World Journal of Neuroscience
Vol.2 No.2(2012), Article ID:19421,4 pages DOI:10.4236/wjns.2012.22013
The prevalence of cognitive dysfunction in the estonian population of the hereditary spastic paraplegia
![]()
1Tallinn University, Tallinn, Estonia
2Estonian MS Centre, West-Tallinn Central Hospital, Tallinn, Estonia
3Tartu University Clinics, Department of Neurology, Tartu, Estonia
4Department of Paediatrics, University of Tartu, Tartu, Estonia
5Department of Neurology and Neurorehabilitation, Children’s Clinic, Tartu University Hospital, Tartu, Estonia
6Institute of Clinical Medicine, Tallinn University of Technology, Tallinn, Estonia
Email: liina.vahter@keskhaigla.ee
Received 5 November 2011; revised 30 January 2012; accepted 16 February 2012
Keywords: Hereditary Spastic Paraplegia; Cognitive Dysfunction
ABSTRACT
The hereditary spastic paraplegias (HSPs) are neurodegenerative disorders of the motor system. The information about the prevalence of the cognitive dysfunction in HSP is inconsistent. The aim of the study was to describe the prevalence of cognitive dysfunction and the cognitive profiles of persons with HSP (pwHSP) compared to healthy controls. Subjects: Participating in the cognition study were 48 persons with HSP from the epidemiological study and 48 healthy controls. Of those with HSP, 81% (39/48) had pure and 19% (9/48) had complex forms. Among pwHSP, 20.8% (10/48) had pathogenic and 14.6% (7/48) had non-pathogenic mutations in the SPAST gene. There were no mutations detected in 31 persons with the SPAST gene. Methods: Neuropsychological test battery, MMSE Results. The results of the neuropsychological tests were significantly lower in persons with HSP than in the controls (Bonferroni correction, p < 0.00625). There were statistically significant differences in subtests measuring consistent long term retrieval (p < 0.001), later recall (p = 0.004) in verbal memory and symbol digit modalities (p = 0.0015). Five persons with HSP had an MMSE score of 24 or less. Conclusions: Our results demonstrate that cognitive dysfunction is present in 16.7% to 33.3% of persons with HSP, depending on the criteria applied. There was cognitive dysfunction in 30% of persons with a known pathogenic mutation in the SPAST gene. The most frequently damaged functions in HSP are consistent long term retrieval and later recall in verbal memory and symbol digit modalities tests that discriminate between controls and pwHSP with dysfunction (1.5 SD) in three or more domains. Dementia in HSP is rare.
1. INTRODUCTION
The hereditary spastic paraplegias (HSPs) are neurodegenerative disorders of the motor system characterized by slowly progressive lower limb spasticity. HSP is classified into “pure” (pHSP) forms when spastic paraparesis exists in isolation and “complex” (cHSP) forms when other clinical symptoms are present, e.g. amyotrophy, mental retardation, eye symptoms, epilepsy, ataxia, dystonia and peripheral neuropathy [1]. pHSPs are inherited most frequently in an autosomal dominant pattern (ADHSP). cHSP forms are rare and most frequently inherited in an autosomal recessive pattern (AR-HSP). Limited information is available about cognitive dysfunction in persons with HSP.
2. COGNITIVE DYSFUNCTION IN HSP
Single case studies have been published about different forms of HSP with either mental retardation [2,3] or cognitive dysfunction [4] or both [5]. There is increasing evidence that cognitive dysfunction is present in persons with HSP [6]. Twelve individuals with “pure” HSP (aged 62 - 70) were described as having a “specific form of cognitive impairment”. A specific pattern of cognitive impairment in an individual (age 57) has been described as the only sign of HSP supporting the hypothesis that spastic paraparesis and cognitive impairment are the result of variable expression of a single gene (rather than a co-incidental occurrence) [7]. It has also been suggested that cognitive dysfunction and dementia are caused by the deletion of exon 17 of the SPAST (also known as SPG4) HSP [8].
