN as a member. However, although TNIP3 has had an NF-κB site characterized near its proximal promoter consistent with its being an LPS-induced gene, a functional STAT3 binding site has not been characterized to date [22] [43] [70] . Furthermore, as secondary response genes, induction of TNIP3 and IL-1RN might be expected to involve the induced expression of a primary response gene which would act in addition to latent transcription factors such as STAT3. Alternatively, a reverse of the mechanism described for IL-1RN might be at work, wherein the LPS-activated NF-κB could confer competence for binding and full transcriptional activity to the IL-10-activated STAT3 (or an unknown IL-10-activated factor). This alternative mechanism would better fit a gene like TNIP3, since it can be induced somewhat by LPS alone but not at all by IL-10 alone. Lastly, the example of synergistic induction of the NOS2 gene in response to crosstalk between IFN-α/β and PRR signaling offers another plausible mechanism. In that case, concerted gene expression occurs through IFN-activated ISGF3 mediating the recruitment of RNA Polymerase II to the NOS2 promoter, which only becomes competent for transcriptional elongation after PRR-activated NF-κB recruits and stabilizes TFIIH [58] .
With regard to possible mechanisms controlling the temporal nature of the IL-10 super-induced genes, we hypothesize that a common thread exists among the primary response genes, which distinguishes them from the secondary response genes in terms of changes that must occur at the gene promoters to allow for an increase in transcription rate. Furthermore, some common mechanistic thread should exist among the early secondary response genes distinguishing their kinetic profiles from the late secondary response genes. Several seminal studies of LPS/TLR-induced gene expression in macrophages have revealed the existence of an analogous temporal transcriptional cascade comprised of early primary response genes, late primary response genes, and secondary response genes [56] [57] [60] - [62] . The LPS-induced early primary response genes were shown to commonly possess promoters containing CpG-islands, which are characterized by unstable nucleosome architecture and open chromatin configuration. In contrast, the late primary response genes and secondary response genes lacked this promoter architecture and required the action of specific chromatin remodeling factors for their stimulus-dependent gene activation [62] [63] . Whether this is a defining feature distinguishing the temporal subsets of IL-10 super-induced genes remains to be tested. Interestingly, our preliminary bioinformatic scan of proximal promoters (−250 to +250 relative to transcription start site) of the IL-10 super-induced genes revealed that 3 out of 4 of the primary response genes possess CpG-island containing promoters (the exception being PTX3), and 10 out of 11 of the secondary response genes lack CpG-islands (the exception being TNFRSF1B). A second mechanism contributing to the temporal gene expression observed for LPS/TLR-induced genes is one wherein the primary response genes have constitutive RNA Pol II binding and transcriptional initiation occurring in the absence of stimulation, but require the stimulus-dependent recruitment of P-TEFb to their promoters in order for transcriptional elongation and RNA splicing to proceed [61] . In contrast, the LPS-induced secondary response genes do not display stimulus-independent constitutive transcriptional initiation, and instead require a more time consuming active recruitment of RNA Pol II to their promoters in response to LPS stimulation. Whether this discriminating temporal feature applies to the IL-10 super-induced primary versus secondary response genes also remains to be tested. Lastly, although all of the IL-10 super-induced genes examined were potentiated by IL-10 through transcriptional up-regulation, mechanisms affecting their mRNA stabilities and/or RNA splicing kinetics may possibly contribute to their distinctive temporal expression profiles. Both mechanisms have been shown to effect the temporal gene expression patterns observed in cells responding to either LPS or TNF [60] [64] [65] . It will be important to resolve the exact interplay between the aforementioned transcriptional and post-transcriptional processes in shaping the temporal gene expression cascade observed in the synergistic response to IL-10 and TLR signaling.
Defining the mechanisms controlling the IL-10 super-induced transcriptional response should be facilitated by examining conserved elements between orthologous genes. For this reason, we extended our characterization of the IL-10 super-induced transcriptional response to include primary mouse macrophages. Several of the genes we characterized using human monocytic cells were also found to be super-induced by IL
-10 in
LPS-stimulated mouse macrophages. Importantly, the three-wave temporal gene expression pattern also appears to be conserved, consisting of primary response genes (DUSP1, SOCS3), early secondary response genes (BATF, ILIRN, TNIP3), and late secondary response genes (CD163, GJA1, S
100A
8). Comparative bioinformatics combined with promoter assays may help in the identification of enhancers and transcription factors being targeted by IL-10 signaling and acting in synergy with TLR signals for the induction of genes in each temporal group.
