World Journal of Neuroscience
Vol.3 No.1(2013), Article ID:27904,10 pages DOI:10.4236/wjns.2013.31005
Pain and memory: Do they share similar mechanisms?
![]()
Department of Neurophysiology, Ivane Beritashvili Experimental Biomedicine Center, Tbilisi, Georgia
Email: tsagareli@biphysiol.ge
Received 26 November 2012; revised 25 December 2012; accepted 3 January 2013
Keywords: Allodynia; Analgesia; Hyperalgesia; Nociception; Synaptic Plasticity; Long-Term Potentiation
ABSTRACT
Pain receptors, nociceptors inputs to the spinal cord and supra spinal structures triggering a prolonged but reversible increase in the excitability and synaptic efficacy of neurons in central nociceptive pathways, is the phenomenon of central sensitization. Key processes for pain memory stabilizing could be considering processes of peripheral and central sensitizations. Mechanical hypersensitivity and allodynia to light touch after central sensitization are pathologic in that they are evoked by Aβ low threshold mechanoreceptors, which normally do not produce painful sensations. Peripheral sensitization allows low-intensity stimuli to produce pain by activating Aδ and C nociceptors whereas central sensitization allows normal lowthreshold Aβ mechanoreceptors to produce pain as a result of changes in sensory processing in the spinal cord. During peripheral and central sensitization, the receptive fields of dorsal horn neurons expand beyond the site of injury into surrounding non-injured tissue. The clinical result of all above changes is hyperalgesia, allodynia, spontaneous pain, referred pain and sympathetically maintained pain. Therefore, these persistent sensory responses to noxious stimuli are a form of memory, the memory for pain. Long lasting synaptic plasticity as the long-term potentialtion at spinal and supra-spinal levels could undergo hyperalgesia and allodynia. The latter could be providing neuronal basis for persistent pain and pain memory. Thus, it will be particularly important to know how to regulate long-lasting plastic changes in spinal cord, thalamus and cortex. Molecular mechanisms of these plastic processes could be main targets for new therapeutic drugs in pain relief.
1. INTRODUCTION
Each of us remembers pain experience in case of illness. Anyone who has ever got electric shock, broken a leg or an arm, or have had a kidney colic remembers it all his life span and never forgets it.
Since 30 years ago, the prevailing view on pain processing in the central nervous system (CNS) was of a largely passive neural relay that conveyed by encoded action potentials, information on the onset, duration, intensity, location and quality of peripheral noxious stimuli, from one site to another. More specifically, the CNS pathway was seen to constitute particular anatomical connections in the spinal cord, brain stem, thalamus and cortex (the “pain pathway”), linking the sensory inflow generated in high threshold primary afferents with those parts of the cortex that leads to the conscious awareness of painful sensations [1].
One of the crucial cellular processes for major symptoms of pain as hyperalgesia and allodynia is considered as a phenomenon of sensitization. Post-injury sensitization of the nervous system has evolved long before vertebrates. Therefore, phylogenetic memories consist of normal withdrawal/avoidance reflexes. Many of neural mechanisms mobilized in vertebrates’ nervous system by painful stimuli are also present in neurons of mollusks. These same mechanisms (ion channels, second messengers, gene expression, etc.) also mediate hyperresponsiveness of the mollusk to an electrical shock delivered at the site of a previous shock a day before. Mollusks and vertebrates evolved from a common ancestor (probably a small worm) 600 million years ago. Therefore, augmentation of responses to successive nociceptive stimuli has been a biological truth for at least this time [2].
2. SENSITIZATION AND SYNAPTIC PLASTICITY IN THE SEA MOLLUSK APLYSIA
More than 35 years ago, Kandel and colleagues offered the simplest model for neuronal and molecular bases of memorization in marine snail Aplysia [3]. They have described a sensitization of the gill withdrawal reflex, which was associated with a facilitation of the synapse between sensory and motor neurons. Six gill motor neurons of the abdominal ganglion are participated in the gill-withdrawal reflex (Figure 1(A)). If its siphon is touched, Aplysia will briskly and reflexively withdraw both its siphon and respiratory organ, the gill. Give the animal a noxious electrical shock to the tail it exhibits an even more powerful gill withdrawal reflex and also withdraws its tail as it prepares to escape. Now if we give the animal painful electrical shock to the tail and few minutes later a weak touch with a fine paintbrush to the siphon, Aplysia will show a much more powerful gill withdrawal as well as a tail withdrawal, again preparatory to escape [4]. The simple neural circuit is responseble for sensitization of this gill-withdrawal reflex (Figure 1(B)). A noxious stimulus to the tail activates tail sensory neurons that excite modulating interneurons. The latter from its part activates siphon sensory and/or modulating neurons result in facilitation of the motor neuron and a much more powerful escape reaction. This is a simplest example of neuronal sensitization and its duration is a function of the number of tail shocks. If we give one, the animal will remember that stimulus for a few minutes, a normal fear reaction. But then if the noxious stimulus is given repeatedl—say five times—however, it produces, a chronic anxiety state that will last for days. This memory or pain state has a completely different molecular basis for short-term plasticity processes (facilitation, habituation); it requires the some gene expression and the synthesis of new proteins [4].
