Open Journal of Molecular and Integrative Physiology
Vol.3 No.3(2013), Article ID:36340,12 pages DOI:10.4236/ojmip.2013.33019
Morphine has latent deleterious effects on the ventilatory responses to a hypoxic-hypercapnic challenge*
![]()
1Pediatric Respiratory Medicine, University of Virginia School of Medicine, Charlottesville, USA
2Division of Biology, Galleon Pharmaceuticals, Horsham, USA
3Department of Anesthesia, University of Iowa College of Medicine, Iowa City, USA
4Department of Pediatrics, Case Western Reserve University, Cleveland, USA
Email: #sjl78@case.edu
Copyright © 2013 Walter J. May et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 25 June 2013; revised 12 July 2013; accepted 23 July 2013
Keywords: Morphine; Hypoxia-Hypercapnia; Minute Ventilation; Arterial Blood Gases; Conscious Rats
ABSTRACT
This study explored the concept that morphine has latent deleterious actions on the ventilatory control systems that respond to a hypoxic-hypercapnic challenge. In this study, we examined the ventilatory responses elicited by hypoxic-hypercapnic challenge in conscious rats at a time when the effects of morphine (10 mg/kg) on arterial blood-gas chemistry and minute ventilation had subsided. Morphine induced pronounced changes in arterial blood-gas chemistry (e.g., an increase in pCO2, decreases in pO2 and sO2) and decreases in minute ventilation. Despite the complete resolution of the morphine-induced changes in arterial blood-gas chemistry and minute ventilation and almost complete resolution of the effects on peak inspiratory flow and peak expiratory flow, subsequent exposure to hypoxic-hypercapnic challenge elicited markedly blunted increases in minute ventilation and in peak inspiratory and expiratory flows. These findings demonstrate that 1) the changes in arterial bloodgas chemistry elicited by morphine parallel changes in minute ventilation rather than PIF and PEF, and 2) morphine has latent untoward effects on the ventilatory responses to hypoxic-hypercapnic challenge. These novel findings raise the possibility that patients deemed to have recovered from the acute ventilatory depresssant effects of morphine may still be susceptible to the latent effects of this opioid analgesic. The mechanisms underlying these latent effects remain to be elucidated.
1. INTRODUCTION
In humans, opioid analgesics such as morphine depress minute ventilation (Vm), disturb arterial blood-gas (ABG) chemistry [1-3], and blunt the ventilatory responses to hypoxic [4-6] and hypercapnic [6] challenges. Studies in rats have also demonstrated that opioids depress Vm and ABG chemistry [3,7-12], blunt the ventilatory responses to hypoxic challenge [13], and depress carotid body chemoafferent responses to hypoxic or hypercapnic challenges [14-18]. It is rightfully assumed that ventilatory control processes perse and the ventilatory responses to hypoxic and/or hypercapnic challenges recover upon the amelioration of the effects of opioids on baseline ventilatory parameters. However, It should be recognized that morphine may have unsuspected actions on ventilatory processes (i.e., when the effects of the opioid itself have resolved) in humans because although the metabolite, morphine-6-glucurodide (M6G), has minimal effects on Vm, it markedly blunts the responses to hypercapnic challenge [19].
At present, the possibility that the ventilatory responses elicited by hypoxic-hypercapnic challenge (H-H) are blunted after the effects of morphine have abated has not been determined in humans or rats. This possibility is important to examine since patients are deemed to have recovered from the acute ventilatory depressant effects of morphine when respiratory rate and ABG chemistry are normal. However, if morphine was still able to depress ventilatory responses during episodes of H-H, patients may be prone to respiratory collapse due to the inability of chemoafferent systems to trigger appropriate ventilatory responses. Accordingly, our objective was to determine the ventilatory responses elicited by exposure to H-H in conscious rats in which the depressant effects of morphine on Vm, ABG chemistry and Alveolar-arterial (A-a) gradient, O2 (A-a) gradient, an index of ventilation-perfusion status in the lung [20,21], had fully subsided.
2. METHODS
2.1. Rats and Surgeries
All studies were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80-23) revised in 1996. The protocols were approved by the Animal Care and Use Committee of the University of Virginia. Adult male Sprague-Dawley rats (Harlan, Madison, WI, USA) received jugular vein and femoral arterial catheters (arterial blood gas studies) or jugular vein catheters only (ventilation studies) under 2% isoflurane anesthesia. The rats were allowed 4 days to recover before use. All studies were performed in a quiet room with relative humidity of 51% ± 3% and room temperature of 21.4˚C ± 0.3˚C.
2.2. Blood Gas Measurements and Determination of Arterial-Alveolar Gradient
The pH, pCO2, pO2 and sO2 of arterial blood samples (100 µL) were measured by a Radiometer Blood-gas machine (ABL800 FLEX). The calculated A-a gradient, measures the difference between alveolar and arterial blood concentrations of O2 [20,21] (see Table 1 for formulae).
2.3. Ventilatory Parameters
Ventilatory parameters were continuously recorded in unrestrained rats using a whole-body plethysmography system (PLY 3223; BUXCO Inc., Wilmington, NC, USA), as described previously [22,23]. The parameters were 1) frequency of breathing (fr), 2) tidal volume (Vt), 3) minute ventilation (Vm = fr × Vt), 4) inspiratory (Ti) and expiratory (Te) times, 5) peak inspiratory (PIF) and expiratory (PEF) flows, 6) end inspiratory pause (EIP), time between end of inspiration and start of expiration, and 7) Vt/Ti, an index of Respiratory Drive [24]. Software constantly corrected digitized values for changes in chamber temperature and humidity and a rejection algorithm was included in the breath-by-breath analysis to exclude motion-induced artifacts. Due to the closeness of body weights in the experimental groups, ventilatory data are presented without corrections for weights.
2.4. Body Temperature Measurements
Body temperature (Bt) was measured via telemetry [25]. In brief, male Sprague Dawley rats were anesthetized with 2% isoflurane and a telemetry probe (ETA-F20, Data Sciences International, Minneapolis, MN) was placed in each peritoneal cavity. A jugular catheter was also implanted. The rats were given four days to recover from surgery. On the day of the study, the rats were placed in the plethysmography chambers, under each of which was placed a telemetry receiver connected via a data exchange matrix to a computer. The Bt signals were transferred to MATLAB (MathWorks, Natick, MA) for analysis.
2.5. Protocols for Arterial Blood Gas Studies
Arterial blood samples (100 µL) were taken before (2 - 3 samples, values averaged) and 5, 15, 30, 45, 60 and 75 min after the bolus injection of vehicle (saline, n = 5 rats; 322 ± 2 g) or (+)-morphine sulfate (10 mg/kg, i.v., n = 5 rats; 321 ± 2 g) from Baxter Healthcare Corporation (Deerfield, IL, USA).
2.6. Protocols for Ventilation Studies
Rats were placed in the plethysmography chambers and given 45 - 60 min to acclimatize. One group (n = 12, 318 ± 3 g) received an injection of vehicle (saline, i.v.). Another group (n = 12, 319 ± 3 g) received (+)-morphine sulfate (10 mg/kg, i.v.). After 75 min, the rats were exposed to H-H via the re-breathing method used to study ventilatory responses in rats [26,27] and the effects of morphine in humans [28]. Air-flow to the chambers was stopped for 55 min allowing the rats to re-breathe their own air, which became progressively lower in O2 and higher in CO2. This lead to progressively lower levels of O2 and higher levels of CO2 in the blood, thereby mimicking clinical scenarios [29,30]. CO2 is a potent arousal stimulus when delivered rapidly, and a gradual increase in chamber CO2 limits the rate of arousal [31]. After 55 min, air-flow (room-air) was returned to the chambers and parameters recorded for 30 min.
2.7. Protocols for Combined Body Temperature and Ventilatory Recordings
The rats were placed in the plethysmography chambers and were allowed 45 - 60 min to acclimatize. One group of rats (n = 6, 314 ± 2 g) received a bolus i.v. injection of vehicle (saline) whereas another group (n = 6, 312 ± 2 g) received an injection of morphine (10 mg/kg, i.v.). The rats then underwent the H-H and return to room-air protocol described above.
2.8. Statistics
Ventilatory data (1 min bins) and derived parameters,
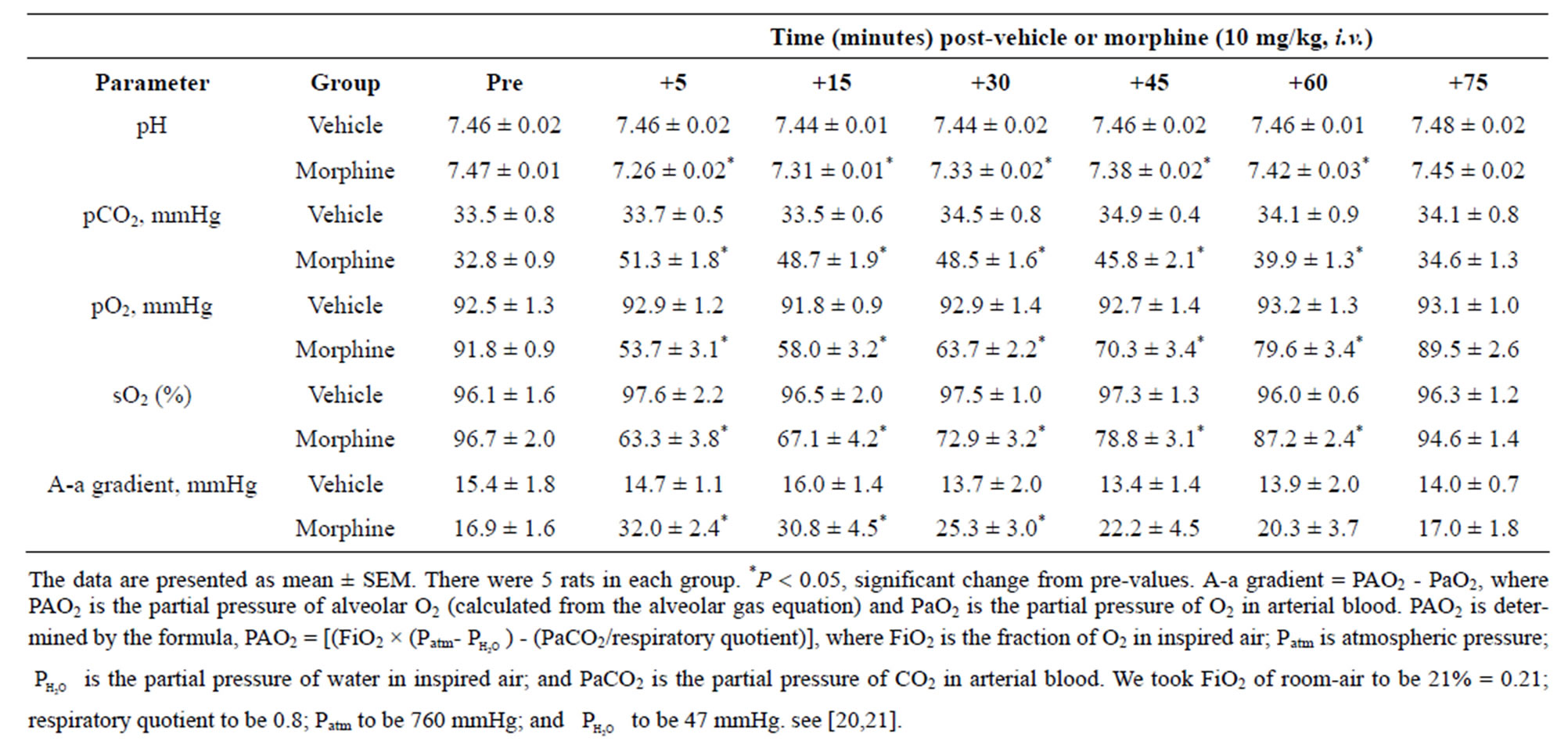
Table 1. Changes in blood gas chemistry and Alveolar-arterial (A-a) gradient elicited by injections of morphine.