Elucidating the regulatory mechanisms should prove valuable in determining the physiological role of IL-10 super-induced genes as a whole (versus the IL-10-induced genes), and for each of the temporal gene modules in the host anti-inflammatory response. For example, if a specific factor responsible for induction of the secondary response genes could be identified, genetic ablation or inhibition of this factor in mice could be used to assess the consequences of lacking expression of these delayed-response genes while keeping primary response gene expression intact during inflammatory responses. The results from experiments using pharmacological protein kinase inhibitors lend support to this prospect and suggest that certain receptor-proximal signaling pathways are differentially involved in the coordinated expression of the different temporal modules. For instance, the induction of the IL-10 super-induced secondary response genes, but not the primary response genes, was dependent on PI3K signaling. Moreover, the early and late secondary response genes appeared to be differentiated based on their dependencies on the MAP kinases, particularly JNK signaling. The JNK pathway appeared to be selectively required to sustain the transcriptional response for the late secondary response genes. It will be important to confirm these findings in experiments employing genetic ablation techniques, and to extend them by linking the PI3K and JNK pathways to specific molecular events taking place at the promoters of genes in the respective temporal transcriptional modules.
From a functional perspective, one can inquire as to why the IL-10 anti-inflammatory response might occur in such a temporal transcriptional cascade. Indeed, some general trends can be observed when considering the functions of the genes by temporal group. It appears that the earliest induced genes (i.e. primary response) are markedly represented by proteins with roles in regulating signal transduction. Both SOCS3 and DUSP1 are known for their important roles as negative regulators of cytokine- and TLR-signaling pathways [71] - [73] . Likewise, as a regulator of G protein-signaling, RGS16 may be involved in modulating macrophage activation, motility, and phagocytosis [74] [75] . With regard to the next wave of gene expression, the early secondary response genes tend to play roles in either regulating signal transduction or suppressing specific inflammatory cytokine response pathways. As a negative regulator of NF-κB signaling, TNIP3 may be involved in the termination of cellular TLR, TNF, and IL-1 responses [43] [76] . As a transcriptional regulator, BATF may be involved in regulating some of the later gene expression in the temporal cascade [20] [77] . With more selective anti-inflammatory effects, IL1RN (also known as IL-1ra) is known to be important in limiting the actions of IL-1 that are initiated by the TLR response [78] . Also with a selective action, TNFRSF1B (also known as TNFR2) can compete with and lessen the pro-inflammatory effects of TNFR1, thereby dampening the cellular responses to TNF [79] [80] . The role IL7R would play in macrophages is less obvious, but its increased expression by lymphocytes may be involved in promoting the effects of IL-10 on lymphocyte activation [16] [81] [82] . As for the most delayed wave of gene expression, the late secondary response genes appear to play lesser roles in regulating inflammatory signal transduction and act more so as secreted effectors modulating late events in the inflammatory response such as lymphocyte activation, resolution of inflammation, and wound healing. As a chemokine for B cells, CXCL13 has been proposed to be part of IL
-10’
s effect in potentiating B cell responses [42] . Expression of the scavenger receptor CD163 appears to play an anti-inflammatory function in the resolution of inflammation allowing for would healing to commence [83] [84] . Likewise, GJA1 (also known as Cnx43), a protein component of gap junctions, may be involved in propagating the anti-inflammatory response and in the promotion of wound healing [85] [86] . S
100A
8 and S
100A
9 have multiple known roles in modulating inflammation, including acting as leukocyte chemoattractants and scavengers of free radicals, thought to be important in the latter stages of the inflammatory response to promote wound healing [87] [88] . On the whole, it appears that the grouping of the genes into their distinct temporal modules does have an underlying basis rooted in functionality relevant to the timing of the anti-inflammatory response.
5. Conclusion
In conclusion, the results presented here reveal that IL-10 induces a multipart temporal cascade of gene expression selectively in TLR-stimulated macrophages. The concerted IL-10 and TLR signals collaborate to induce this targeted gene expression at the level of increased transcription rate. The synergistic induction of transcription occurs in a regulated sequence to coordinate expression of genes in distinct temporal modules. This signaling system may have evolved as a way to impart specific anti-inflammatory actions of IL
-10 in
the appropriate situation and at the proper time during immune responses. This study should provide a groundwork for future research aimed at elucidating the molecular mechanisms governing this synergistic and temporal transcriptional response and determining its physiological role in the anti-inflammatory response.
Acknowledgements
We thank all of the past and present members of the Weaver laboratory for helpful discussions in the design and execution of experiments. We also thank
John
Steiert
,
Paula
Steiert
, and
Courtney
Coleman
for their advice and critical reading of the manuscript. This work was supported by funds awarded by the Biology Department and the Graduate College of Missouri State University and the Missouri State University Foundation.
References
- Martinez,
F.O.
, Sica, A., Mantovani, A. and Locati, M. (2008) Macrophage Activation and Polarization. Frontiers in Bioscience, 13, 453-461. http://dx.doi.org/10.2741/2692
- Martinez,
F.O.