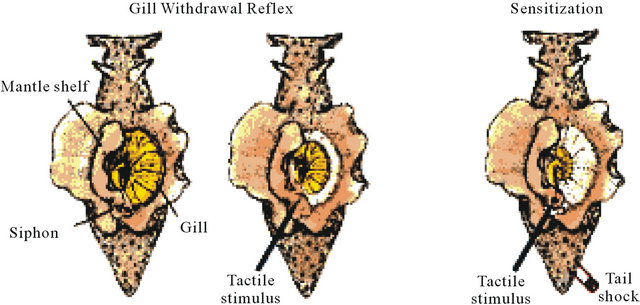
(A)
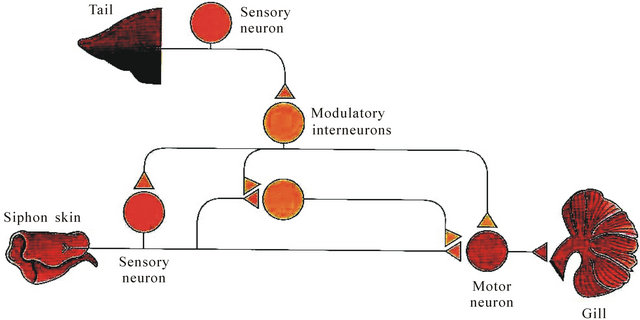
(B)
Figure 1. (A) A dorsal view of Aplysia showing the gill, the animal’s respiratory organ. The gill lies in the mantle cavity and is normally covered partially by the mantle shelf. A light touch to the siphon with a fine paintbrush (left) causes the siphon to contract and the gill to withdraw under the protection of the mantle shelf. Here, the mantle shelf is shown to retracted for a better view of the gill. Sensitization of the gill-withdrawal reflex, produced by applying a noxious stimulus to another part of the body, such as tail, leads to an enhancement of the withdrawal reflex of both siphon and the gill (right); (B) The neural circuit for sensitization of the gill-withdrawal reflex in Aplysia. A noxious stimulus to the tail activates tail sensory neurons that excite modulating interneurons. Their signals to the sensory neurons of the siphon enhance transmitter release (single neurons are shown for simplicity). Modified from [4].
3. PERIPHERAL AND CENTRAL SENSITIZATIONS IN MAMMALIAN NERVOUS SYSTEM
Neuronal sensitization described by Kandel [4] is probably similar to the phenomena of central hyperalgesia and mechanical allodynia in human pain states. Usually, hyperalgesia means an increased response to a stimulus, which is normally painful while allodynia means pain due to a stimulus that does not normally provoke pain.
Key processes for pain memory stabilizing are peripheral and central sensitizations. Peripheral sensitization is induced by neuro-humoral alterations at the site of injury and nearby. Biochemicals released by tissue injury (prostaglandins, bradykinin, histamine, serotonin, etc.) excite pain receptors (nociceptors) and/or increase their sensitivity at the injured site (primary hyperalgesia). These pro-inflammatory agents sensitize adjacent Aδ and C nociceptors (secondary hyperalgesia). Increased transduction produces continuous nociceptive input that can induce primary and secondary hyperalgesia and allodynia.
Central sensitization takes place within the dorsal horn of spinal cord, the brain stem, and the cerebral cortex.
Amplification of nociceptive inputs in the spinal cord produces secondary hyperalgesia around the site of injury. Mechanical hypersensitivity and allodynia to light touch after central sensitization are pathologic in that they are evoked by Aβ low threshold mechanoreceptors, which normally do not produce painful sensations. Peripheral sensitization allows low-intensity stimuli to produce pain by activating Aδ and C nociceptors whereas central sensitization allows normal low-threshold Aβ mechanoreceptors to produce pain as result of changes in sensory processing in the spinal cord. Figures 2 and 3 summarize sensory processing under normal circumstances and the changes that result from induction of central sensitization.