Some specific patterns of the cognitive dysfunction of persons with HSP, based on families analyzed in genetic studies, have been described. The scores in tests for orientation, memory, language expression and comprehension were significantly lower in one study of 19 families with 41 SPAST-positive carriers. Also, all subjects had lower total Cambridge Cognitive Examination (CAMCOG) scores than control subjects. The authors concluded that mild, age-related cognitive impairment is a common feature in these families [9]. In another study, carriers of the pathogenic SPAST mutations were not demented but had a subclinical cognitive impairment primarily affecting executive functions [10]. On the other hand, the definition of cognitive dysfunction is unclear. In different studies, cognitive dysfunction is defined either as impairment –1 or –2 SD in subtests of neuropsychological battery. The definition of cognitive impairment is even more complicated, as different groups have defined cognitive dysfunction as lower scores in one to three or more subtests of the neuropsychological test batteries [11-13]. Therefore, the exact definition of cognitive dysfunction is unclear and the proportion of persons with diagnosed cognitive dysfunction depends on the wide range of definitions used in different studies.
Age-related cognitive decline in persons with HSP Cognitive dysfunction was more severe in SPAST gene mutation carriers older than 50 years and correlated with the progression of the disease, not with age [10]. In addition, in family members of persons diagnosed with ADpHSP, evidence of late onset cognitive impairment was detected. The pattern of cognitive dysfunction was identified as subcortical and was similar for all family members. The presence of cognitive impairment appeared to be related to age and not to the severity of motor symptoms [14,15].
Dementia and HSP Statistically significant differences in Mini-Mental State Examination (MMSE) scores between affected (mean 27.05) and at-risk (mean 28.46) subjects (p = 0.0086) in four families with 35 subjects have been identified. The differences in MMSE scores between affected patients and controls were still significant (p = 0.018) after two outliers with an MMSE score under 24 were excluded from the analysis. The same authors detected cognitive impairment in family members under the age of 50 years [16]. Seven of 11 persons with a pathogenic SPAST gene mutation older than 45 years were considered to have dementia [17]. The association of late onset spastic paraparesis and dementia, without other pathological findings, in two families has not been reported elsewhere and probably represents a distinct entity [18].
In conclusion, according to the literature, it seems that cognitive dysfunction is common in pwHSP with the rare patient presenting advanced impairment satisfying the criteria of dementia. Also, characteristic cognitive dysfunction has been described in otherwise unaffected family members. Therefore, cognitive dysfunction seems to be part of HSP clinical manifestation and related to gene expression. However, the exact prevalence, profile and severity of cognitive dysfunction in the HSP population are unclear.
3. OBJECTIVES
The aim of the study was to describe the prevalence of cognitive dysfunction and the cognitive profiles of patients with HSP as compared to healthy controls.
4. METHODS
A clinical neuropsychologist screened all participants for cognitive dysfunction with the Brief Repeatable Neuropsychological Test Battery (BRB-N) [19] and MMSE [20]. The BRB-N consists of six subtests: Buschke selective reminding test (SRT) (measuring verbal memory), 10/36 spatial recall test (measuring visuospatial memory), symbol digit modalities test (measuring information processing speed), delayed recall of SRT, delayed recall of 10/36 spatial recall test, and word list generation (category “animals”). Cognitive dysfunction was defined using comparable methods, with a score of 1, 1.5 or 2 SD below mean in one, two, three or more tests measuring different neuropsychological domains.
MMSE assesses orientation, attention, immediate and short-term recall, language, and the ability to follow simple verbal and written commands [19]. The cut-off score of 24 or less was used to categorize persons with dementia. The test results of persons with pathogenic, nonpathogenic and no mutations in the SPAST gene [21] were analyzed separately.
The study was approved by the Ethics Review Committee on Human Research of the University of Tartu.
5. PARTICIPANTS
As part of a nationwide epidemiological study, 59 persons with a clinically definite HSP diagnosis were identified in Estonia [22]. Of these, 48 persons agreed to participate in the cognition substudy, with 20.8% (10/48) having pathogenic and 14.6% (7/48) having non-pathogenic mutations in the SPAST gene. There were no mutations detected in the SPAST gene of 64.6% (31/48) of participants. The detailed description of the SPAST pathogenic and non-pathogenic gene mutations found in this cochort is published elsewhere [21]. In addition, 48 sociodemographically-matched healthy controls participated in the study (Table 1).
6. STATISTICAL ANALYSIS
Mean and standard deviations (SD) were computed for continuous variables, and count and percentages were computed for categorical variables. Differences in 8 neuropsychological tests between the HSP patients and the controls were assessed using the unpaired Student’s t-test or Mann-Whitney U test when the assumption of approximate normality did not hold (using the Bonferroni correction, p < 0.00625). Spearman’s rank correlation coefficients were computed. Free software R (version 2.2.0) was used for statistical analysis. A significance level of 0.05 was used.