, Helming, L. and
Gordon
, S. (2009) Alternative Activation of Macrophages: An Immunologic Functional Perspective. Annual Review of Immunology, 27, 451-483. http://dx.doi.org/10.1146/annurev.immunol.021908.132532
- Medzhitov, R. (2008) Origin and Physiological Roles of Inflammation. Nature, 454, 428-435. http://dx.doi.org/10.1038/nature07201
- Serbina,
N.V.
, Jia, T., Hohl,
T.M.
and Pamer, E.G. (2008) Monocyte-Mediated Defense against Microbial Pathogens. Annual Review of Immunology, 26, 421-452. http://dx.doi.org/10.1146/annurev.immunol.26.021607.090326
-
Broz
, P. and Monack, D.M. (2013) Newly Described Pattern Recognition Receptors Team up against Intracellular Pathogens. Nature Reviews Immunology, 13, 551-565. http://dx.doi.org/10.1038/nri3479
- Onoguchi, K., Yoneyama, M. and Fujita, T. (2011) Retinoic Acid-Inducible Gene-I-Like Receptors. Journal of Interferon & Cytokine Research, 31, 27-31. http://dx.doi.org/10.1089/jir.2010.0057
- Palm, N.W. and Medzhitov, R. (2009) Pattern Recognition Receptors and Control of Adaptive Immunity. Immunological Reviews, 227, 221-233. http://dx.doi.org/10.1111/j.1600-065X.2008.00731.x
- Takeuchi, O. and
Akira
, S. (2010) Pattern Recognition Receptors and Inflammation. Cell, 140, 805-820. http://dx.doi.org/10.1016/j.cell.2010.01.022
- Arpaia, N. and
Barton
, G.M. (2011) Toll-Like Receptors: Key Players in Antiviral Immunity. Current Opinion in Virology, 1, 447-454. http://dx.doi.org/10.1016/j.coviro.2011.10.006
- Barton, G.M. and Medzhitov, R. (2002) Toll-Like Receptors and Their Ligands. Current Topics in Microbiology and Immunology, 270, 81-92. http://dx.doi.org/10.1007/978-3-642-59430-4_5
- Blasius,
A.L.
and Beutler, B. (2010) Intracellular Toll-Like Receptors. Immunity, 32, 305-315. http://dx.doi.org/10.1016/j.immuni.2010.03.012
- Kawai, T. and
Akira
, S. (2010) The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-Like Receptors. Nature Immunology, 11, 373-384. http://dx.doi.org/10.1038/ni.1863
-
O’Neill
, L.A. and
Bowie
,
A.G.
(2007) The Family of Five: TIR-Domain-Containing Adaptors in Toll-Like Receptor Signalling. Nature Reviews Immunology, 7, 353-364. http://dx.doi.org/10.1038/nri2079
- Takeda, K. and
Akira
, S. (2005) Toll-Like Receptors in Innate Immunity. International Immunology, 17, 1-14. http://dx.doi.org/10.1093/intimm/dxh186
-
Yamamoto
, M. and
Akira
, S. (2005) TIR Domain-Containing Adaptors Regulate TLR Signaling Pathways. Advances in Experimental Medicine and Biology, 560, 1-9. http://dx.doi.org/10.1007/0-387-24180-9_1
-
Moore
,
K.W.
,
de Waal
Malefyt, R., Coffman,
R.L.
and
O’Garra
, A. (2001) Interleukin-10 and the Interleukin-10 Receptor. Annual Review of Immunology, 19, 683-765. http://dx.doi.org/10.1146/annurev.immunol.19.1.683
- Sabat, R., Grutz, G., Warszawska, K., Kirsch, S.,
Witte
, E., Wolk, K. and Geginat, J. (2010) Biology of Interleukin-10. Cytokine & Growth Factor Reviews, 21, 331-344. http://dx.doi.org/10.1016/j.cytogfr.2010.09.002
- Saraiva, M. and
O’Garra
, A. (2010) The Regulation of IL-10 Production by Immune Cells. Nature Reviews Immunology, 10, 170-181. http://dx.doi.org/10.1038/nri2711
-
Murray
, P.J. (2006) Understanding and Exploiting the Endogenous Interleukin-10/STAT3-Mediated Anti-Inflamma- tory Response. Current Opinion in Pharmacology, 6, 379-386. http://dx.doi.org/10.1016/j.coph.2006.01.010
- Grutz, G. (2005) New Insights into the Molecular Mechanism of Interleukin-10-Mediated Immunosuppression. Journal of Leukocyte Biology, 77, 3-15.
-
Williams
,
L.M.
, Ricchetti, G., Sarma, U., Smallie, T. and Foxwell, B.M. (2004) Interleukin-10 Suppression of Myeloid Cell Activation—A Continuing Puzzle. Immunology, 113, 281-292. http://dx.doi.org/10.1111/j.1365-2567.2004.01988.x
-
Hutchins
,
A.P.