Vt/Ti and Cumulative Response (cumulative arithmetic changes from pre-values) determined from the 1 min bins, were taken for analysis. All data are presented as mean ± SEM and were analyzed by one-way or two-way ANOVA followed by Student’s modified t test with Bonferroni corrections for multiple comparisons between means using the error mean square terms from the ANOVAs [32]. A value of P < 0.05 denoted statistical significance.
3. RESULTS
3.1. Arterial Blood Gases and Alveolar-Arterial Gradients
The injection of morphine (10 mg/kg, i.v.) elicited decreases in pH, pO2, and sO2 and increases in pCO2 and A-a gradient (Table 1). The morphine-induced effects were prominent at 5 and 15 min, began to subside within 45 - 60 min, and were completely resolved by 75 min.
3.2. Ventilatory Responses Elicited by Morphine
There were no between group differences in any resting ventilatory parameter prior to the injection of vehicle or morphine (Table 2). The injection of vehicle had minor effects on ventilation (Figures 1-3). Morphine elicited minor initial changes in fr (Figure 1; Table 2, column “Initial max”). As time progressed, fr in morphinetreated (MOR) rats rose to higher levels than in vehicle-treated (VEH) rats (Table 2, column “Pre-H-H”). Morphine elicited an immediate decrease in Vt that lasted for 45 - 50 min (Figure 1, Table 2). Taken together, morphine decreased Vm for about 30 - 40 min (Figure 1, Table 2). Morphine elicited a prompt increase in Ti, a decrease in Te and an increase in EIP (Figure 2, Table 2). These responses were still evident at 75 min. Morphine elicited a fall in Vt/Ti that was maximal between 15 - 30 min and which had resolved by 75 min (Table 3). Morphine produced prompt decreases in PIF and PEF that were still evident at 75 min (Figure 3, Table 2).
3.3. Ventilatory Responses during and Following Hypoxic-Hypercapnic Challenge
In VEH rats, H-H elicited gradual increases in fr, Vt and Vm that were associated with gradual decreases in Ti and Te but not EIP (Figures 1 and 2, Tables 2 and 4). H-H also elicited gradual increases in PIF and PEF (Figure 3, Tables 2 and 4). H-H elicited markedly smaller increases in fr in MOR rats even though resting fr was actually a little higher than in VEH rats (Figure 1, Tables 2 and 4). The increases in Vt elicited by H-H in MOR rats were similar to those in VEH rats (Figure 1, Tables 2 and 4). As such, H-H elicited a substantially smaller increase in Vm in MOR rats primarily because of the depressed fr response (Figure 1, Tables 2 and 4). In MOR rats, H-H elicited gradual and robust decreases in Ti, Te and EIP (Figure 2, Tables 2 and 4). In contrast, H-H elicited substantially smaller increases in Vt/Ti (Table 3) and in PIF and PEF (Figure 3, Tables 2 and 4) in MOR rats than in VEH rats. In VEH rats, ventilatory parameters returned to and/or closely approached pre H-H levels upon return to room air (Figures 1-3, Tables 2-4). In MOR rats, fr remained above pre-H-H levels whereas Te remained below pre-H-H levels upon return to room-air (Figures 1 and 2, Tables 2 and 4). The other parameters essentially recovered to and/or closely approached pre-H-H levels

Table 2. Changes in baseline parameters during various stages of the experimental protocols.
(Figures 2 and 3, Tables 2-4).
3.4. Body Temperature
As seen in Figure 3 (bottom panel), morphine elicited a gradual increase in Bt, which was still evident at the time of exposure to H-H (+0.52˚C ± 0.06˚C, P < 0.05). H-H gradually decreased Bt in VEH rats. This decrease began within 30 - 35 min and was still evident at the end of H-H challenge (i.e., at 55 min, −0.39˚C ± 0.06˚C, P < 0.05). H-H also gradually decreased Bt in MOR rats. This decrease began within 25 - 30 min and was still evident at +55 min (−0.57˚C ± 0.09˚C, P < 0.05). As a consequence, Bt values in VEHor MOR rats were similar to each other from 30 - 55 min of H-H challenge. Bt values diverged in the two groups upon return to room-air. Specifically, Bt returned to pre-values in VEH rats whereas Bt declined in MOR rats (i.e., +30 min, −0.32 ± 0.06˚C from +55 min post-H-H values, P < 0.05). The ventilatory responses that occurred during this study were indistinguishable from those recorded in rats without telemetry devices (data not shown).
4. DISCUSSION
The novel findings of this study are that the ventilatory responses to H-H were depressed in MOR rats although the effects of morphine on ABG chemistry and A-a gradient had fully subsided before exposure to H-H. As will be discussed, numerous types of patients may be especially susceptible to the latent effects of morphine and/or it metabolites, for which there is no current therapy.
4.1. Body Temperature
As expected, morphine elicited a minor gradual hyperthermia (+0.5˚C) that was sustained for 55 min [33,34]. Hyperthermia of 1˚C or above stimulates ventilation [35] whereas smaller increases in Bt do not [36,37]. Hypoxia substantially decreases Bt in man and animals [38]. Hypercapnia elicits relatively minor decreases in Bt [39,40] and addition of CO2 to the environment attenuates the falls in Bt elicited by hypoxia [41]. In our VEH rats, H-H elicited a minor hypothermia (0.4˚C). In MOR rats, H-H also elicited a decrease in Bt with final values falling equal to those in VEH rats. These minor changes in Bt would have minimal direct effects on ventilation and ABG chemistry.