7. RESULTS
Baseline demographic characteristics of the HSP and control groups are described in Table 1. There were no significant differences in baseline sociodemographic covariates between HSP subjects and controls.
The prevalence of cognitive dysfunction depended widely on the definitions applied (Table 2).
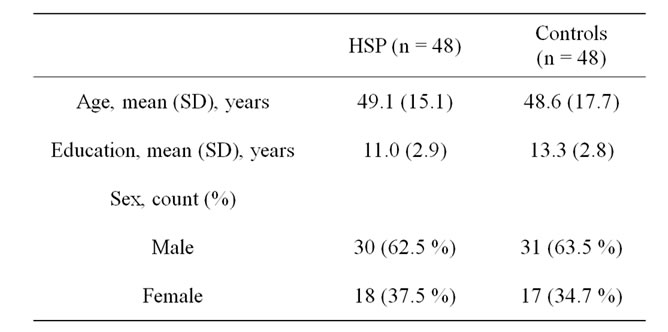
Table 1. Baseline demographic characteristics of the HSP and control group.
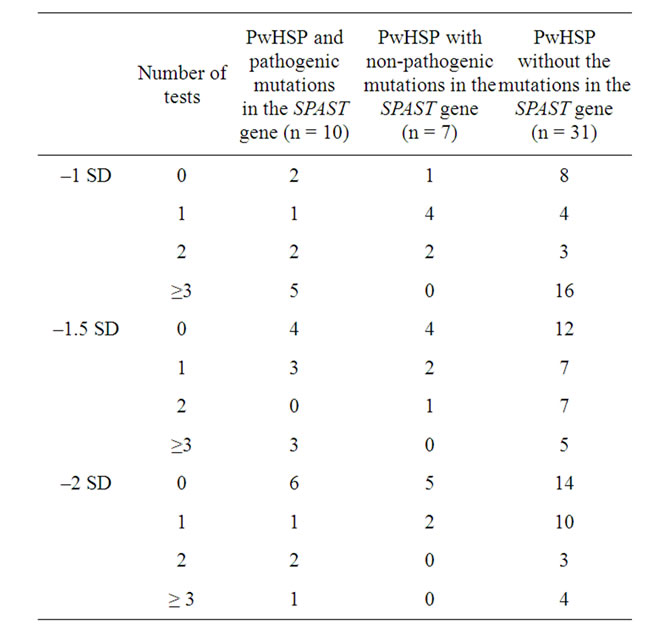
Table 2. Prevalence of cognitive dysfunction in persons with HSP and control group using different criteria.
Cognitive dysfunction was present in 77.0% (37/48) of the HSP group when the most conservative limits were applied (–1 SD in at least one or more subtests) as opposed to 10.4% (5/48) if a more liberal definition was applied (with –2 SD in three or more subtests). In the study group, 33.3% (16/48) of pwHSP had cognitive dysfunction scoring 1.5 SD below mean in two or more subtests and 16.7% (8/48) lower than 1.5 SD in three or more neuronpsychological domains. Of pwHSP, 10.4% (5/48) had an MMSE score of 24 or less. The results of the neuropsychological tests of persons with HSP and the control group are described in Table 3. The results of the neuron-psychological tests were significantly lower in persons with HSP than in the controls (p < 0.001). There were statistically significant differences in subtests measuring consistent long term retrieval (p < 0.001), later recall (p = 0.004) in verbal memory and symbol digit modalities (p = 0.0015).