, Diez, D. and Miranda-Saavedra, D. (2013) The IL-10/STAT3-Mediated Anti-Inflammatory Response: Recent Developments and Future Challenges. Briefings in Functional Genomics, 12, 489-498. http://dx.doi.org/10.1093/bfgp/elt028
- Smallie, T., Ricchetti, G., Horwood, N.J., Feldmann, M.,
Clark
,
A.R.
and
Williams
,
L.M.
(2010) IL-10 Inhibits Transcription Elongation of the Human TNF
Gene
in Primary Macrophages. Journal of Experimental Medicine, 207, 2081- 2088. http://dx.doi.org/10.1084/jem.20100414
-
Murray
,
P.J.
(2005) The Primary Mechanism of the IL-10-Regulated Antiinflammatory Response Is to Selectively Inhibit Transcription. Proceedings of the National Academy of Sciences of the United States of America, 102, 8686-8691. http://dx.doi.org/10.1073/pnas.0500419102
- Biswas, R., Datta, S.,
Gupta
,
J.D.
, Novotny, M., Tebo, J. and
Hamilton
, T.A. (2003) Regulation of Chemokine mRNA Stability by Lipopolysaccharide and IL-10. Journal of Immunology, 170, 6202-6208. http://dx.doi.org/10.4049/jimmunol.170.12.6202
- Kontoyiannis, D., Kotlyarov, A., Carballo, E., Alexopoulou, L., Blackshear,
P.J.
, Gaestel, M.,
Davis
, R., Flavell, R. and Kollias, G. (2001) Interleukin-10 Targets p38 MAPK to Modulate ARE-Dependent TNF mRNA Translation and Limit Intestinal Pathology. EMBO Journal, 20, 3760-3770. http://dx.doi.org/10.1093/emboj/20.14.3760
-
Kim
,
H.S.
,
Armstrong
, D.,
Hamilton
,
T.A.
and Tebo,
J.M.
(1998) IL-10 Suppresses LPS-Induced KC mRNA Expression via a Translation-Dependent Decrease in mRNA Stability. Journal of Leukocyte Biology, 64, 33-39.
- Correa, I., Veny, M., Esteller, M., Pique,
J.M.
, Yague, J., Panes, J. and Salas, A. (2009) Defective IL-10 Production in Severe Phenotypes of Crohn’s Disease. Journal of Leukocyte Biology, 85, 896-903. http://dx.doi.org/10.1189/jlb.1108698
- Glocker,
E.O.
, Kotlarz, D.,
Klein
, C., Shah, N. and Grimbacher, B. (2011) IL-10 and IL-10 Receptor Defects in Humans. Annals of the New York Academy of Sciences, 1246, 102-107. http://dx.doi.org/10.1111/j.1749-6632.2011.06339.x
- Parkes, M., Satsangi, J. and
Jewell
, D. (1998) Contribution of the IL-2 and IL-10
Genes
to Inflammatory Bowel Disease (IBD) Susceptibility. Clinical and Experimental Immunology, 113, 28-32. http://dx.doi.org/10.1046/j.1365-2249.1998.00625.x
- Shah, N., Kammermeier, J., Elawad, M. and Glocker, E.O. (2012) Interleukin-10 and Interleukin-10-Receptor Defects in Inflammatory Bowel Disease. Current Allergy & Asthma Reports, 12, 373-379. http://dx.doi.org/10.1007/s11882-012-0286-z
-
Kuhn
, R., Lohler, J., Rennick, D., Rajewsky, K. and Muller, W. (1993) Interleukin-10-Deficient Mice Develop Chronic Enterocolitis. Cell, 75, 263-274. http://dx.doi.org/10.1016/0092-8674(93)80068-P
- Spencer, S.D.,
Di Marco
, F., Hooley, J., Pitts-Meek, S., Bauer, M.,
Ryan
, A.M., Sordat, B.,
Gibbs
,
V.C.
and Aguet, M. (1998) The Orphan Receptor CRF2-4 Is an Essential Subunit of the Interleukin 10 Receptor. Journal of Experimental Medicine, 187, 571-578. http://dx.doi.org/10.1084/jem.187.4.571
- Pils,
M.C.
,
Pisano
, F., Fasnacht, N., Heinrich,
J.M.
, Groebe, L., Schippers, A., Rozell, B.,
Jack
,
R.S.
and Muller, W. (2010) Monocytes/Macrophages and/or Neutrophils Are the Target of IL
-10 in
the LPS Endotoxemia Model. European Journal of Immunology, 40, 443-448. http://dx.doi.org/10.1002/eji.200939592
- Takeda, K., Clausen,
B.E.