4.2. Morphine-Induced Changes in Arterial Blood Gas Chemistry and Alveolar-Arterial Gradient
The morphine-induced changes in ABG chemistry and A-a gradient were consistent with hypoventilation and mismatch of ventilation-perfusion. Specifically, morphine decreased pH, increased pCO2, decreased pO2 and sO2, and increased A-a gradient. The mechanisms/sites of action by which activation of ORs may depress ABG chemistry include central and peripherally-mediated depression of ventilatory drive), chest-wall rigidity, increases in upper/lower airway resistances, inhibition of
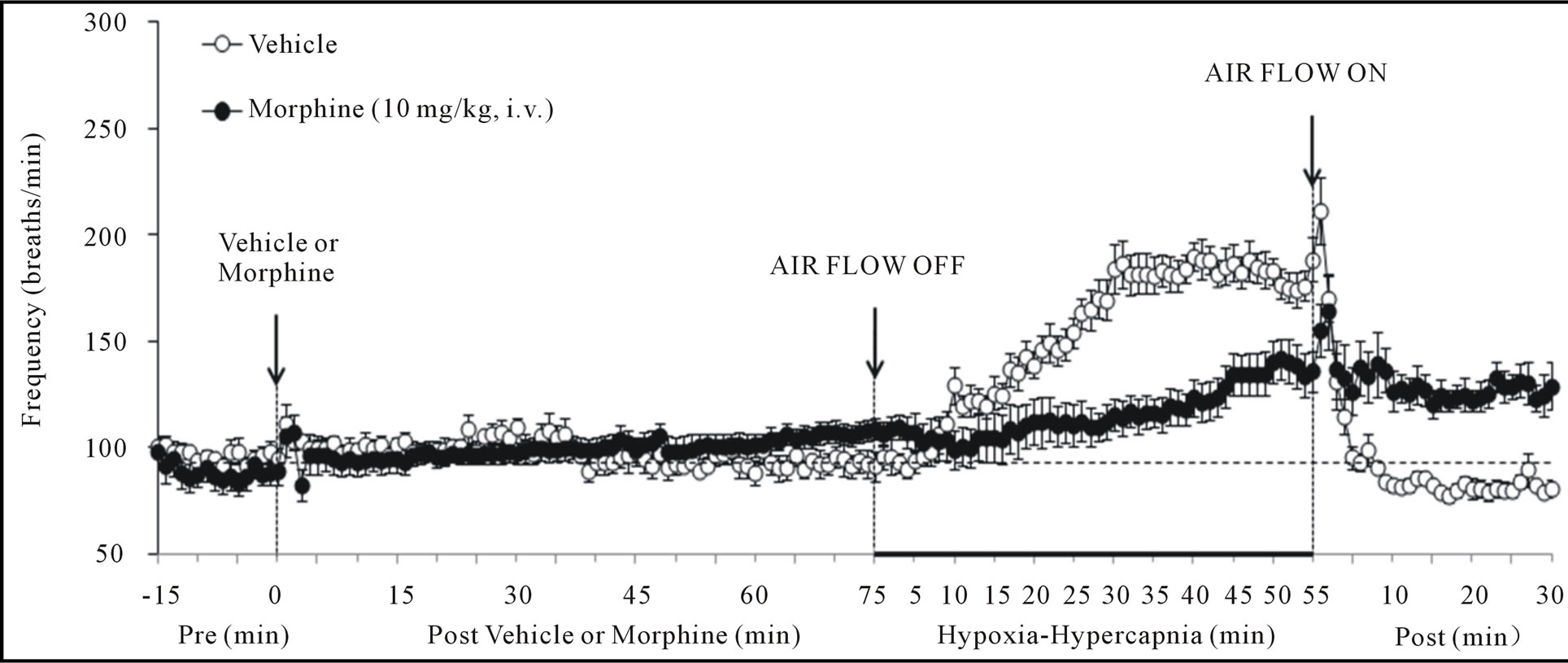
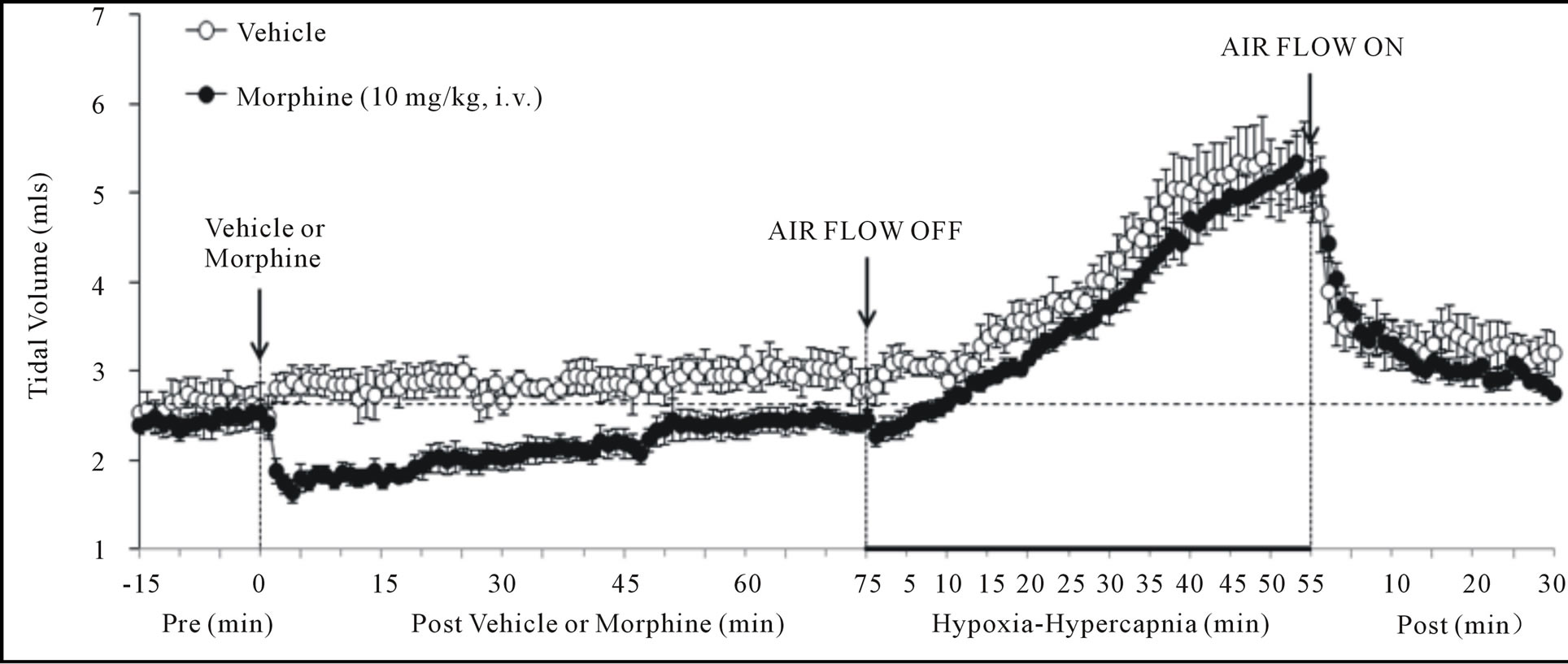
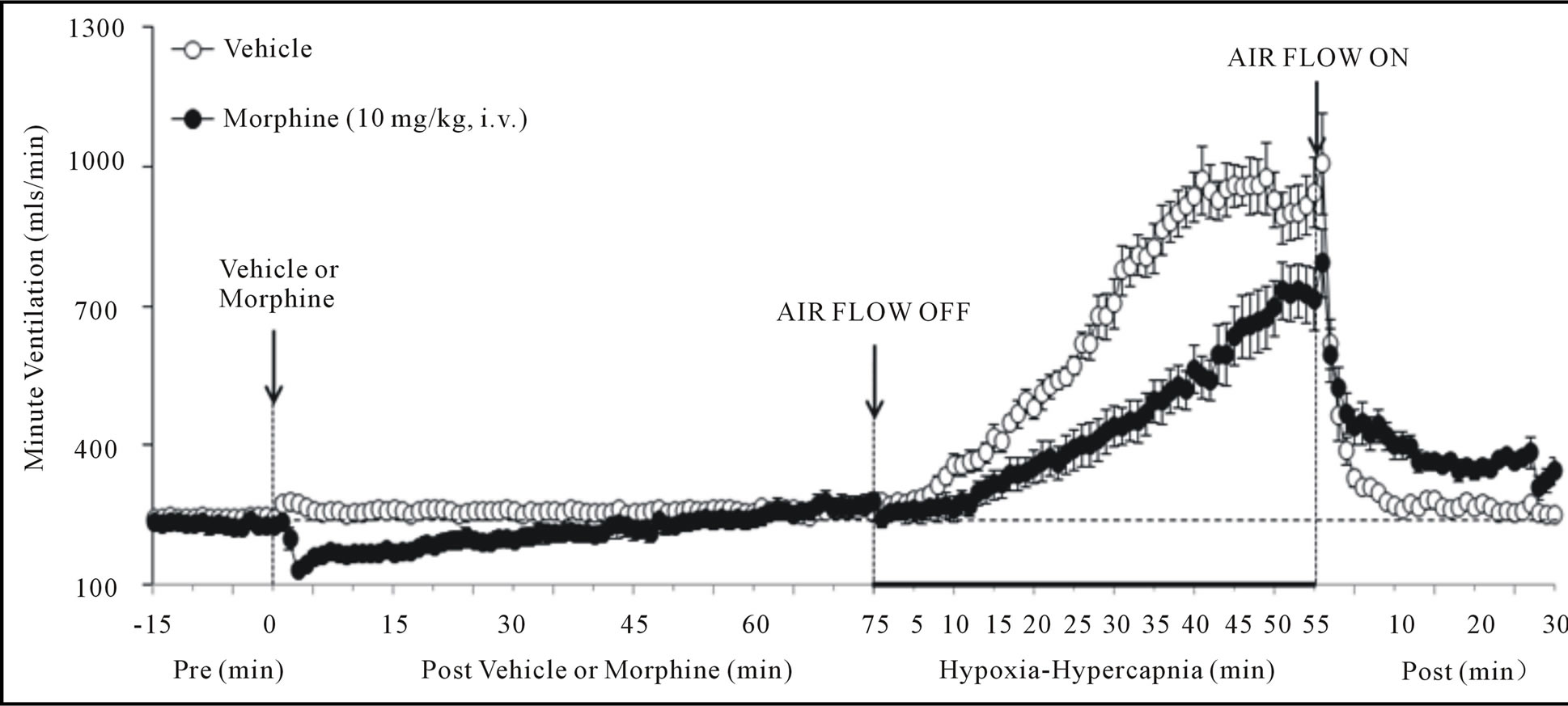
Figure 1. Changes in frequency of breathing (top panel), tidal volume (middle panel) and minute ventilation (lower panel) elicited by injections of vehicle or morphine (10 mg/kg, i.v.) and subsequent exposure to hypoxia-hypercapnia (air flow off) and return to room-air (air flow on). The data are presented as mean ± SEM. There were 6 rats in each group.