The presence of cognitive dysfunction in the group with pathogenic mutations in the SPAST gene [21] was similar whether cognitive dysfunction was defined by deficits in either two or three neuropsychological domains —in 30.0% (3/10). All three persons were from different HSP families. Other family members participating in the study were cognitively intact (1/2, 2/3, 4/5 respectively). The results of the neuropsychological tests of persons with HSP with pathogenic mutation in the SPAST gene are described in Table 3. Cognitive dysfunction was present either in long term retrieval, later recall in verbal memory and/or in symbol digit modalities in all persons with cognitive dysfunction. The HSP group with pathogenic mutations in the SPAST gene had statistically significantly lower mean scores in the test measuring consistent long term retrieval in verbal memory (p = 0.0018), but the multiple comparison test of the neuropsychological tests revealed that the group profiles of the groups with and without SPAST mutations and the control group did not have a significant difference. Among those with cognitive dysfunction and pathogenic mutations in the SPAST gene, 1/10 (10%) patients scored less than 24 on the MMSE. Persons with non-pathogenic mutations in the SPAST gene as a group did not present cognitive dysfunction. The results of the neuropsychological tests of persons with HSP with non-pathogenic mutations in the SPAST gene and the control group are described in Table 3. There were no statistically significant differences detected in comparison with any subtests of the neuropsychological battery. Cognitive dysfunction (lower scores in later recall and symbol digit modalities subtests) was present in one (14.2%, 1/7) HSP person with nonpathogenic mutations in SPAST, and none scored lower in three neuropsychological domains. None of the persons scored below 24 points on the MMSE.
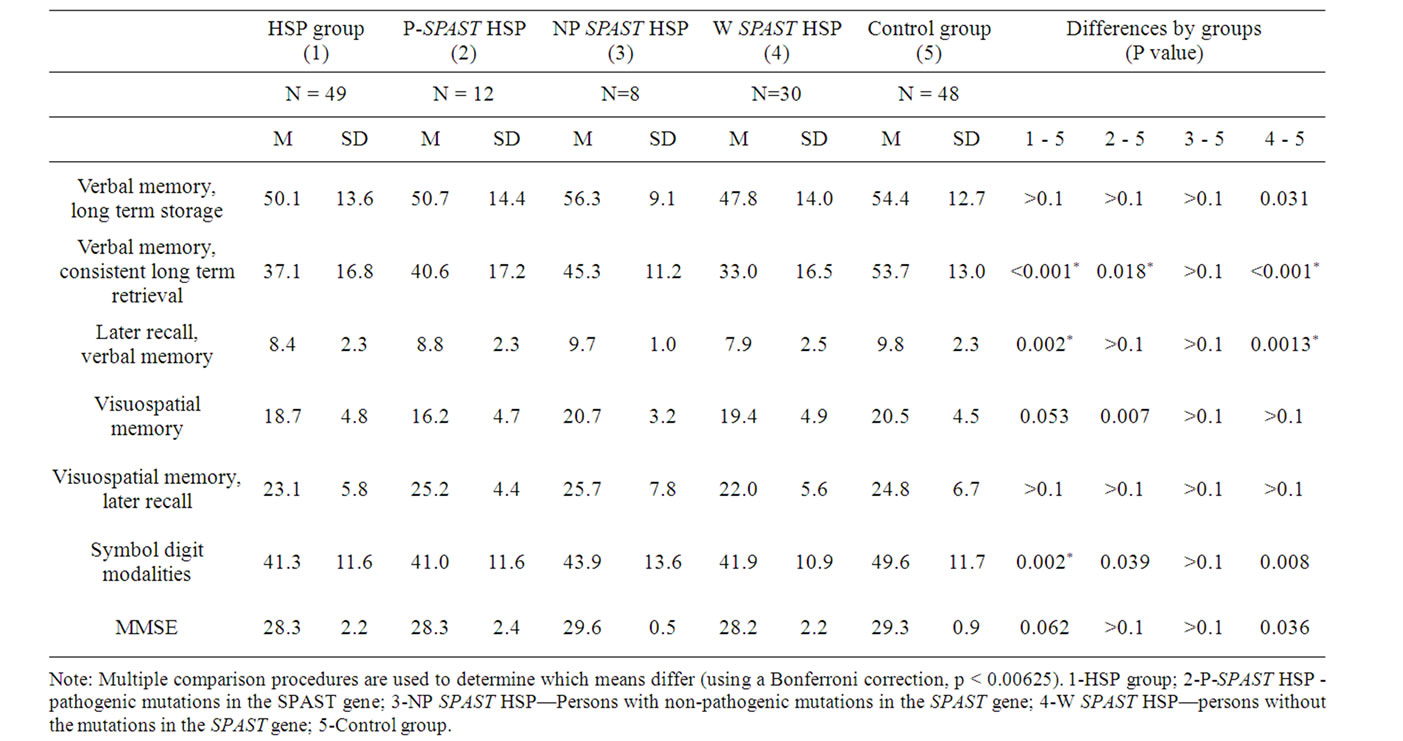
Table 3. Results of the neuropsychological tests of the persons with HSP as a group, subgroups and controls.