Central sensitization was concerned instead, with the facilitation that manifested after the end of the conditioning stimuli, and that once triggered remained autonomous for some time, or only required a very low level of nociceptor input to sustain it. Furthermore, central sensitization represented a condition where input in one set of nociceptor sensory fibers (the conditioning input) amplified subsequent responses to other non-stimulated nonnociceptor or nociceptor fibers (the test input). This form
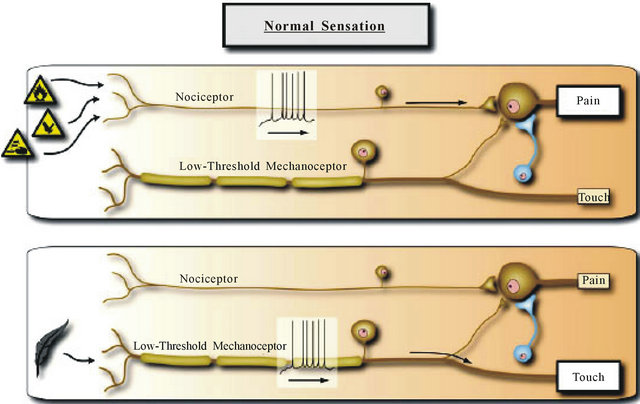
Figure 2. Normal sensation. The somatosensory system is organized such that the highly specialized primary sensory neurons that encode low intensity stimuli only activate those central pathways that lead to innocuous sensations, while high intensity stimuli that activate nociceptors only activate the central pathways that lead to pain and the two parallel pathways do not functionally intersect. This is mediated by the strong synaptic inputs between the particular sensory inputs and pathways and inhibitory neurons that focus activity to these dedicated circuits. Reproduced from [1].
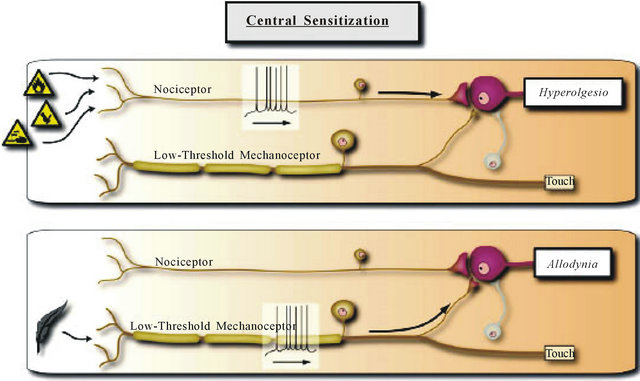
Figure 3. Central sensitization. With the induction of central sensitization in somatosensory pathways with increases in synaptic efficacy and reductions in inhibition, a central amplification occurs enhancing the pain response to noxious stimuli in amplitude, duration and spatial extent, while the strengthening of normally ineffective synapses recruits subliminal inputs such that inputs in low threshold sensory inputs can now activate the pain circuit. The two parallel sensory pathways converge. Reproduced from [1].
of facilitation is termed heterosynaptic potentiation to distinguish it from homosynaptic potentiation where the test and conditioning input are the same [5].
During peripheral and central sensitization, the recaptive fields of dorsal horn neurons expand beyond the site of injury into surrounding non-injured tissue. The clinical result of all above changes is hyperalgesia, allodynia, spontaneous pain, referred pain and sympathetically maintained pain. Therefore, these persistent sensory responses to noxious stimuli are a form of memory, the memory for pain.
4. THE CENTRAL SENSITIZATION AND LONG-TERM POTENTIATION
It is one of interesting questions of this topic—is central sensitization of nociceptive transmission in pain pathways an example of long-term potentiation (LTP)? Analysis of the molecular mechanisms underlying the generation and maintenance of central sensitization and LTP indicates that, although there are differences between the synaptic plasticity contributing to memory and pain, there are also striking similarities.
The LTP has been most intensively studied in hippocampus where it is considered a fundamental cellular model of learning and memory formation [6-8]. The role of LTP in baso-lateral amygdala as a simplified form of memory in fear has been also established [9-12]. The LTP has also been shown in pain pathways at synapses between primary afferent Aδ- or C-fibers and spinal dorsal horn neurons and is one of the proposed cellular mechanisms of pain amplification [13,14]. The potential consequences of the LTP in pain pathways are manifold: 1) after the LTP is established, supra-threshold excitatory input will now evoke stronger excitation of nociceptive neurons. Thus, the LTP at nociceptive synapses may underlie hyperalgesia; 2) previously sub-threshold excitatory input, from the subliminal fringe of a neurons’ receptive field may now elicit action potential firing. This may widen painful areas and contribute to hyperalgesia as well; and 3) some spinal dorsal horn neurons receive sub-threshold input from somatotopically inappropriate body areas, for example, the contralateral body half, the LTP anywhere along this subliminal input path may cause mirror image pain or radiating pain [13].