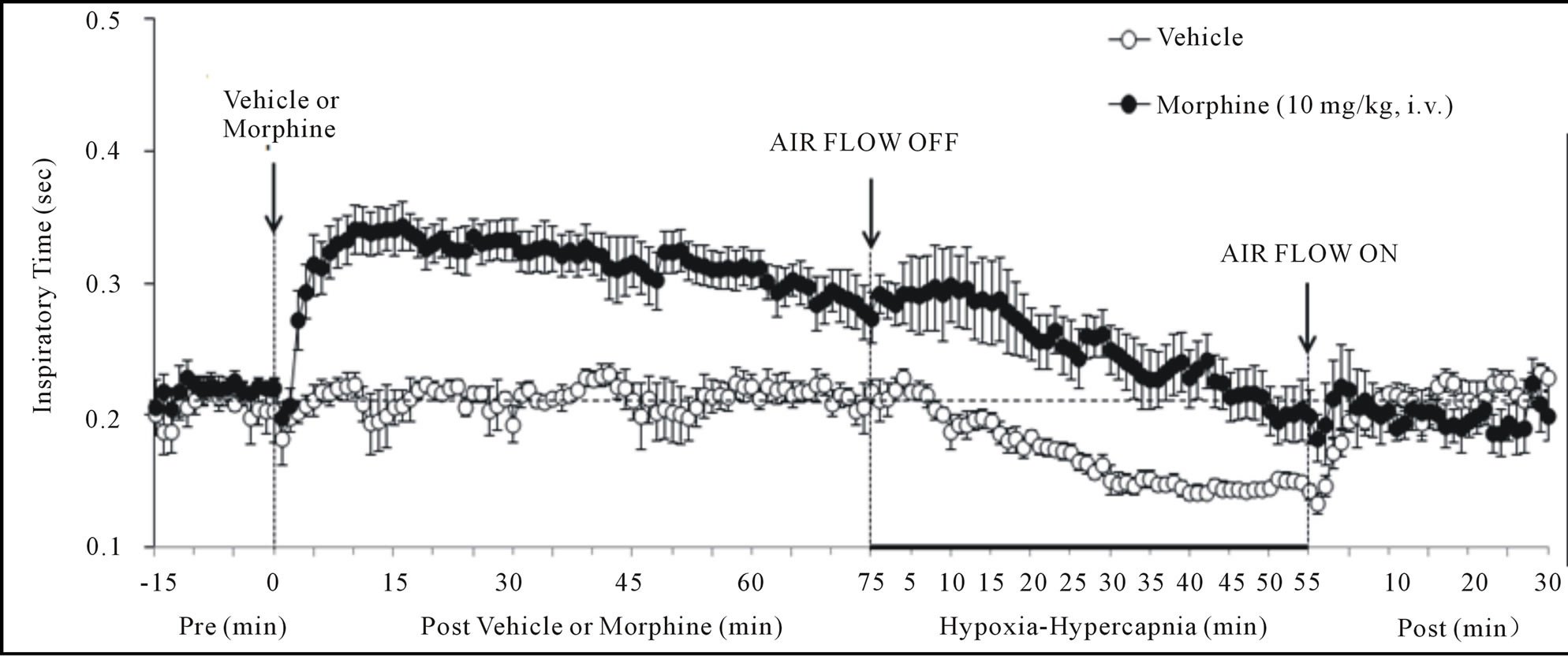
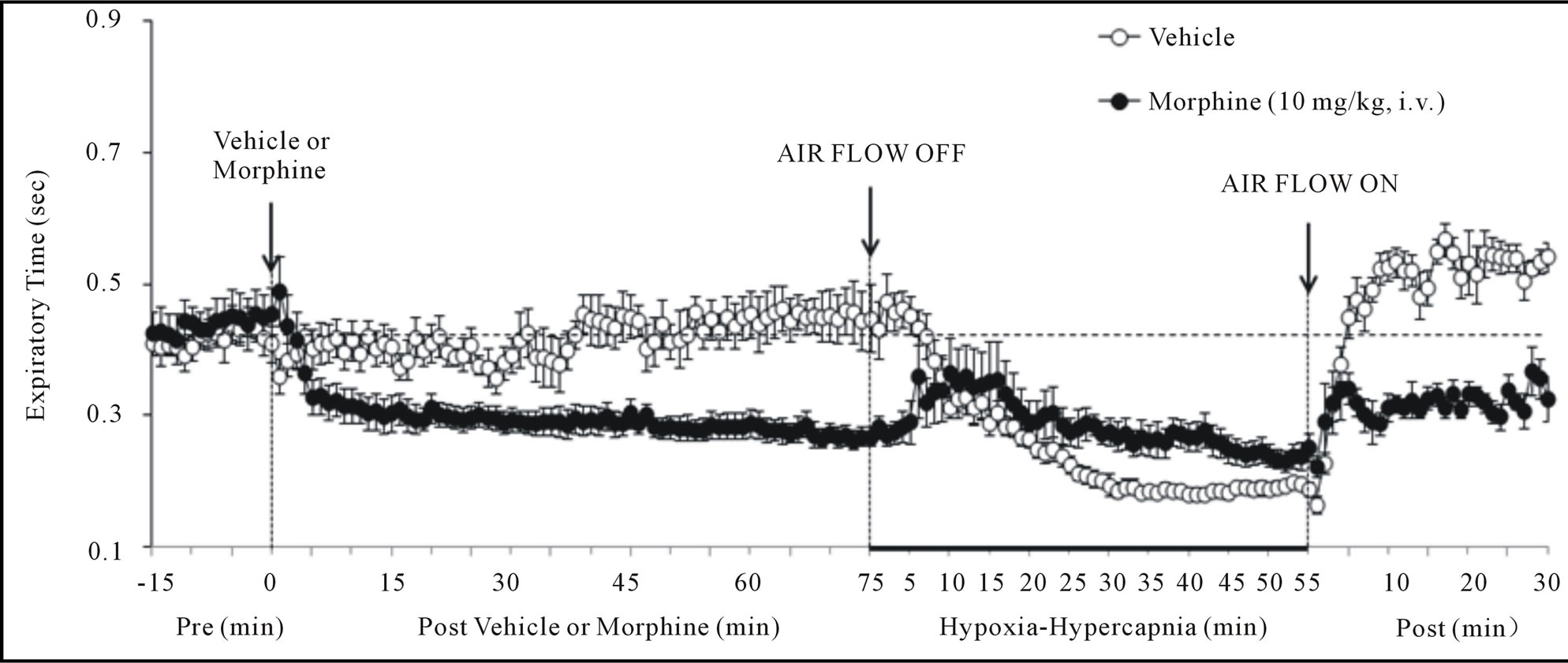
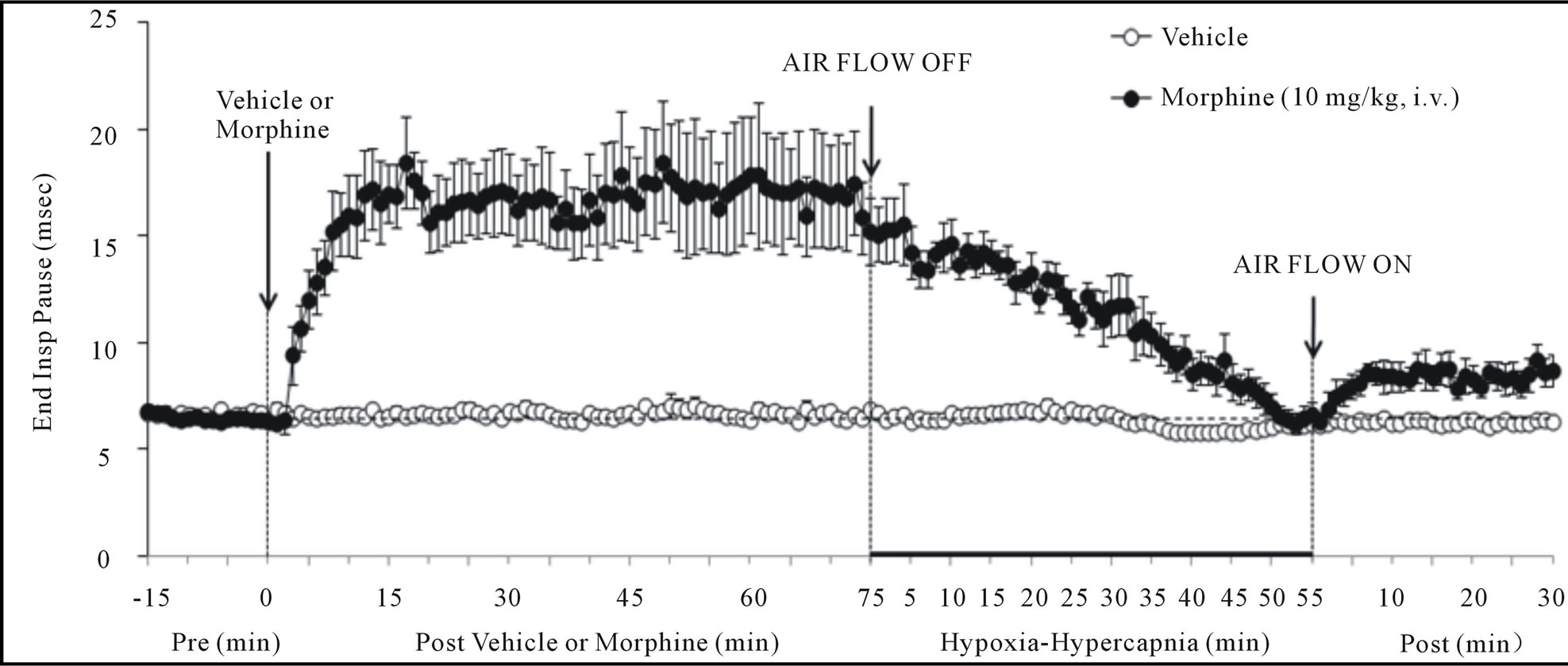
Figure 2. Changes in inspiratory time (upper panel), expiratory time (middle panel) and end inspiratory pause (lower panel) elicited by injections of vehicle or morphine (10 mg/kg, i.v.) and subsequent exposure to hypoxia-hypercapnia (air flow off) and return to room-air (air flow on). The data are presented as mean ± SEM. There were 6 rats in each group.