, Kaisho, T., Tsujimura, T., Terada, N.,
Forster
, I. and
Akira
, S. (1999) Enhanced Th1 Activity and Development of Chronic Enterocolitis in Mice Devoid of Stat
3 in
Macrophages and Neutrophils. Immunity, 10, 39-49. http://dx.doi.org/10.1016/S1074-7613(00)80005-9
- Welte, T., Zhang, S.S., Wang, T., Zhang, Z., Hesslein, D.G., Yin, Z., Kano, A., Iwamoto, Y., Li, E., Craft, J.E., Bothwell, A.L., Fikrig, E., Koni, P.A., Flavell, R.A. and Fu, X.Y. (2003) STAT3 Deletion during Hematopoiesis Causes Crohn’s Disease-Like Pathogenesis and Lethality: A Critical Role of STAT
3 in
Innate Immunity. Proceedings of the National Academy of Sciences of the United States of America, 100, 1879-1884. http://dx.doi.org/10.1073/pnas.0237137100
- Pestka, S., Krause,
C.D.
, Sarkar, D.,
Walter
,
M.R.
, Shi, Y. and Fisher, P.B. (2004) Interleukin-10 and Related Cytokines and Receptors. Annual Review of Immunology, 22, 929-979. http://dx.doi.org/10.1146/annurev.immunol.22.012703.104622
-
Donnelly
,
R.P.
, Dickensheets, H. and Finbloom,
D.S.
(1999) The Interleukin-10 Signal Transduction Pathway and Regulation of
Gene
Expression in Mononuclear Phagocytes. Journal of Interferon & Cytokine Research, 19, 563-573. http://dx.doi.org/10.1089/107999099313695
- Weber-Nordt,
R.M.
,
Riley
,
J.K.
, Greenlund,
A.C.
,
Moore
,
K.W.
, Darnell,
J.E.
and Schreiber, R.D. (1996) Stat3 Recruitment by Two Distinct Ligand-Induced, Tyrosine-Phosphorylated Docking Sites in the Interleukin-10 Receptor Intracellular Domain. Journal of Biological Chemistry, 271, 27954-27961. http://dx.doi.org/10.1074/jbc.271.44.27954
- Jung, M., Sabat, R., Kratzschmar, J., Seidel, H., Wolk, K., Schonbein, C., Schutt, S., Friedrich, M., Docke, W.D., Asadullah, K., Volk, H.D. and Grutz, G. (2004) Expression Profiling of IL-10-Regulated Genes in Human Monocytes and Peripheral Blood Mononuclear Cells from Psoriatic Patients during IL-10 Therapy. European Journal of Immunology, 34, 481-493. http://dx.doi.org/10.1002/eji.200324323
-
Lang
, R., Patel, D.,
Morris
,
J.J.
, Rutschman,
R.L.
and
Murray
, P.J. (2002) Shaping Gene Expression in Activated and Resting Primary Macrophages by IL-10. Journal of Immunology, 169, 2253-2263. http://dx.doi.org/10.4049/jimmunol.169.5.2253
- Perrier, P., Martinez,
F.O.
, Locati, M., Bianchi, G., Nebuloni, M., Vago, G., Bazzoni, F., Sozzani, S., Allavena, P. and Mantovani, A. (2004) Distinct Transcriptional Programs Activated by Interleukin-10 with or without Lipopolysaccharide in Dendritic Cells: Induction of the B Cell-Activating Chemokine, CXC Chemokine Ligand 13. Journal of Immunology, 172, 7031-7042. http://dx.doi.org/10.4049/jimmunol.172.11.7031
- Weaver,
B.K.
, Bohn, E., Judd,
B.A.
,
Gil
,
M.P.
and Schreiber, R.D. (2007) ABIN-3: A Molecular Basis for Species Divergence in Interleukin-10-Induced Anti-Inflammatory Actions. Molecular and Cellular Biology, 27, 4603-4616. http://dx.doi.org/10.1128/MCB.00223-07
-
Williams
, L., Jarai, G., Smith, A. and Finan, P. (2002) IL-10 Expression Profiling in Human Monocytes. Journal of Leukocyte Biology, 72, 800-809.
- Antoniv,
T.T.
, Park-Min,
K.H.
and Ivashkiv, L.B. (2005) Kinetics of IL-10-Induced
Gene
Expression in Human Macrophages. Immunobiology, 210, 87-95. http://dx.doi.org/10.1016/j.imbio.2005.05.003
-
El Kasmi
,
K.C.
, Smith, A.M.,
Williams
, L., Neale, G., Panopoulos, A.D., Watowich,
S.S.
, Hacker, H., Foxwell,
B.M.
and
Murray
,
P.J.
(2007) Cutting Edge: A Transcriptional Repressor and Corepressor Induced by the STAT3-Regulated Anti-Inflammatory Signaling Pathway. Journal of Immunology, 179, 7215-7219. http://dx.doi.org/10.4049/jimmunol.179.11.7215
-
Hutchins
,
A.P.