In superficial spinal lamina I, the LTP is selectively induced in a group of nociceptive specific neurons which express the neurokinin 1 (NK1) receptor for substance P and which project to the brain [13]. This group of neurons plays a key role in hyperalgesia following inflamemation and nerve injury. In spinal cord, the LTP can be induced in different ways, for example, by electrical nerve stimulation, by nerve injury, or peripheral noxious events. Stimulation within the innervation territory of a sensory nerve, for example, with chemical irritants (Figures 4(A) and (B)), or by inflammation may also trigger the LTP at C-fiber synapses. All conditioning stimuli tested so far which induce the LTP at Aδ-synapses and/ or C-fiber synapses may also cause hyperalgesia in experimental animals. The LTP of C-fiber-evoked spinal field potentials can also be induced pharmacologically. In spinalized rats, topical application of N-methyl-D-aspartate (NMDA), substance P or neurokinin A induces

Figure 4. Mean time courses of C-fiber-evoked field potentials recorded extracellulary in superficial spinal dorsal horn in response to electrical stimulation of left sciatic nerve of deeply anaesthetized adult rats with spinal cords and afferent nerves intact (A) and (B). Subcutaneous injections of transient receptor potential vanilloid 1 channel agonist capsaicin (1%, 100 ml, n = 5, (A) or formalin (5%, 100 ml, n = 6); (B) Into the glabrous skin at the ipsilateral hind paw, within the innervation territory of the sciatic nerve at time zero (arrows) induced LTP (closed circles), while injections of the respective solvents (open circles) had no effects (n = 3 in each group). Conditioning electrical low frequency stimulation (LFS) (2 Hz, 2 min at C-fiber intensity) of sciatic nerve at time zero (arrow) also induced LTP (n = 28); (C) which was prevented by NMDA receptor antagonist MK-801 (3 mg∙kg−1, i.v.-infusion over 30 min: horizontal bar, n = 5); (D) A second conditioning LFS four hours later (arrow) was partially effective in inducing LTP. Nitric oxide synthase (NOS) inhibitor L-NMMA (100 mg∙kg−1∙h−1, i.v., horizontal bar, n = 5); (E) Also blocked LTP induction. This block was fully reversible as shown by a second LFS 3 h later (arrow). Similarly, LFS of dorsal roots at C-fiber strength induces robust LTP at synapses between Cfibre afferents and lamina I neurons with a projection to the midbrain periaqueductal gray (PAG) in a rat spinal cord-dorsal root preparation (n = 18); (F) Reproduced from [13].
the LTP [13].
It is very interesting that the LTP of C-fiber-evoked field potentials in vivo is not affected by deep (surgical) level of anesthesia with urethane, isoflurane, or sevoflurane in mature rats. But the LTP is, however, prevented by low-dose intravenous infusion of μ-opioid receptor agonist fentanyl [13].
In addition, Sandkühler has studied the LTP in pain pathways in vitro slices [13]. He has induced the LTP by natural, low-frequency afferent barrage evoked by inflammation of peripheral tissue and by low-frequency stimulation of dorsal root afferents (Figures 4(C)-(E)).
On the one hand, induction protocols signal transduction pathways and pharmacology of the LTP and some forms of hyperalgesia are virtually identical, rendering the LTP in spinal dorsal horn an attractive cellular model of hyperalgesia [13]. On the other hand, homosynaptic potentiation at synapses on dorsal horn neurons can occur experimentally in response to brief duration, highfrequency nociceptor stimulation. The potentiation is restricted to the activated synapse and is persistent. However nociceptors do not usually fire at high frequentcies, and therefore homosynaptic potentiation may be limited to very intense stimuli producing a spatially and modality constrained, if long lasting, facilitation. Because heterosynaptic potentiation can be initiated by low-frequency nociceptor inputs, it is most prominent feature of synaptic plasticity in the dorsal horn [14]. We can conclude, thus, as a number of studies provide convergent evidence that the LTP at spinal synapses of nociceptive Aδ- or C-fibers facilitates downstream processing of pain-related information. This includes action potential firing in superficial and deep dorsal horn neurons and pain perception in human subjects [13,15].
The second example of the LTP of sensory excitatory synapses in adult mouse spinal cord slices [16] is shown in the Figure 5.
5. SYNAPTIC PLASTICITY IN THE ANTERIOR CINGULATE CORTEX
For the last decade, the anterior cingulate cortex (ACC) is one of the most frequently activated regions in pain research [17-19]. Nociceptive responses are transmitted to cingulate cortex from the midline, mediodorsal and intralaminar thalamic nuclei (MITN). Several lines of evidence indicate that these nuclei provide the primary source of nociceptive information to the ACC [17-19]. The Figure 6 shows thalamo-cingulate responses during formalin injection to the hind paw of rats [17].