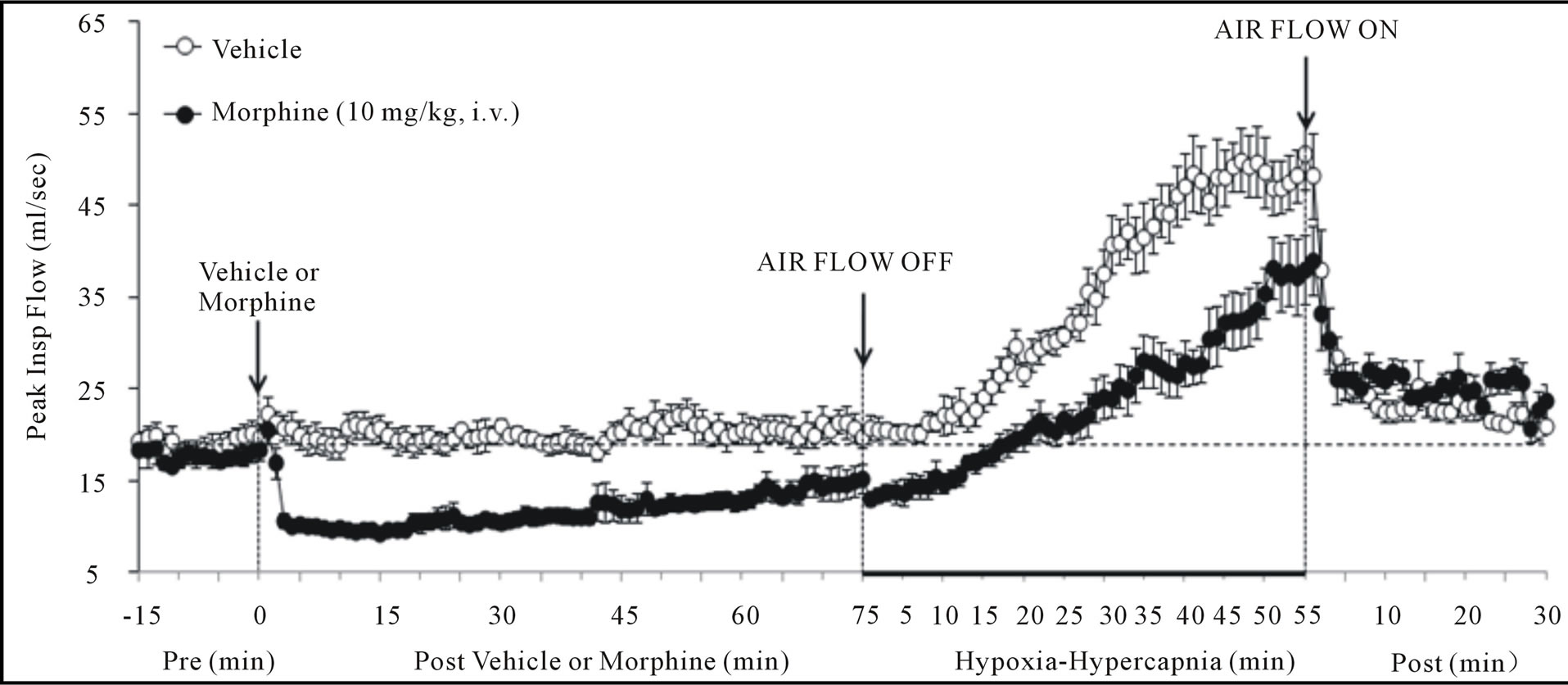
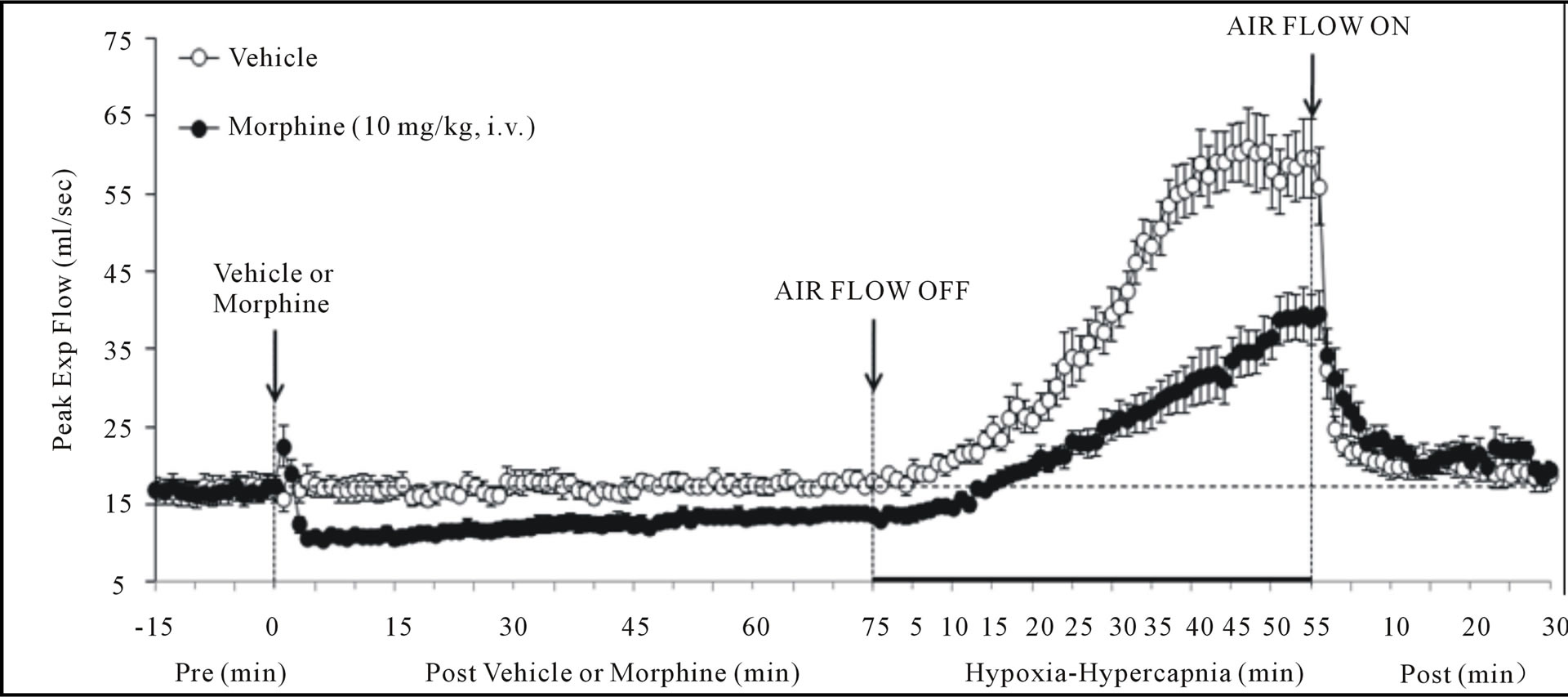
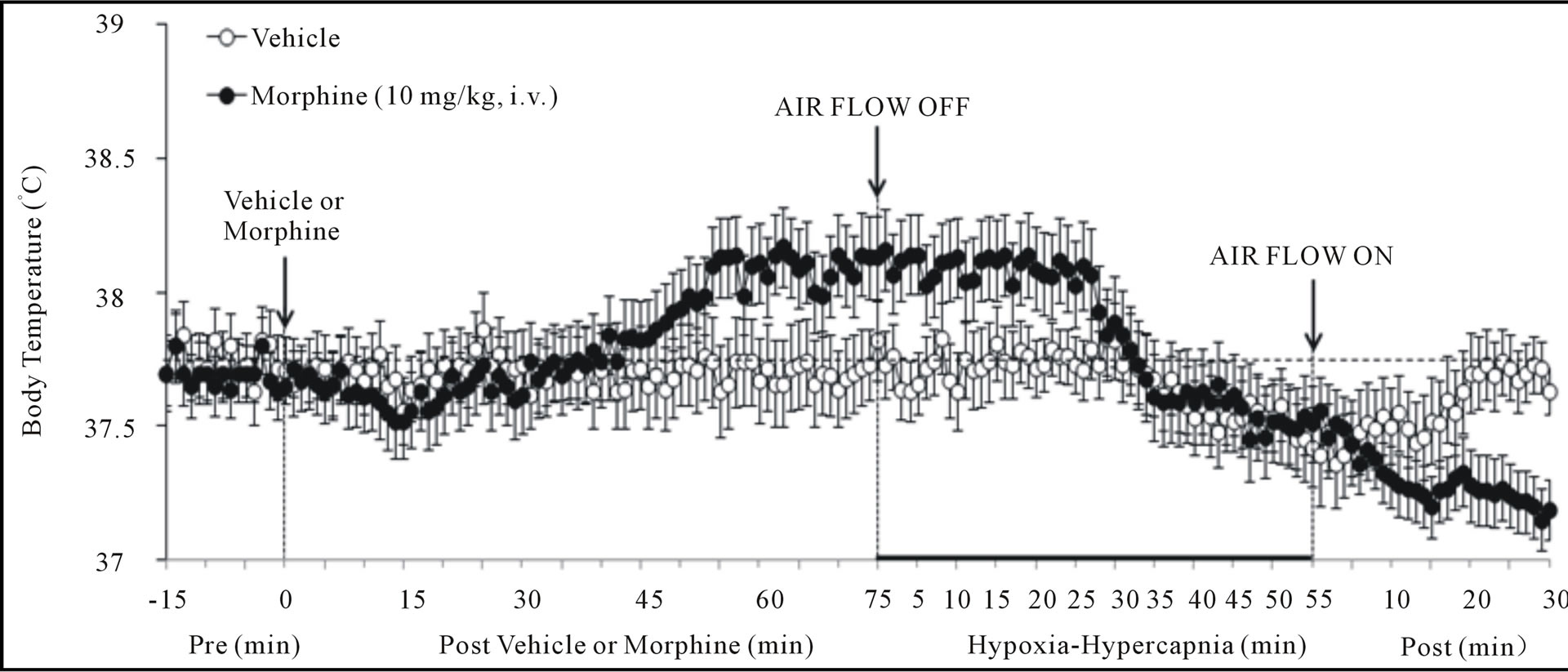
Figure 3. Changes in peak inspiratory flow (upper panel), peak expiratory flow (middle panel) and body temperature (bottom panel) elicited by injections of vehicle or morphine (10 mg/kg, i.v.) and subsequent exposure to hypoxia-hypercapnia (air flow off) and return to room-air (air flow on). The data are presented as mean ± SEM. There were 6 rats in each group.