, Poulain, S. and Miranda-Saavedra, D. (2012) Genome-Wide Analysis of STAT3 Binding in Vivo Predicts Effectors of the anti-Inflammatory Response in Macrophages. Blood, 119, e110-119. http://dx.doi.org/10.1182/blood-2011-09-381483
- Smith, A.M., Qualls,
J.E.
,
O’Brien
, K., Balouzian, L.,
Johnson
,
P.F.
, Schultz-Cherry, S., Smale,
S.T.
and
Murray
,
P.J.
(2011) A Distal Enhancer in Il12b Is the Target of Transcriptional Repression by the STAT3 Pathway and Requires the Basic Leucine Zipper (B-ZIP) Protein NFIL3. Journal of Biological Chemistry, 286, 23582-23590. http://dx.doi.org/10.1074/jbc.M111.249235
- Kuwata, H., Watanabe, Y., Miyoshi, H.,
Yamamoto
, M., Kaisho, T., Takeda, K. and
Akira
, S. (2003) IL-10-Inducible Bcl-3 Negatively Regulates LPS-Induced TNF-Alpha Production in Macrophages. Blood, 102, 4123-4129. http://dx.doi.org/10.1182/blood-2003-04-1228
- Celada, A.,
Gray
,
P.W.
, Rinderknecht, E. and Schreiber, R.D. (1984) Evidence for a Gamma-Interferon Receptor That Regulates Macrophage Tumoricidal Activity. Journal of Experimental Medicine, 160, 55-74. http://dx.doi.org/10.1084/jem.160.1.55
-
Lois
, C., Hong,
E.J.
, Pease, S., Brown,
E.J.
and
Baltimore
, D. (2002) Germline Transmission and Tissue-Specific Expression of Transgenes Delivered by Lentiviral Vectors. Science, 295, 868-872. http://dx.doi.org/10.1126/science.1067081
-
Zhou
, L., Nazarian,
A.A.
and Smale, S.T. (2004) Interleukin-10 Inhibits Interleukin-12 p40 Gene Transcription by Targeting a Late Event in the Activation Pathway. Molecular and Cellular Biology, 24, 2385-2396. http://dx.doi.org/10.1128/MCB.24.6.2385-2396.2004
- Antoniv,
T.T.
and Ivashkiv, L.B. (2011) Interleukin-10-Induced
Gene
Expression and Suppressive Function Are Selectively Modulated by the PI3K-Akt-GSK3 Pathway. Immunology, 132, 567-577. http://dx.doi.org/10.1111/j.1365-2567.2010.03402.x
-
Arthur
,
J.S.
and Ley, S.C. (2013) Mitogen-Activated Protein Kinases in Innate Immunity. Nature Reviews Immunology, 13, 679-692. http://dx.doi.org/10.1038/nri3495
- Troutman, T.D., Bazan, J.F. and Pasare, C. (2012) Toll-Like Receptors, Signaling Adapters and Regulation of the Pro- Inflammatory Response by PI3K. Cell Cycle, 11, 3559-3567. http://dx.doi.org/10.4161/cc.21572
- Medzhitov, R. and Horng, T. (2009) Transcriptional Control of the Inflammatory Response. Nature Reviews Immunology, 9, 692-703. http://dx.doi.org/10.1038/nri2634
- Smale, S.T. (2012) Transcriptional Regulation in the Innate Immune System. Current Opinion in Immunology, 24, 51- 57. http://dx.doi.org/10.1016/j.coi.2011.12.008
- Farlik, M., Reutterer, B., Schindler, C., Greten, F., Vogl, C., Muller, M. and Decker, T. (2010) Nonconventional Initiation Complex Assembly by STAT and NF-kappaB Transcription Factors Regulates Nitric Oxide Synthase Expression. Immunity, 33, 25-34. http://dx.doi.org/10.1016/j.immuni.2010.07.001
- Qiao, Y., Giannopoulou, E.G., Chan,
C.H.
, Park,
S.H.
, Gong, S.,
Chen
, J., Hu, X., Elemento, O. and Ivashkiv, L.B. (2013) Synergistic Activation of Inflammatory
Cytokine
Genes
by Interferon-Gamma-Induced Chromatin Remodeling and Toll-Like Receptor Signaling. Immunity, 39, 454-469. http://dx.doi.org/10.1016/j.immuni.2013.08.009
- Hao, S. and
Baltimore
, D. (2009) The Stability of mRNA Influences the Temporal Order of the Induction of
Genes
Encoding Inflammatory Molecules. Nature Immunology, 10, 281-288. http://dx.doi.org/10.1038/ni.1699
- Hargreaves, D.C., Horng, T. and Medzhitov, R. (2009) Control of Inducible
Gene
Expression by Signal-Dependent Transcriptional Elongation. Cell, 138, 129-145. http://dx.doi.org/10.1016/j.cell.2009.05.047
- Ramirez-Carrozzi,
V.R.