Cognitive dysfunction defined in two or more neuronpsychological domains was present in 38.7% (12/31) and in three or more cognitive domains in 16.2% (5/31) of persons without the mutations in the SPAST gene. The results of the neuropsychological tests of persons of HSP without the mutations in the SPAST gene and the control group are described in Table 3.
Persons with HSP without the mutations in the SPAST gene performed less well than the controls (p < 0.001). There were statistically significant differences in subtests measuring consistent long term retrieval (p < 0.001) and later recall (p = 0.001) in verbal memory. All but one patient exhibited lower scores in these two tests but not in the symbol modalities subtest. One patient exceptionally demonstrated lower scores only in immediate and later recall in visual-spatial memory subtests. Of this group, 13.0% (4/31) demonstrated an MMSE score below 24. All five persons with cognitive dysfunction demonstrated lower scores in either consistent long term retrieval or the later recall subtest of verbal memory.
The most sensitive tests to detect cognitive impairment in the HSP group were the consistent long term retrieval and later recall in verbal memory subtest and symbol digit modalities subtest. All patients with cognitive dysfunction, defined as failing in at least three cognitive tests, demonstrated cognitive dysfunction in one of these three tests. In the group of patients with cognitive dysfunction defined as lower scores in two or more tests, only one patient scored lower in immediate and later recall in visuospatial memory.
Comparing the results of the neuropsychological profile of persons with HSP with pathogenic mutation in the SPAST gene with the groups having non-pathogenic or no mutations in the SPAST gene, then multiple analysis did not reveal significant differences (p = 0.53) between the cognitive profiles.
Age-related cognitive decline in HSP Results of the neuropsychological tests in different age groups of the HSP population and control group are described in Table 4. The two age subgroups from HSP and the control group consisted of persons either up to 44 years or 45 years and older people. On the group level there was a statistically significant difference in the results of the consistent long term retrieval (verbal memory) subtest (p < 0.001) in the age group up to 44 years. Two persons from the group aged up to 44 years had cognitive decline. Both of them had low scores in subtests meassuring verbal memory (long term retrieval and consistent long term retrieval). Among persons in both groups older than 45 years, there was a statistically significant difference in the symbol digit modalities subtest (p = 0.035). Seven persons aged over 45 years scored lower in verbal memory subtests. In addition, there was decline detected in the symbol digit modalities subtest.
8. DISCUSSION
To our knowledge, this is the largest HSP cohort studied
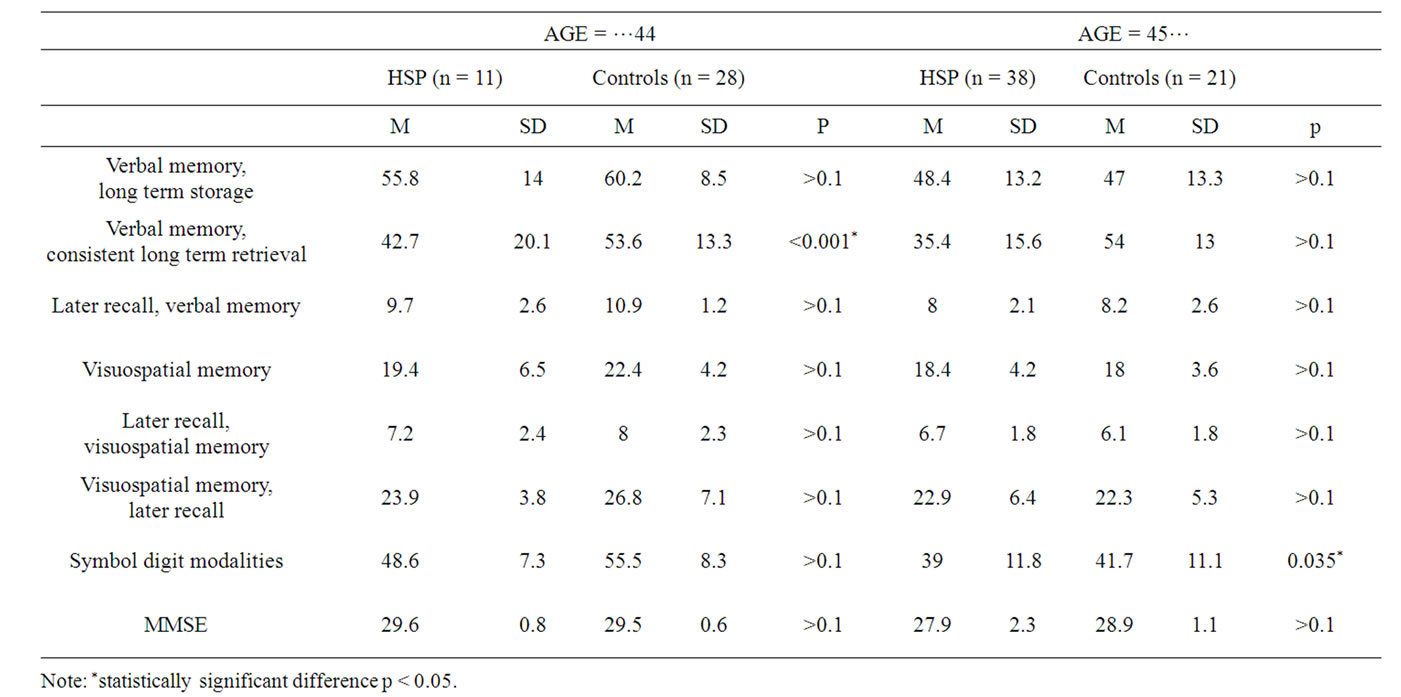
Table 4. Results of the neuropsychological tests in different age groups of the HSP population and control group.
for cognitive function with neuropsychological testing. This cohort is unique as it includes 81% (48/59) of persons with HSP identified in the nation-wide epidemiological study [21]. Our study group includes persons with HSP with pathogenic, nonpathogenic and no mutations in the SPAST gene.