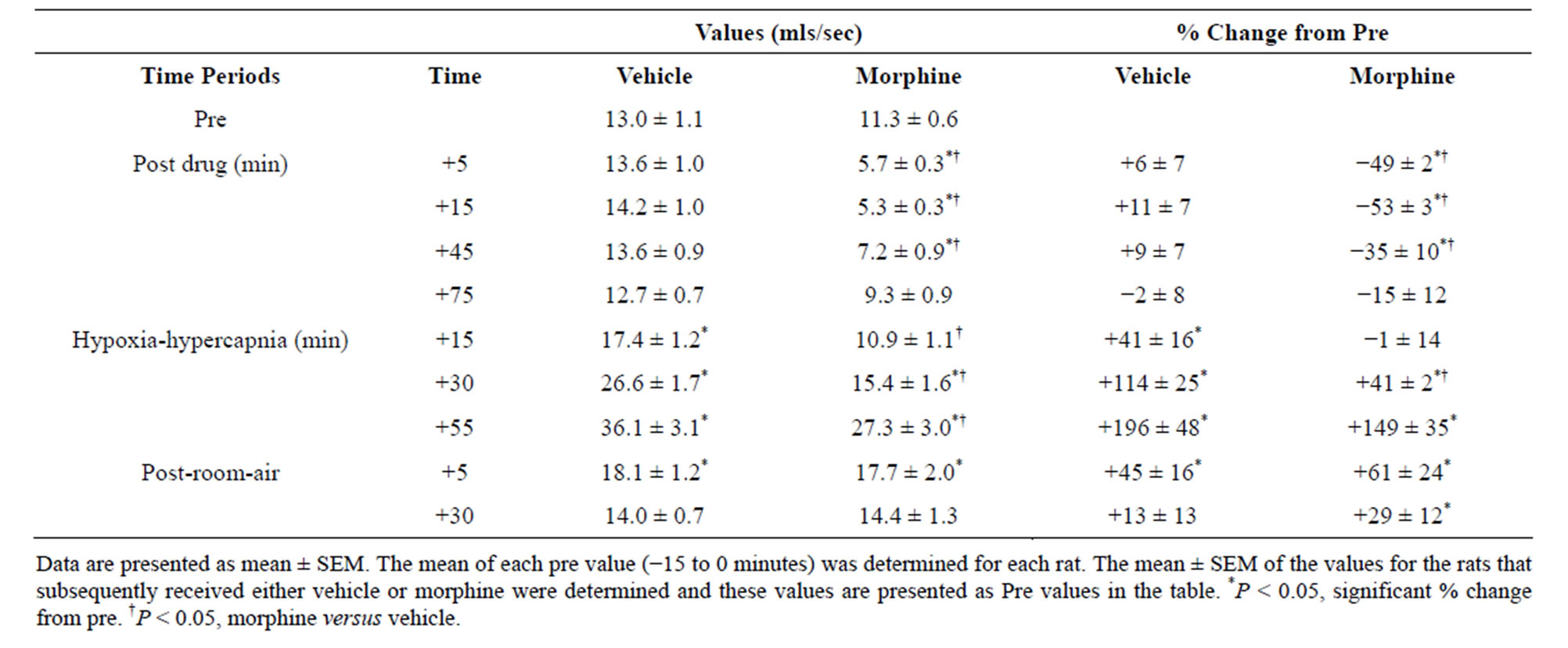
Table 3. Tidal volume/inspiratory time values during key times of the experiment.
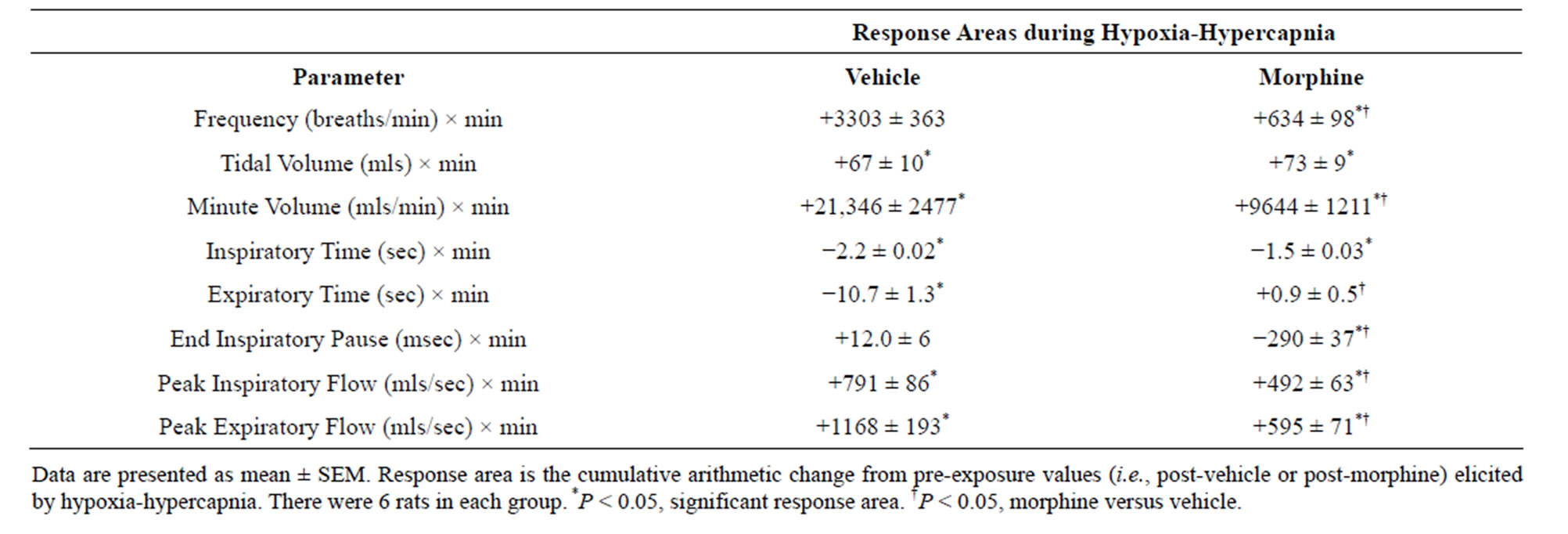
Table 4. Cumulative responses during exposure to hypoxia-hypercapnia in vehicleor morphine-injected rats.
carotid body sensitivity, and an increase pulmonary vascular resistance [3]. The ability of morphine to increase A-a gradient may be due to 1) a direct increase in pulmonary vascular resistance (thereby diminishing arterial blood flow to alveoli), 2) exacerbation of the hypoxic pulmonary vasoconstriction as a result of morphine-induced reduction in ventilation and the concomitant decreases in arterial blood pO2, and/or 3) an increase in airway resistance. Indeed, morphine and other opiates increase pulmonary vascular resistance in humans [42,43] and animals [7,10]. Morphine may also have negatively affected ABG via increases in upper and/or lower airway resistance [8,9]. Whatever the mechanism, it appears that a perfusion-ventilation mismatch is a major mechanism by which morphine diminished arterial pO2.
4.3. Morphine-Induced Changes in Ventilatory Parameters
Morphine elicited 1) a prompt decrease in Vm via reductions in Vt (but not fr) that had resolved by 75 min (the time of H-H exposure), 2) prompt increases in Ti and EIP but a prompt decrease in Te (i.e., active expiration was faster) that were still evident at 75 min, 3) prompt decreases in PIF and PEF still evident at 75 min, and 4) a prompt decrease in Vt/Ti (Respiratory Drive [24]) that had resolved by 75 min. As such, our data suggest that a principal mechanism by which morphine affects ABG chemistry is a diminished Vt, which is likely to involve central and/or peripherally-mediated decreases in neural drive to the internal intercostals and diaphragm [44,45]. The concomitant decreases in PIF and PEF suggests that morphine affected the mechanical efficiency of chest muscles driving ventilation. Despite the lack of inhibitory effect of morphine on fr, morphine lengthened Ti and EIP and so had definite effects on inspiratory control mechanisms. On the other hand, morphine decreased Te. This may be due to diminished Vt on inspiration requiring less time to exhale although direct stimulatory effects of morphine on pathways driving expiration cannot be discounted. In summary, the negative effects of morphine on ABG chemistry in our rats involved 1) reductions in the mechanical efficiency of breathing, 2) a possible increase in airway resistance, and 3) a decrease in pulmonary gas exchange. In contrast, it appears that the negative effects of morphine on ventilatory parameters still present at 75 min (i.e., increased Ti and EIP, decreases in PIF and PEF) did not translate into changes in ABG chemistry.
4.4. Ventilatory Responses during and Following Hypoxic-Hypercapnic Challenge
Exposure of VEH rats to H-H elicited gradual and sustained changes in ventilation including 1) an increase in Vm via increases in fr and Vt, 2) decreases in Ti and Te with no change in EIP, 3) increases in PIF and PEF, and 4) an increase in Vt/Ti. Accordingly, H-H elicited active and sustained increases in inspiratory and expiratory drive. The mechanisms driving the effects of H-H on ventilatory drive will involve carotid body responses to hypoxia and H+ ions, central sensors of CO2/H+ ions [30,45], and the direct synergistic action between hypoxia and hypercapnia at the level of the carotid bodies [45]. Morphine blunted the H-H-induced increases in fr and Vt/Ti. Since these parameters were not diminished before exposure to H-H, it appears that morphine elicited insidious effects on central ventilatory control structures such as the retrotrapezoid nucleus (RTN), which controls inspiratory and expiratory drive, receives major polysynaptic inputs from the carotid bodies, and is exquisitely responsive to changes in CO2 via intrinsic pHsensitive mechanisms [46]. Indeed, the activity of RTN neurons in anesthetized rats is modestly depressed by morphine, which also causes a downward shift in the relationship between RTN neuron activity and end-tidal CO2 (the level of end-tidal CO2 necessary for the unit to be active was elevated by morphine) [47]. Morphine did not blunt the increases in Vt elicited by H-H. Without reference to the other effects of morphine, this finding suggests that morphine did not affect central neural drive to the chest or diaphragm. However, exposure to H-H elicited smaller increases in PIF and PEF in MOR rats. It should be remembered that resting PIF and PEF were still depressed in MOR rats (−22% ± 6% and −17% ± 4%, respectively, P < 0.05 for both values) immediately prior to H-H. Accordingly, whereas morphine did not affect the ability of the ventilatory system to achieve a Vt equal to that in VEH rats, it did affect the dynamics of active inspiratory and active expiratory ventilation, compromising the system’s ability to reach peak performance. Our finding that morphine diminished the H-H responses when its effects on ABG chemistry had resolved, extend evidence that opioids inhibit central reactivity to hypercapnia in humans and animals [3,6,12,19] and depress responses of carotid body chemoafferents to hypoxia and hypercapnia (see Introduction) when the effects of opiates on ventilatory function are in full effect.
In VEH rats, all parameters returned to pre-H-H levels upon return to room-air. In MOR rats, fr and Vm remained elevated whereas Te remained decreased for fully 30 min after return to room-air. These novel findings suggest that morphine and/or metabolites inhibit re-adjustment of ventilatory parameters when room-air is available. This possibility is supported by the finding that EIP (increased by morphine, values equal to pre-morphine levels at the end of the H-H exposure) actually rose again upon return of room-air. It could be argued that morphine and/or M3G altered the activities of signaling elements in the carotid body and/or brain such that these elements were not responsive to the return to normal environmental levels of O2 and CO2.