Short-term synaptic plasticity shapes the postsynaptic response to bursts of impulses and is crucial for plastic changes of central neurons after strong noxious stimulation. The hypothesis guiding studies of the thalamo-

Figure 5. (A) Diagram of a spinal slice showing the placement of a whole-cell patch clamp recording and a stimulation electrodes in the superficial dorsal horn of the adult mouse spinal cord; (B) Schematic illustrating the induction protocol consisting 80 pulses at 2 Hz while holding at +30 mV (paired training); (C) An example of spinal LTP induced by paired training protocol in a superficial dorsal horn neuron; (D) Summary result of the LTP experiments under control conditions (n = 9 neurons). The excitatory postsynaptic current (EPSC) responses were averaged over five-minute intervals. Reproduced from [16].
cingulate circuit is that the nociceptive mediodorsal and intralaminar thalamic nuclei input generates short-term plasticity in the ACC and may be a precursor to longerterm pain processing events. Thus, the specific shortterm facilitation of the thalamo-cingulate pathway will likely enable the enhancement of the transferring of the abnormal thalamic bursting activities to the cingulate cortex. These activities will result in a resonant interacttion between the thalamus and cingulate cortex and thus sustained nociceptive activities. Such short-term modifications in cortical synapses appear to cingulate afferent signals and this regulation is likely important to the transition from acute nociceptive stimulation to chronic pain conditions associated with persistent peripheral noxious stimulation [17].
Here, we have to emphasize that the short-term plasticity (STP) time scale is relatively short, about seconds and minutes, and thus it cannot produce entirely the processes underlying chronic pain conditions. It is crucial to note that the STP plays a transitional role in transferring the nociceptive signal mediating acute traumatic injury to the formation of long lasting changes in the ACC.
Wei and Zhuo [18] found very interesting data in an in vivo setting, that amputation of a central digit of the hindpaw caused long-lasting changes in the ACC (Figure 7). Because amputation caused local damage to skin as well as nerves innervating the digit, the performed amputation on the hindpaw contralateral to the one to which stimulation was delivered. Therefore, any changes
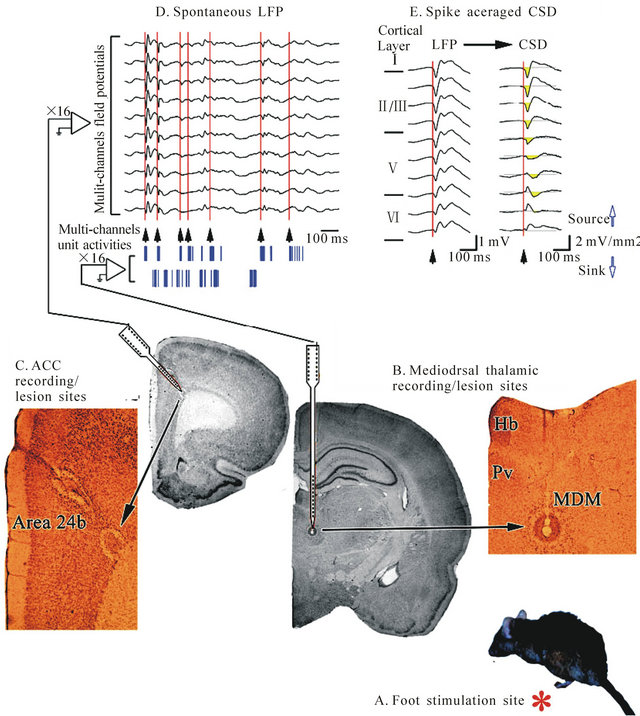
Figure 6. Thalamocingulate responses during formalin injection to the hind paw (A, red asterisk). Multichannel unit activities and local field potentials were recorded from the MITN (B) and ACC (C) respectively; (D) Aligned multichannel thalamic unit activities and ACC spontaneous local field potentials showing that bursting activity and local field potential in both structures is correlated; (E) The initial time of bursting (arrowheads aligned at the red lines) of MITN unit activities were aligned. Abbreviations: MITN, mediodorsal and intralaminar thalamic nuclei; MDM, mediodorsal thalamic nucleus; Pv, paraventricular thalamic nucleus; Hb, habenular nucleus. Reproduced from [17].
in synaptic responses were not simply due to the alteration of peripheral excitability caused by amputation. After amputation of a central digit of the hindpaw, they observed a rapid enhancement of sensory responses to peripheral electrical shocks delivered to the normal hindpaw. The potentiation was long lasting; evoked responses remained enhanced for at least 2 hours. In contrast, in animals receiving sham treatment, field excitatory postsynaptic potentials (EPSPs) were not significantly affected at 2 hours after the treatment and the enhancement observed in rats following amputation was significantly greater than that in sham treated animals [18].