So far published data on the cognitive function of persons with HSP is limited to descriptions of cognitive functions in a single family or single-case studies identified through affected family members [4-7]. Therefore we believe that our study represents the cognitive function of a broader clinical spectrum of HSP.
The definition of cognitive dysfunction is used widely as impairment –1 to –2 SD in either two or three cognitive tests of the neuropsychological test batteries [11-13]. We have analyzed our data in several ways, demonstrating that the number of persons with defined cognitive dysfunction varies significantly according to different definitions. Cognitive dysfunction is present in the HSP cohort if the person fails in two domains by 33.3%, but 16.7% if we define cognitive dysfunction as failing in tests of three domains.
Cognitive decline was not uniformly divided between different HSP subgroups. The highest prevalence of cognitive dysfunction was present in patients with HSP without mutations of the SPAST gene. Of this group, 38.7% scored less than 1.5 SD in at least three cognitive domains, as opposed to the group with pathogenic mutations of the SPAST gene in which similarly defined cognitive dysfunction was present in 30%. Interestingly only 14.2% of participants had cognitive dysfunction if they had nonpathogenic SPAST mutations. So, surprisingly, nonpathogenic SPAST mutations seemed to have a protective effect—the test results of this group did not differ from healthy controls.
Previously reported single case studies have described the profile of cognitive dysfunction in HSP [4,5]. Lower results in subtests measuring orientation, memory, language expression and comprehension [7] and dysfunction of executive functions [10] have been described. Our results demonstrate significant differences between persons with HSP as a group and demographically-matched healthy controls in subtests measuring consistent long term retrieval (p < 0.001) and later recall (p = 0.002) in verbal memory and symbol digit modalities (p = 0.0015), confirming the results of earlier descriptions.
According to our analysis, we would recommend a battery of tests describing three cognitive domains in persons with HSP. Subtests measuring consistent long term retrieval and later recall in verbal memory and symbol digit modalities would identify all persons with cognitive dysfunction in three or more domains. However, using only these tests, one person with cognitive dysfunction in two domains would have been missed from the group of patients without pathogenic mutations in the SPAST gene. According to our results, the testing should start with tests for consistent long term retrieval, later recall in verbal memory and symbol digit modalities tests as these tests identify all patients with cognitive dysfunction in at least three domains. However, if these tests are within normal limits and there is clinical suspicion for milder impairment, further testing is recommended. According to our data cognitive dysfunction in familial cases does not uniformly represent intrafamilial variability of the disease. It seems that even among different family members cognitive dysfunction is not always present and the cognitive profile is different.