4.5. Mechanisms Responsible for the Delayed Effects of Morphine—Morphine Metabolites
Bhargava et al. [48] found that 1) plasma concentrations of morphine after injection of a 10 mg/kg dose in rats reached »1000 ng/ml in less than 1 min and were »100 ng/ml after 60 min, 2) the hyperthermic effects paralleled the rise and decay in plasma morphine concentrations, and 3) plasma morphine concentrations at 360 min were still »100 ng/ml although Bt had normalized. As such, it is unlikely that plasma levels of morphine present at +75 min are entirely responsible for the depressed ventilatory responses to H-H although tissue sequestration may have delayed effects beyond the peak increases in plasma levels. It is also possible that these concentrations of morphine are insufficient to affect central/peripheral drive whereas they can blunt the mechanisms responsible for sensing/expressing ventilatory responses to H-H. In rats, (+)-morphine is metabolized to morphine-3-glucuronide (M3G) [49]. Maximal plasma levels of M3G occur in 30 min of morphine administration and the plasma elimination half-life is greater than 200 min [50,51]. M3G has a low affinity for opiate receptors [52-54] and minimal analgesic or respiratory depressant activity [52,55]. However, M3G is not inert [see 50] since central injections of M3G cause behavioral excitation [56-58], excessive grooming and epiliptoform discharges [56,59] whereas intrathecal injections elicit hyperaesthesia and allodynia [57]. The findings that the responses to M3G were exacerbated by the opioid receptor antagonist, naloxone [56,59], suggest that the responses were mediated by non-opioid receptors. The development of drugs that inhibit the effects of M3G may lead to a better understanding of the mechanisms by which morphine exerts its latent deleterious effects on ventilatory control processes.
5. CONCLUSION
Opioid-induced hypoventilation, upper airway obstructtion, and destabilization of breathing during sleep are major clinical concerns [60,61]. Our study suggests that patients may be under threat from opioids that blunt the ventilatory responses to hypoxia and/or hypercapnia even when the effects of opioids on ABG chemistry had subsided. Adults [60] and children [61] with obstructive sleep apnea (OSA) are at higher risk for respiratory complications post-operatively when opioids are routinely given. Moreover, humans with OSA or Obesity Hypoventilation Syndrome [29,30] would be especially prone to respiratory collapse following opioid administration due to the inability of chemoafferent systems to trigger appropriate ventilatory responses. These insidious effects of opioids may be exerted especially when the patient is asleep [62]. In humans, (+)-morphine is primarily metabolized to M6G via UDP-glucuronosyltransferase 2B7 [49,63]. Similar to morphine, M6G elicits analgesia and respiratory depression in animals via activation of µ- opioid receptors [50,64,65]. However, M6G minimally affects resting ventilation in humans yet blunts the ventilatory response to hypercapnia challenge [19]. As reviewed by Kilpatrick and Smith [65], further work is required to better understand the activity profile of M6G in humans, including whether M6G and morphine differentially access areas of the brain involved in respiratory control or whether µ-opioid receptors activated by these opioids differ in their regulation or pharmacology. Moreover, it is evident that the pharmacological actions of M3G in rats [49-51] need further evaluation with respect to the ability of this metabolite to affect ventilatory responses to hypoxic and hypercapnic challenges.
REFERENCES
- Shook, J.E., Watkins, W.D. and Camporesi E.M. (1990) Differential roles of opioid receptors in respiration, respiratory disease, and opiate-induced respiratory depresssion. American Review of Respiratory Disease, 142, 895- 909. doi:10.1164/ajrccm/142.4.895
- Cashman, J.N. and Dolin, S.J. (2004) Respiratory and haemodynamic effects of acute postoperative pain management: Evidence from published data. British Journal of Anaesthesia, 93, 212-223. doi:10.1093/bja/aeh180
- Dahan, A., Aarts, L. and Smith, T.W. (2010) Incidence, reversal, and prevention of opioid-induced respiratory depression. Anesthesiology, 112, 226-238. doi:10.1097/ALN.0b013e3181c38c25
- Berkenbosch, A., Teppema, L.J., Olievier, C.N. and Dahan, A. (1997) Influences of morphine on the ventilatory response to isocapnic hypoxia. Anesthesiology, 86, 1342- 1349. doi:10.1097/00000542-199706000-00016
- Dahan, A., Sarton, E., Teppema, L. and Olievier, C. (1998) Sex-related differences in the influence of morphine on ventilatory control in humans. Anesthesiology, 88, 903- 913. doi:10.1097/00000542-199804000-00009
- Sarton, E., Teppema, L. and Dahan, A. (1999) Sex differences in morphine-induced ventilatory depression reside within the peripheral chemoreflex loop. Anesthesiology, 90, 1329-1338. doi:10.1097/00000542-199905000-00017
- Schurig, J.E., Cavanagh, R.L. and Buyniski, J.P. (1978) Effect of butorphanol and morphine on pulmonary mechanics, arterial blood pressure and venous plasma histamine in the anesthetized dog. Archives Internationales de Pharmacodynamie et de Thérapie, 233, 296-304.
- Willette, R.N., Krieger, A.J. and Sapru, H.N. (1982) Opioids increase laryngeal resistance and motoneuron activeity in the recurrent laryngeal nerve. European Journal of Pharmacology, 80, 57-63. doi:10.1016/0014-2999(82)90177-7
- Willette, R.N., Barcas, P.P., Krieger, A.J. and Sapru, H.N. (1983) Pulmonary resistance and compliance changes evoked by pulmonary opiate receptor stimulation. European Journal of Pharmacology, 91, 181-188. doi:10.1016/0014-2999(83)90463-6
- Hakim, T.S., Grunstein, M.M. and Michel, R.P. (1992) Opiate action in the pulmonary circulation. Pulmonary Pharmacology, 5, 159-165. doi:10.1016/0952-0600(92)90036-G
- Weinger, M.B. and Bednarczyk, J.M. (1994) Atipamezole, an alpha 2 antagonist, augments opiate-induced muscle rigidity in the rat. Pharmacology, Biochemistry and Behavior, 49, 523-529. doi:10.1016/0091-3057(94)90064-7
- Campbell, C., Weinger, M.B. and Quinn, M. (1995) Alterations in diaphragm EMG activity during opiate-induced respiratory depression. Respiratory Physiology, 100, 107-117. doi:10.1016/0034-5687(94)00119-K
- Zhang, Z., Xu, F., Zhang, C. and Liang, X. (2009) Opioid μ-receptors in medullary raphe region affect the hypoxic ventilation in anesthetized rats. Respiratory Physiology and Neurobiology, 168, 281-288. doi:10.1016/j.resp.2009.07.015
- McQueen, D.S. and Ribeiro, J.A. (1980) Inhibitory actions of methionine-enkephalin and morphine on the cat carotid chemoreceptors. British Journal of Pharmacology, 71, 297-305.
- McQueen, D.S. and Ribeiro, J.A. (1981) Effects of betaendorphin, vasoactive intestinal polypeptide and cholecystokinin octapeptide on cat carotid chemoreceptor activity. Quarterly Journal of Experimental Physiology, 66, 273-284.
- Zimpfer, M., Beck, A., Mayer, N., Raberger, G. and Steinbereithner, K. (1983) Effects of morphine on the control of the cardiovascular system by the carotid-sinus-reflex and by the carotid chemoreflex. Anaesthesist, 32, 60-66.
- Kirby, G.C. and McQueen, D.S. (1986) Characterization of opioid receptors in the cat carotid body involved in chemosensory depression in vivo. British Journal of Pharmacology, 88, 889-898. doi:10.1111/j.1476-5381.1986.tb16263.x
- Mayer, N., Zimpfer, M., Raberger, G. and Beck, A. (1989) Fentanyl inhibits the canine carotid chemoreceptor reflex. Anesthesia and Analgesia, 69, 756-762. doi:10.1213/00000539-198912000-00012
- Peat, S.J., Hanna, M.H., Woodham, M., Knibb, A.A. and Ponte, J. (1991) Morphine-6-glucuronide: Effects on ventilation in normal volunteers. Pain, 45, 101-104. doi:10.1016/0304-3959(91)90170-3
- Torda, T.A. (1981) Alveolar-arterial oxygen tension difference: A critical look. Anaesthia and Intensive Care, 9, 326-330.
- Stein, P.D., Goldhaber, S.Z. and Henry, J.W. (1995) Alveolar-arterial oxygen gradient in the assessment of acute pulmonary embolism. Chest, 107, 139-143. doi:10.1378/chest.107.1.139
- Kanbar, R., Stornetta, R.L., Cash, D.R., Lewis, S.J. and Guyenet, P.G. (2010) Photostimulation of phox2b medullary neurons activates cardiorespiratory function in conscious rats. American Journal of Respiratory Critical Care Medicine, 182, 1184-1194. doi:10.1164/rccm.201001-0047OC
- Young, A.P., Gruber, R.B., Discala, J.F., May, W.J., Palmer, L.A. and Lewis, S.J. (2013) Co-activation of μ- and δ-opioid receptors elicits tolerance to morphine-induced ventilatory depression via generation of peroxynitrite. Respiratory Physiology and Neurobiology, 186, 255-264. doi:10.1016/j.resp.2013.02.028
- Moss, I.R., Brown, K.A. and Laferriere, A. (2006) Recurrent hypoxia in rats during development increases subsequent respiratory sensitivity to fentanyl. Anesthesiology, 105, 715-718. doi:10.1097/00000542-200610000-00017
- Fairchild, K.D., Saucerman, J.J., Raynor, L.L., Sivak, J.A., Xiao, Y., Lake, D.E. and Moorman, J.R. (2009) Endotoxin depresses heart rate variability in mice: Cytokine and steroid effects. American Journal of Physiology— Regulatory, Integrative and Comparative Physiology, 297, R1019-R1027. doi:10.1152/ajpregu.00132.2009
- Hayashi, F., Yoshida, A., Fukuda, Y. and Honda, Y. (1982) CO2-ventilatory response of the anesthetized rat by rebreathing technique. Pflugers Archives, 393, 77-82. doi:10.1007/BF00582395
- Pauluhn, J. and Thiel, A. (2007) A simple approach to validation of directed-flow nose-only inhalation chambers. Journal of Applied Toxicology, 27, 160-167. doi:10.1002/jat.1188
- Bourke, D.L. and Warley, A. (1989) The steady-state and rebreathing methods compared during morphine administration in humans. Journal of Physiology (Lond), 419, 509-517.