These in vivo results, thus, provide direct evidence that excitatory synaptic transmission within the ACC undergo long-term potentiation after peripheral injury [18]. At the same time, LTP may not completely mimic synaptic mechanisms of persistent pain. These findings suggest that it may require different forms of the LTP to mimic the pathological mechanisms of persistent pain. It is thus easy to believe that different forms of memory are likely

Figure 7. Sensory response of the ACC to peripheral stimulation in adult rats. (A) diagram of in vivo recording from the ACC in an anaesthetized rat; animals were maintained in a lightly anaesthetized state by halothane. The recording electrode was placed into the ACC contralateral to the peripheral stimulation electrode. Amputation (the removal of the third digit of the hindpaw) was performed on the nonstimulated hindpaw. During amputation, a higher concentration of halothane was used; (B) representative traces of EPSPs 5 min before amputation (Pre) and 115 - 120 min after (Post) sham treatment (a) or amputation (b). In b, the latency of sensory responses was not changed after the amputation, while the EPSP slope was increased; (c), amputation of a single digit of the contra-lateral hindpaw (indicated by an arrow) caused long-lasting enhancement of sensory responses (closed circles). Sensory responses were not significantly changed in sham-treated animals (open circles). The test stimulation frequency was 0.01 Hz; (C) traces of synaptic responses to electrical stimulation applied to the hindpaw at a low intensity (5.0 V) and a higher intensity (25.0 V). An arrow indicates the time of hindpaw electrical stimulation. Modified from [18].
mediated by different mechanisms [16,19].
The short-term and long-term plastic changes in the ACC may facilitate the acute nociceptive activities to become persistent. Enhanced and maintained nociceptive activities may have an adverse effect in the cingulate cortex. Studies have shown that excessive activation of NMDA receptors, which are the most widely and densely distributed of the glutamate receptor subtypes in the cingulate cortex, plays an important role in the pathophysiology of acute CNS injury syndromes. Thus, it has been suggested to use NMDA antagonists in chronic conditions like neuropathic pain [16,19].
As we mentioned above, Sandkühler has described that in adult rats, conditioning low frequency stimulation (LFS) of sciatic nerve fibers at C-fiber intensity induced the LTP of spinal C-fiber-evoked field potentials [13]. He and colleagues has just recently found [20] that a brief intravenous infusion of a high dose of the ultrashort-acting μ-opioid receptor agonist remifentanil acutely depressed potentiated responses (Figure 8).
Furthermore, subcutaneous injections of capsaicin quickly led to mechanical hyperalgesia at the injected hindpaw. The same dosage regimen of remifentanil that caused opioid induces reduction or depotentiation significantly attenuated capsaicin induced hyperalgesia (Figure 9(A)). Protein phosphatase 1 (PP1) inhibitor calyculin a fully blocked the attenuation of hyperalgesia by remifentanil, suggesting that depotentiation at nociceptive C-fibers may erase a memory trace of pain (Figure 9(B)).
These findings demonstrate that activation of spinal μ-opioid receptor triggers distinct, bidirectional, and state dependent synaptic plasticity in naïve versus potentiated C-fiber synapses. The presently identified reversal of synaptic LTP in nociceptive pathways provides a rationale for novel therapeutic strategies to cure rather than temporarily dampen some forms of pain with opioids [20].

Figure 8. Acute opioid administration induces depotentiation of spinal LTP. Area of C-fiber-evoked field potentials was normalized to baseline values before LTP induction (dotted lines) and plotted versus time (min). Data are expressed as mean ± SEM. Insets show originnal traces of field potentials recorded at indicated time points; calibration bars indicate 50 ms and 0.2 mV. (A) Mean time course of LTP of C-fiber-evoked field potentials. LFS (time point zero, arrow) induced LTP in all animals tested (n = 13, P < 0.001); (B) Sixty min post-LFS, a high-dose remifentanil infusion (450 mg·kg−1·hour−1 intravenously (i.v.)) was started by bolus injection (30 mg·kg−1) and continuously infused for 1 hour (black horizontal bar). After washout of the drug, LTP was depotentiated (n = 25, P < 0.001). Modified from [20].