Our results demonstrate that dementia, defined as scoring 24 or less on MMSE, was present in 5/48 (10.4%) of persons with HSP. Although pwHSP with pathological mutations in the SPAST gene showed more consistant cognitive dysfunction as a group, the number of patients with an MMSE score below 24 was comparable (10% vs. 13%) with persons with or without mutations in the SPAST gene.
Several studies have demonstrated that cognitive decline is age-dependent [7,10,14,16]. In our study, age was correlated with performance in subtests measuring long term storage and later recall in verbal memory and symbol digit modalities. Two persons from the group aged less than 45 years and seven persons aged over 45 years scored lower in verbal memory subtests. In addition there was decline detected in the symbol digit modalities subtest in older age groups. Our studied group was cross-sectional and too small to identify the importance of age-related cognitive decline in HSP. However, tests measuring verbal memory may be suggested as a sensitive marker for detecting age-related cognitive decline in this population.
The major limitation of our study is the cross-sectional analysis of cognitive functions.
9. CONCLUSION
Our results demonstrate that cognitive dysfunction is present in 16.7% to 33.3% of persons with HSP depending on criteria applied. Of persons with known pathogenic mutation in the SPAST gene, 30% had cognitive dysfunction. The most frequently damaged functions in HSP are consistent long term retrieval and later recall in verbal memory and symbol digit modalities tests that discriminate between controls and pwHSP with dysfunction (1.5 SD) in three or more domains. Dementia in HSP is rare.
10. ACKNOWLEDGEMENTS
This study was supported by Estonian Science Foundation research grant no. ETF5680 and targeted financing TARLA 2695. No conflict of interests has been detected.
REFERENCES
- Harding, A.E. (1983) Classification of the hereditary ataxias and paraplegias. Lancet, 21, 1151-1155. doi:10.1016/S0140-6736(83)92879-9
- Okuda, B., Iwamoto, Y. and Tachibana, H. (2002) Hereditary spastic paraplegia with thin corpus callosum and cataract: A clinical description of two siblings. Acta Neurologica Scandinavica, 106, 222-224. doi:10.1034/j.1600-0404.2002.01210.x
- Iwabuchi, K., Yagishita, S., Amano, N. and Kosaka, K. (1991) A new type of complicated form of hereditary spastic paraplegia showing mental deterioration, quadriplegia with muscular atrophy, sensory disturbance, extrapyramidal disorders, and epilepsy. Rinsho Shinkeigaku, 31, 945-952.
- Okubo, S., Ueda, M., Kamiya, T., Mizumura, S., Terashi, A. and Katayama, Y. (2000) Neurological and neuroradiological progression in hereditary spastic paraplegia with a thin corpus callosum. Acta Neurologica Scandinavica, 102, 196-199. doi:10.1034/j.1600-0404.2000.102003196.x
- Maruta, K. and Kondo, I. (2001) A family of hereditary spastic paraplegia with dementia, ataxia, and dystonia. Rinsho Shinkeigaku, 41, 683-690.
- Uttner, I., Baumgartner, A., Sperfeld, A.D. and Kassubek, J. (2007) Cognitive performance in pure and complicated hereditary spastic paraparesis: A neuropsychological and neuroimaging study. Neuroscience Letters, 419, 158-161. doi:10.1016/j.neulet.2007.04.031
- Byrne, P.C., Webb, S., McSweeney, F., Burke, T., Hutchinson, M. and Parfrey, N.A. (1998) Linkage of AD HSP and cognitive impairment to chromosome 2p: Haplotype and phenotype analysis indicates variable expression and low or delayed penetrance. European Journal of Human Genetics, 6, 275-282. doi:10.1038/sj.ejhg.5200185
- Murphy, S., Gorman, G., Beetz, C., Byrne, P., Dytko, M., McMonagle, P., Kinsella, K., Farrell, M. and Hutchinson, M. (2009) Dementia in SPG4 hereditary spastic paraplegia: Clinical, genetic, and neuropathologic evidence. Neurology, 73, 378-384. doi:10.1212/WNL.0b013e3181b04c6c
- Byrne, P.C., McMonagle, P., Webb, S.F., Fitzgerald, B., Parfrey, N.A. and Hutchinson M. (2000) Age-related cognitive decline in hereditary spastic paraparesis linked to chromosome 2p. Neurology, 54, 1510-1517.