, Braas, D., Bhatt,
D.M.
, Cheng,
C.S.
, Hong, C., Doty,
K.R.
, Black,
J.C.
,
Hoffmann
, A.,
Carey
, M. and Smale,
S.T.
(2009) A Unifying Model for the Selective Regulation of Inducible Transcription by CpG Islands and Nucleosome Remodeling. Cell, 138, 114-128. http://dx.doi.org/10.1016/j.cell.2009.04.020
- Ramirez-Carrozzi,
V.R.
, Nazarian,
A.A.
, Li,
C.C.
, Gore,
S.L.
, Sridharan, R., Imbalzano,
A.N.
and Smale, S.T. (2006) Selective and Antagonistic Functions of SWI/SNF and Mi-2beta Nucleosome Remodeling Complexes during an Inflammatory Response. Genes & Development, 20, 282-296. http://dx.doi.org/10.1101/gad.1383206
- Hao, S. and
Baltimore
, D. (2013) RNA Splicing Regulates the Temporal Order of TNF-Induced
Gene
Expression. Proceedings of the National Academy of Sciences of the United States of America, 110, 11934-11939. http://dx.doi.org/10.1073/pnas.1309990110
- Pandya-Jones, A., Bhatt,
D.M.
,
Lin
,
C.H.
, Tong,
A.J.
, Smale,
S.T.
and Black, D.L. (2013) Splicing Kinetics and Transcript Release from the Chromatin Compartment Limit the Rate of Lipid A-Induced
Gene
Expression. RNA, 19, 811- 827. http://dx.doi.org/10.1261/rna.039081.113
- Auernhammer,
C.J.
, Bousquet, C. and Melmed, S. (1999) Autoregulation of Pituitary Corticotroph SOCS-3 Expression: Characterization of the Murine SOCS-3 Promoter. Proceedings of the National Academy of Sciences of the United States of America, 96, 6964-6969. http://dx.doi.org/10.1073/pnas.96.12.6964
- Ehret,
G.B.
, Reichenbach, P., Schindler, U., Horvath,
C.M.
,
Fritz
, S., Nabholz, M. and Bucher, P. (2001) DNA Binding Specificity of Different STAT Proteins. Comparison of in Vitro Specificity with Natural Target Sites. Journal of Biological Chemistry, 276, 6675-6688. http://dx.doi.org/10.1074/jbc.M001748200
-
Qin
, H., Roberts, K.L., Niyongere, S.A., Cong, Y., Elson,
C.O.
and Benveniste, E.N. (2007) Molecular Mechanism of Lipopolysaccharide-Induced SOCS-3
Gene
Expression in Macrophages and Microglia. Journal of Immunology, 179, 5966-5976. http://dx.doi.org/10.4049/jimmunol.179.9.5966
- Tamassia, N., Castellucci, M., Rossato, M., Gasperini, S., Bosisio, D., Giacomelli, M., Badolato, R., Cassatella, M.A. and Bazzoni, F. (2010) Uncovering an IL-10-Dependent NF-{kappa}B Recruitment to the IL-1ra Promoter That Is Impaired in STAT3 Functionally Defective Patients. FASEB Journal, 24, 1365-1375. http://dx.doi.org/10.1096/fj.09-145573
- Verstrepen, L., Adib-Conquy, M., Kreike, M., Carpentier, I., Adrie, C., Cavaillon, J. and Beyeart, R. (2008) Expression of the NF-κB Inhibitor ABIN
-3 in
Response to TNF and Toll Like Receptor 4 Is Itself Regulated by NF-κB. Journal of Cell and Molecular Medicine, 12, 316-329. http://dx.doi.org/10.1111/j.1582-4934.2007.00187.x
- Kubo, M., Hanada, T. and Yoshimura, A. (2003) Suppressors of Cytokine Signaling and Immunity. Nature Immunology, 4, 1169-1176. http://dx.doi.org/10.1038/ni1012
-
Alexander
,
W.S.
and
Hilton
,
D.J.
(2004) The Role of Suppressors of Cytokine Signaling (SOCS) Proteins in Regulation of the Immune Response. Annual Review of Immunology, 22, 503-529. http://dx.doi.org/10.1146/annurev.immunol.22.091003.090312
- Liu, Y., Shepherd, E.G. and Nelin, L.D. (2007) MAPK Phosphatases—Regulating the Immune Response. Nature Reviews Immunology, 7, 202-212. http://dx.doi.org/10.1038/nri2035
-
Shankar
,
S.P.
,
Wilson
, M.S., DiVietro,
J.A.
, Mentink-Kane,
M.M.
, Xie, Z.,
Wynn
,
T.A.
and Druey,
K.M.