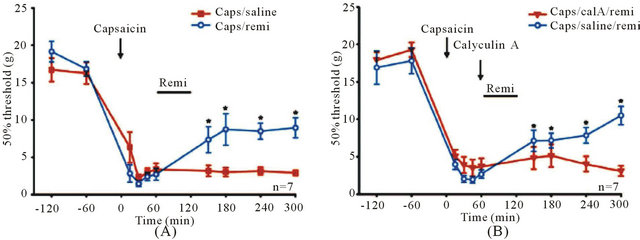
Figure 9. Capsaicin-induced mechanical hyperalgesia is reduced after opioid administration. Capsaicin injection (time point zero, arrow) significantly reduced mechanical withdrawal thresholds in the ipsilateral paw of awake, drug-free rats. (A) One group of animals received a 1-hour high dose remifentanil infusion (horizontal bar) 60 min after capsaicin injection (blue circles, n = 7). A control group was treated with an intravenous saline infusion (red squares, n = 7). After wash-out of the opioid, mechaniccal thresholds were elevated significantly compared with thresholds before opioid treatment at 150 min (P = 0.05), 180 min (P = 0.003), 240 min (P = 0.004), and 300 min (P = 0.002), indicating partial reversal of hyperalgesia; (B) Opioid-induced reduction of mechanical hyperalgesia is blocked by the PP1 inhibitor calyculin A. After capsaicin injection, calyculin A (300 nM, 10 ml) was injected intrathecally 10 min before a 1-hour high dose remifentanil infusion (red triangles, n = 7). Intrathecal injections of saline served as control (blue circles, n = 7). After wash out of the opioid, mechanical thresholds were elevated significantly compared with thresholds before opioid treatment in the control group at 150 min (P = 0.021), 180 min (P = 0.019), 240 min (P = 0.003), and 300 min (P < 0.001). No effect of the opioid treatment on mechanical thresholds could be observed in the calyculin A-treated group (P = 0.523). One-way repeated measure ANOVA or repeated measure ANOVA on ranks was used for statistical comparisons. Reproduced from [20].
6. CENTRAL SENSITIZATION IN EXPERIMENTAL AND CLINICAL PAIN STATES
The data obtained in human volunteer studies demonstrate that induction of use-dependent central facilitation in nociceptive central pathways increases pain sensitivity and, therefore, contribute to clinical pain syndromes. Experimental studies in human volunteers are necessarily restricted to use non-injurious conditioning inputs, and therefore are limited to studying only the activity-dependent components of pain hypersensitivity elicited by sensory inputs, and not those transcription dependent and structural changes that manifest after inflammation or nerve injury, which may have different mechanisms, time courses and presentations [1,21].
These human experimental studies indicate that if a patient has dynamic tactile allodynia, secondary punctuate/pressure hyperalgesia, temporal summation and sensory aftereffects, central sensitization may well be involved. Any sensory experience greater in amplitude, duration and spatial extent than that would be expected from a defined peripheral input under normal circumstances qualifies as potentially reflecting a central amplification due to increased excitation or reduced inhibition. These changes could include a reduction in threshold, exaggerated response to a noxious stimulus, pain after the end of a stimulus, and a spread of sensitivity to normal tissue. Moreover, there are some features of patient’s symptoms, which are more likely to indicate central rather than peripheral contribution to pain hypersensitivity. These include pain mediated by low threshold Aβ fibers (determined by nerve block or electrical stimulation), a spread of pain sensitivity to areas with no demonstrable pathology, after sensations, enhances temporal summation, and the maintenance of pain by low frequency stimuli that normally do not evoke any ongoing pain. However, given the caveats about the lack of absolute diagnostic criteria for identifying the presence of central sensitization in patients, a large number of studies have nevertheless putatively identified this phenomenon as contributing to patients’ pain phenotype [1,21].
7. SUMMARY AND CONCLUSIONS
Concluding this review, it should be emphasized that long lasting synaptic plasticity at spinal and supra-spinal levels could undergo hyperalgesia and allodynia. The latter could be providing neuronal basis for persistent pain and then pain memory. Thus, it will be particularly important to know how to regulate long-lasting plastic changes in spinal cord, thalamus and cortex. These sensory changes represent the manifestation of central sensitization, and extensive experimental medicine and clinical investigations over the past twenty years have revealed it to be an important component of the pain hypersensitivity present many patients. While considerable progress has been made in the cellular and molecular mechanism responsible for central sensitization, much remains still to be learned, particularly which genetic and environmental contributors increase the risk of developing central sensitization in particular systems, exactly what triggers and sustains the phenomenon, and what is responsible in some individuals for its persistence [1,21].