- Lévy, P., Bonsignore, M.R. and Eckel, J. (2009) Sleepdisordered breathing and metabolic consequences. European Respiratory Journal, 34, 243-260. doi:10.1183/09031936.00166808
- Dempsey, J.A., Veasey, S.C., Morgan, B.J. and O’Donnell, C. (2010) Pathophysiology of sleep apnea. Physiology Reviews, 90, 47-112. doi:10.1152/physrev.00043.2008
- Fewell, J.E. and Konduri, G.G. (1988) Repeated exposure to rapidly developing hypoxemia influences the interaction between oxygen and carbon dioxide in initiating arousal from sleep in lambs. Pediatric Research, 24, 28- 33. doi:10.1203/00006450-198807000-00008
- Wallenstein, S., Zucker, C.L. and Fleiss, J.L. (1980) Some statistical methods useful in circulation research. Circulation Research, 47, 1-9. doi:10.1161/01.RES.47.1.1
- Quock, R.M., Vaughn, L.K., Barlament, J. and Wojcechowskyj, J.A. (1985) Sex and strain differences in morphine-induced temperature effects in WKYs and SHRs. Brain Research Bulletin, 14, 323-326. doi:10.1016/0361-9230(85)90192-3
- Jorenby, D.E., Keesey, R.E. and Baker, T.B. (1988) Characterization of morphine’s excitatory effects. Behavioral Neuroscience, 102, 975-985. doi:10.1037/0735-7044.102.6.975
- Kaminski, R.P., Forster, H.V., Klein, J.P., Pan, L.G., Bisgard, G.E. and Hamilton, L.H. (1982) Effect of elevated PICO2 on metabolic rate in humans and ponies. Journal of Applied Physiology, 52, 1623-1628.
- Whipp, B.J. and Wassermann, K. (1970) Effect of body temperature on the ventilatory response to exercise. Respiratory Physiology, 8, 354-360. doi:10.1016/0034-5687(70)90042-3
- Henry, J.G. and Bainton, C.R. (1974) Human core temperature increase as a stimulus to breathing during moderate exercise. Respiratory Physiology, 21, 183-191. doi:10.1016/0034-5687(74)90093-0
- Biancardi, V., Da Silva, L.T., Bícego, K.C. and Gargaglioni, L.H. (2010) Role of Locus coeruleus noradrenergic neurons in cardiorespiratory and thermal control during hypoxia. Respiratory Physiology and Neurobiology, 170, 150-156. doi:10.1016/j.resp.2009.12.004
- Barros, R.C. and Branco, L.G. (1998) Effect of nitric oxide synthase inhibition on hypercapnia-induced hypothermia and hyperventilation. Journal of Applied Physiology, 85, 967-972.
- Mortola, J.P. and Seifert, E.L. (2002) Circadian patterns of breathing. Respiratory Physiology and Neurobiology, 131, 91-100. doi:10.1016/S1569-9048(02)00040-X
- Gautier, H., Bonora, M. and Trinh, H.C. (1993) Ventilatory and metabolic responses to cold and CO2 in intact and carotid body-denervated awake rats. Journal of Applied Physiology, 75, 2570-2579.
- Popio, K.A., Jackson, D.H., Ross, A.M., Schreiner, B.F. and Yu, P.N. (1978) Hemodynamic and respiratory effects of morphine and butorphanol. Clinical Pharmacology and Therapeutics, 23, 281-287.
- Mitaka, C., Sakanishi, N., Tsunoda, Y. and Mishima, Y. (1985) Comparison of hemodynamic effects of morphine, butorphanol, buprenorphine and pentazocine on ICU patients. The Bulletin of Tokyo Medical and Dental University, 32, 31-39.
- Rankin, J. and Dempsey, J.A. (1967) Respiratory muscles and the mechanisms of breathing. American Journal of Physical Medicine, 46, 198-244.
- Teppema, L.J. and Dahan, A. (2010) The ventilatory response to hypoxia in mammals: Mechanisms, measurement, and analysis. Physiology Reviews, 90, 675-754. doi:10.1152/physrev.00012.2009
- Guyenet, P.G., Stornetta, R.L., Abbott, S.B., Depuy, S.D., Fortuna, M.G. and Kanbar, R. (2010) Central CO2-chemoreception and integrated neural mechanisms of cardiovascular and respiratory control. Journal of Applied Physiology, 108, 995-1002. doi:10.1152/japplphysiol.00712.2009
- Fortuna, M.G., Stornetta, R. L., West, G.H. and Guyenet, P.G. (2009) Activation of the retrotrapezoid nucleus by posterior hypothalamic stimulation. Journal of Physiology, 587, 5121-5138. doi:10.1113/jphysiol.2009.176875
- Bhargava, H.N., Villar, V.M., Gulati, A. and Chari, G. (1991) Analgesic and hyperthermic effects of intravenously administered morphine in the rat are related to its serum levels. Journal of Pharmacology and Experimental Therapeutics, 258, 511-516.
- Hasegawa, Y., Kishimoto, S., Shibatani, N., Nomura, H., Ishii, Y., Onishi, M., Inotsume, N., Takeuchi, Y. and Fukushima, S. (2010) The pharmacokinetics of morphine and its glucuronide conjugate in a rat model of streptozotocin-induced diabetes and the expression of MRP2, MRP3 and UGT2B1 in the liver. Journal of Pharmacy and Pharmacology, 62, 310-314. doi:10.1211/jpp.62.03.0004
- Christrup, L.L. (1997) Morphine metabolites. Acta Anaesthesiologica Scandinavica, 41, 116-122. doi:10.1111/j.1399-6576.1997.tb04625.x
- Zheng, M., McErlane, K.M. and Ong, M.C. (1998) Highperformance liquid chromatography-mass spectrometrymass spectrometry analysis of morphine and morphine metabolites and its application to a pharmacokinetic study in male Sprague-Dawley rats. Journal of Pharmaceutical and Biomedical Analysis, 16, 971-980. doi:10.1016/S0731-7085(97)00094-0
- Christensen, C.B. and Jargensen, L.N. (1987) Morphine- 6-glucuronide has high affinity for the opioid receptor. Pharmacology and Toxicology, 60, 75-76. doi:10.1111/j.1600-0773.1987.tb01724.x
- Pasternak, G.W., Bodnar, R.J., Clark, J.A. and Inturrisi, C.E. (1987) Morphine-6-glucuronide, a potent mu agonist. Life Sciences, 41, 2845-2849. doi:10.1016/0024-3205(87)90431-0
- Paul, D., Standifer, K.M., Inturrisi, C.E. and Pasternak, G.W. (1989) Pharmacological characterization of morphine-6 beta-glucuronide, a very potent morphine metabolite. Journal of Pharmacology and Experimental Therapeutics, 251, 477-483.
- Shimomura, K., Kamata, O., Ueki, S., Ida, S. and Oguri, K. (1971) Analgesic effect of morphine glucuronides. Tohoku Journal of Experimental Medicine, 105, 45-52. doi:10.1620/tjem.105.45
- Labella, F.S., Pinsky, C. and Havlicek, V. (1979) Morphine derivatives with diminished opiate receptor potency show enhanced central excitatory activity. Brain Research, 174, 263-271. doi:10.1016/0006-8993(79)90849-7
- Yaksh, T.L., Harty, G.J. and Onofrio, B.M. (1986) High doses of spinal morphine produce a nonopiate receptor mediated hyperesthesia: Clinical and theoretic implications. Anesthesiology, 64, 590-597. doi:10.1097/00000542-198605000-00008
- Smith, M.T., Watt, J.A. and Cramond, T. (1990) Morphine-3-glucuronide—A potent antagonist of morphine analgesia. Life Sciences, 47, 579-585. doi:10.1016/0024-3205(90)90619-3
- Bartlett, S.E., Cramond, T. and Smith, M.T. (1994) The excitatory effects of morphine-3-glucuronide are attenuated by LY274614, a competitive NMDA receptor antagonist and by midazolam, an antagonist at the benzodiazepine site on the GABAA receptor complex. Life Sciences, 54, 687-694. doi:10.1016/0024-3205(94)00552-4
- Kryger, M.H. (2000) Management of obstructive sleep apnea-hypopnea syndrome: Overview. In: Kryger, M.H., Roth, T., Dement, W.C. and Saunders, W.B., Eds., Principles and Practice of Sleep Medicine, 3rd Edition, Elsevier, Philadelphia, pp. 940-954.
- Brown, K.A., Laferriere, A., Lakheeram, I. and Moss, I.R. (2006) Recurrent hypoxemia in children is associated with increased analgesic sensitivity to opiates. Anesthesiology, 105, 665-669. doi:10.1097/00000542-200610000-00009
- Wang, D. and Teichtahl, H. (2007) Opioids, sleep architecture and sleep-disordered breathing. Sleep Medicine Reviews, 11, 35-46. doi:10.1016/j.smrv.2006.03.006
- Stain, F., Barjavel, M.J., Sandouk, P., Plotkine, M., Scherrmann, J.M. and Bhargava, H.N. (1995) Analgesic response and plasma and brain extracellular fluid pharmacokinetics of morphine and morphine-6-beta-D-glucuronide in the rat. Journal of Pharmacology and Experimental Therapeutics, 274, 852-857.
- Romberg, R., Sarton, E., Teppema, L., Matthes, H.W., Kieffer, B.L. and Dahan, A. (2003) Comparison of morphine-6-glucuronide and morphine on respiratory depressant and antinociceptive responses in wild type and mu-opioid receptor deficient mice. British Journal of Anaesthesia, 91, 862-870. doi:10.1093/bja/aeg279
- Kilpatrick, G.J. and Smith, T.W. (2005) Morphine-6-glucuronide: Actions and mechanisms. Medicinal Research Reviews, 25, 521-544.doi:10.1002/med.20035
NOTES
*This study was supported by grants from Galleon Pharmaceuticals (S.J.L).
Conflicts of Interest: None.
#Corresponding author.