(2012) RGS16 Attenuates Pulmonary Th2/Th17 Inflammatory Responses. Journal of Immunology, 188, 6347-6356. http://dx.doi.org/10.4049/jimmunol.1103781
- Shi, G.X., Harrison, K., Han, S.B., Moratz, C. and Kehrl, J.H. (2004) Toll-Like Receptor Signaling Alters the Expression of Regulator of G Protein Signaling Proteins in Dendritic Cells: Implications for G Protein-Coupled Receptor Signaling. Journal of Immunology, 172, 5175-5184. http://dx.doi.org/10.4049/jimmunol.172.9.5175
- Wullaert, A., Verstrepen, L.,
Van Huffel
, S., Adib-Conquy, M., Cornelis, S., Kreike, M., Haegman, M.,
El Bakkouri
, K., Sanders, M., Verhelst, K., Carpentier, I., Cavaillon,
J.M.
, Heyninck, K. and Beyaert, R. (2007) LIND/ABIN-3 Is a Novel Lipopolysaccharide-Inducible Inhibitor of NF-kappaB Activation. Journal of Biological Chemistry, 282, 81-90. http://dx.doi.org/10.1074/jbc.M607481200
- Echlin,
D.R.
, Tae,
H.J.
, Mitin, N. and Taparowsky, E.J. (2000) B-ATF Functions as a Negative Regulator of AP-1 Mediated Transcription and Blocks Cellular Transformation by Ras and Fos. Oncogene, 19, 1752-1763. http://dx.doi.org/10.1038/sj.onc.1203491
- Dinarello, C.A. (2011) A Clinical Perspective of IL-1beta as the Gatekeeper of Inflammation. European Journal of Immunology, 41, 1203-1217. http://dx.doi.org/10.1002/eji.201141550
- Erickson, S.L., de Sauvage, F.J., Kikly, K., Carver-Moore, K., Pitts-Meek, S., Gillett, N., Sheehan, K.C., Schreiber, R.D., Goeddel, D.V. and Moore, M.W. (1994) Decreased Sensitivity to Tumour-Necrosis Factor but Normal T-Cell Development in TNF Receptor-2-Deficient Mice. Nature, 372, 560-563. http://dx.doi.org/10.1038/372560a0
- Peschon,
J.J.
, Torrance,
D.S.
, Stocking,
K.L.
, Glaccum,
M.B.
, Otten, C.,
Willis
,
C.R.
, Charrier, K., Morrissey, P.J., Ware, C.B. and Mohler, K.M. (1998) TNF Receptor-Deficient Mice Reveal Divergent Roles for p55 and p
75 in
Several Models of Inflammation. Journal of Immunology, 160, 943-952.
- Corfe, S.A. and
Paige
,
C.J.
(2012) The Many Roles of IL
-7 in
B Cell Development; Mediator of Survival, Proliferation and Differentiation. Seminars in Immunology, 24, 198-208. http://dx.doi.org/10.1016/j.smim.2012.02.001
- Huang,
H.Y.
and Luther, S.A. (2012) Expression and Function of Interleukin
-7 in
Secondary and Tertiary Lymphoid Organs. Seminars in Immunology, 24, 175-189. http://dx.doi.org/10.1016/j.smim.2012.02.008
- Etzerodt, A. and Moestrup, S.K. (2013) CD163 and Inflammation: Biological, Diagnostic, and Therapeutic Aspects. Antioxidants & Redox Signaling, 18, 2352-2363. http://dx.doi.org/10.1089/ars.2012.4834
- Fabriek,
B.O.
, Dijkstra,
C.D.
and
van den Berg
, T.K. (2005) The Macrophage Scavenger Receptor CD163. Immunobiology, 210, 153-160. http://dx.doi.org/10.1016/j.imbio.2005.05.010
- Chanson, M., Derouette,
J.P.
,
Roth
, I., Foglia, B., Scerri, I., Dudez, T. and Kwak,
B.R.
(2005) Gap Junctional Communication in Tissue Inflammation and Repair. Biochimica et Biophysica Acta, 1711, 197-207. http://dx.doi.org/10.1016/j.bbamem.2004.10.005
- Churko,
J.M.
and Laird, D.W. (2013) Gap Junction Remodeling in Skin Repair Following Wounding and Disease. Physiology (Bethesda), 28, 190-198. http://dx.doi.org/10.1152/physiol.00058.2012
- Averill,
M.M.
, Kerkhoff, C. and Bornfeldt, K.E. (2012) S
100A
8 and S
100A
9 in
Cardiovascular Biology and Disease. Arteriosclerosis, Thrombosis, and Vascular Biology, 32, 223-229. http://dx.doi.org/10.1161/ATVBAHA.111.236927
- Perera, C., McNeil,
H.P.
and Geczy, C.L. (2010) S100 Calgranulins in Inflammatory Arthritis. Immunology & Cell Biology, 88, 41-49. http://dx.doi.org/10.1038/icb.2009.88
NOTES

*These authors contributed equally to this work.
#Corresponding author.