Nevertheless, the identification of the contribution of plastic processes in central sensitization to many “unexplained” clinical pain conditions has both provided a mechanistic explanation, and offered a target for developing new therapeutic drugs in pain relief.
REFERENCES
- Woolf, C.J. (2011) Central sensitization: Implications for the diagnosis and treatment of pain. Pain, 152, S2-S15. doi:10.1016/j.pain.2010.09.030
- Walters, E.T. (2009) Evolutionary aspects of pain. In: Basbaum, A.I. and Bushnell, M.C., Eds., Science of Pain, Elsevier, San Diego, 175-184.
- Brunelli, M., Castellucci, V. and Kandel, E.R. (1976) Synaptic facilitation and behavioral sensitization in Aplysia: Possible role of serotonin and cyclic AMP. Science, 194, 1178-1181. doi:10.1126/science.186870
- Squire, L.R. and Kandel, E.R. (2009) Memory: From mind to molecules. 2nd Edtion, Roberts & Company, Greenwood Village.
- Thompson, S.W., Woolf, C.J., Sivilotti, L.G. (1993) Small-caliber afferent inputs produce a heterosynaptic facilitation of the synaptic responses evoked by primary afferent A-fibers in the neonatal rat spinal cord in vitro. Journal of Neurophysiology, 69, 2116-28.
- Bliss, T.V.P. and Collingridge, G.L. (1993) A synaptic model of memory: Long-term potentiation in the hippocampus. Nature, 361, 31-39. doi:10.1038/361031a0
- Malenka, R.C. and Nicoll, R.A. (1999) Long-term potentiation—A decade of progress? Science, 285, 1870-1874. doi:10.1126/science.285.5435.1870
- Neves, G., Cook, S.F. and Bliss, T.V. (2008) Synaptic plasticity, memory and the hippocampus: A neural network approach to causality. Nature Reviews of Neuroscience, 9, 65-75. doi:10.1038/nrn2303
- LeDoux, J.E. (2000) Emotional circuits in the brain. Annual Reviews of Neuroscience, 23, 155-184. doi:10.1146/annurev.neuro.23.1.155
- Maren, S. (2005) Synaptic mechanisms of associative memory in the amygdala. Neuron, 47, 783-786. doi:10.1016/j.neuron.2005.08.009
- Phelps, E.A. and Le Doux, J.E. (2005) Contribution of the amygdala to emotion processing: From animal models to human behavior. Neuron, 48, 175-187. doi:10.1016/j.neuron.2005.09.025
- Sigurdsson, T., Doyère, V., Cain, C.K. and LeDoux, J.E. (2007) Long-term potentiation in the amygdala: A cellular mechanism of fear learning and memory. Neuropharmacology, 51, 215-227. doi:10.1016/j.neuropharm.2006.06.022
- Sandkühler, J. (2009) Long-term potentiation in pain pathways. In: Basbaum, A.I. and Bushnell, M.C., Eds., Science of Pain, Elsevier, San Diego, 401-406.
- Woolf, C.J. and Salter, M.W. (2006) Plasticity and pain: Role of the dorsal horn. In: McMahon, S.B. and Koltzenburg, M., Eds., Wall & Melzack’s Textbook of Pain, Elsevier, London, 91-105.
- Willis, W.D. (2002) Long-term potentiation in spinothalamic neurons. Brain Research Reviews, 40, 202-214. doi:10.1016/S0165-0173(02)00202-3
- Zhuo, M. (2007) Neuronal mechanism for neuropathic pain. Molecular Pain, 3, 14-23. doi:10.1186/1744-8069-3-14
- Shyu, B.-C. and Vogt, B.A. (2009) Short-term synaptic plasticity in the nociceptive thalamic anterior cingulate cortex. Molecular Pain, 5, 51-71. doi:10.1186/1744-8069-5-51
- Wei, F. and Zhuo, M. (2001) Potentiation of sensory responses in the anterior cingulate cortex following digit amputation in the anaesthetized rat. The Journal of Physiology, 532, 823-833. doi:10.1111/j.1469-7793.2001.0823e.x
- Zhuo, M. (2007) A synaptic model for pain: Long-term potentiation in the anterior cingulate cortex. Molecules and Cells, 23, 259-271.
- Drdla-Schutting, R., Benrath, J., Wunderbaldinger, G. and Sandkühler, J. (2012) Erasure of a spinal memory trace of pain by a brief, high-dose opioid administration. Science, 335, 235-238. doi:10.1126/science.1211726
- Latremoliere, A. and Woolf, C.J. (2009) Central sensitization: A generator of pain hypersensitivity by central neural plasticity. The Journal of Pain, 10, 895-926. doi:10.1016/j.jpain.2009.06.012