- Tallaksen, C.M., Guichart-Gomez, E., Verpillat, P., Hahn-Barma, V., Ruberg, M., Fontaine, B., Brice, A., Dubois, B. and Durr, A. (2003) Subtle cognitive impairment but no dementia in patients with spastin mutations. Archives of Neurology, 60, 1113-1118. doi:10.1001/archneur.60.8.1113
- Valentino, P., Cerasa, A., Chiriaco, C., Nisticò, R., Pirritano, D., Gioia, M., Lanza, P., Canino, M., Del Giudice, F., Gallo, O., Condino, F., Torchia, G. and Quattrone, A. (2009) Cognitive deficits in multiple sclerosis patients with cerebellar symptoms. Multiple Sclerosis Journal, 15, 854-859. doi:10.1177/1352458509104589
- Amato, M.P., Ponziani, G., Siracusa, G. and Sorbi, S. (2001) Cognitive dysfunction in early-onset multiple sclerosis: A reappraisal after 10 years. Archive of Neurology, 58, 1602-1606. doi:10.1001/archneur.58.10.1602
- Patti, F., Amato, M.P., Trojano, M., Bastianello, S., Tola, M.R., Goretti, B., Caniatti, L., Di Monte, E., Ferrazza, P., Brescia Morra, V., Lo Fermo, S., Picconi, O., Luccichenti, G. and COGIMUS Study Group. (2009) Cognitive impairment and its relation with disease measures in mildly disabled patients with relapsing-remitting multiple sclerosis: baseline results from the Cognitive Impairment in Multiple Sclerosis (COGIMUS) study. Multiple Sclerosis Journal, 15, 779-788. doi:10.1177/1352458509105544
- Webb, S., Patterson, V. and Hutchinson, M. (1997) Two families with autosomal recessive spastic paraplegia, pigmented maculopathy and dementia. Journal of Neurology, Neurosurgery & Psychiatry, 63, 628-632. doi:10.1136/jnnp.63.5.628
- Webb, S. and Hutchinson, M. (1998) Cognitive impairment in families with pure autosomal dominant hereditary spastic paraparesis. Brain, 121, 923-929. doi:10.1093/brain/121.5.923
- Reid, E., Grayson, C., Rubinsztein, D.C., Rogers, M. and Rubinsztein, J.S. (1999) Subclinical cognitive impairment in autosomal dominant “pure” hereditary spastic paraplegia. Journal of Medical Genetics, 36, 797-798.
- McMonagle, P., Byrne, P. and Hutchinson, M. (2004) Further evidence of dementia in SPG4-linked autosomal dominant hereditary spastic paraplegia. Neurology, 62, 407-410.
- Lizcano-Gil, L.A., Garcia-Cruz, D., del Pilar BernalBeltran, M. and Hernandez, A. (1997) Association of late onset spastic paraparesis and dementia: Probably an autosomal dominant form of complicated paraplegia. American Journal of Medical Genetics, 68, 1-6. doi:10.1002/(SICI)1096-8628(19970110)68:1<1::AID-AJMG1>3.0.CO;2-V
- Rao, S.M. and the Cognitive Function Study Group of the National Multiple Sclerosis Society. (1990) A manual for brief repeatable battery of the neuropsychological tests in multiple sclerosis. Medical College of Wisconsin, Milwaukee, WI.
- Folstein, M.F., Folstein, S.E. and McHugh, P.R. (1975) Mini-mental state: A practical method for grading the state of patients for the clinician. Journal of Psychiatric Research, 12, 189-198. doi:10.1016/0022-3956(75)90026-6
- Braschinsky, M., Tamm, R., Beetz, C. Sachez-Ferrero, E., Raukas, E., Lüüs, S.M., Gross-Paju, K., Boillot, C., Canzian, F., Metspalu, A. and Haldre, S. (2010) Unique spectrum of SPAST variants in Estonian HSP patients: Presence of benign missense changes but lack of exonic rearrangements. BMC Neurology, 9, 10-17.
- Braschinsky, M., Lüüs, S.-M., Gross-Paju, K. and Haldre, S. (2009) The prevalence of hereditary spastic paraplegia and the occurence of SPG4 mutations in Estonia. Neuroepidemiology, 32, 89-93. doi:10.1159/000177033