Open Journal of Molecular and Integrative Physiology
Vol.2 No.1(2012), Article ID:17239,4 pages DOI:10.4236/ojmip.2012.21005
Interaction between peroxisomes and mitochondria in fatty acid metabolism
![]()
Laboratory of Metabolic Biochemistry and Nutrition, Inserm U866, Université de Bourgogne, Dijon, France
Email: jean.demarquoy@u-bourgogne.fr
Received 7 November 2011; revised 14 September 2011; accepted 22 October 2011
Keywords: Peroxisome; Mitochondria; Fatty Acid Metabolism; Beta Oxidation
ABSTRACT
Peroxisomes and mitochondria are ubiquitously found organelles. They both are dynamic structures able to divide, to fuse and to undergo autophagic processes. Their activities are dependent on proteins that are, for most (mitochondria) or all (peroxisome) of them, synthesized in the cytosol from the nuclear genome. Nevertheless, the membrane structures and the DNA content differ between these two organelles. Mitochondria possess a small circular genome while peroxisomes don’t. The control of their dynamic is dependent on specific factors even if some of those are able to affect both. These two organelles are metabolically connected: they are both involved in lipid metabolism. They are both able to beta oxidize fatty acids and are implicated in ROS production. However, their precise function in these metabolic pathways and their physiological functions are different. While mitochondrial metabolism is closely related to energy production, peroxisome does not seem to be associated with energy production but with the production of bioactive molecules and in detoxification processes.
1. INTRODUCTION
Fatty acids represent a complex family of bioactive molecules. Even if they share the same kind of backbone, differences in their length, the number of double bounds and their location give to these molecules completely different metabolic fate and physiological activities. The metabolism of fatty acids may take place in several cell compartments but beta oxidation of these lipids is restricted to only 2 compartments: the mitochondria and the peroxisomes. The objective of this review is to indicate the respective properties of these two organelles and to identify similarities and differences in their metabolic activities.
Mitochondrial beta oxidation is clearly linked to energy production and ATP needs for the cell. It has been shown that while energy is required by the cell, mitochondrial metabolism increases, and apart from very non-physiological states, no increase in mitochondrial beta oxidation can be observed if no need in energy is necessary. However, a metabolic link between mitochondria and peroxisomes has been proposed. Very long chain fatty acids will be oxidized in the peroxisome but as peroxisomal oxidation is not complete and leads to shortened fatty acids, it has been hypothesized that these shortened fatty acids can be taken in charge by mitochondrial metabolism and be fully oxidized. This has never been clearly shown but seems possible. On the other hand, peroxisomal metabolism can also be regarded as a specific metabolism that will detoxify specific fatty acids as branched fatty acids or be involved in the synthesis of specific fatty acids.
To better understand the respective properties of each organelle, peroxisome and mitochondrial structure, renewal and physiology are precised in this manuscript. At last, fatty acid metabolisms in those two cell organelles are described and compared.
2. THE STRUCTURE OF MITOCHONDRIA AND PEROXISOMES
Mitochondria and peroxisomes are organelles present in virtually any cell types. Their sizes are comparable, ranging from 0.1 to 1 µm. Peroxisomes are limited by a single membrane and do not contain any DNA molecule, their protein content derives from nuclear genes. Mitochondria are limited by a double membrane system and contain a short DNA sequence. Mitochondrial DNA (mtDNA) is circular and encodes for several proteins of the respiratory chain [1]. This is a 16 kbase DNA containing 37 genes. Mutations in the mitochondrial DNA lead to various serious diseases including neurological disorders and metabolic diseases [2]. Although, most of the mitochondrial proteins are encoded by nuclear genes and are imported into the mitochondria [3].
Mitochondrial alterations seem also involved in the aging process probably in connection with the ROS metabolism (see paragraph on ROS) [4,5].
2.1. Mitochondria
The number and the shape of mitochondria in the cell are variable, mitochondria can take various shapes depending on the nature of the cell and its metabolism. They may appear as small spherical structures, as bean-shaped organelles or as elongated structures.
Mitochondria are dynamic organelles that can fuse, divide, move or be broken up. The structure of the mitochondria appeared to be associated with the cell metabolic status and the mechanisms of fission (division) and fusion that may occur in the cell [6]. Depending on the energy requirements of the cells, mitochondria change their structure, their shape and their function through alterations in the fission and fusion processes [7]. These changes allow for recruiting mitochondria where and when needed and to install new networks inside the cell.
Furthermore, it seems that these mechanisms of fission and fusion are critical for maintaining not only the structure but also the function and the stability of the mitochondria [8,9].
2.1.1. Fusion vs Fission in Mitochondria
In the cell, under regular conditions fusion and fission are well balanced. This participates in maintaining a normal mitochondrial number, adequate function and structure allowing the cell to respond to changing physicological conditions. It has been proposed that even if fusion and fission participate at equilibrating mitochondrial content and function, fusion will allow mitochondria to fuse their content and then enabling mtDNA repair, protein complementation and balance in metabolites. On the other hand, fission may participate in mtDNA segregation and may help removing altered mitochondria through mitophagy process.
Fusion requires several proteins, today three proteins have been identified as essential for mitochondrial fusion [10]. In mammals, there are two mitofusins (Mfn1 and Mfn2) and an enzyme called OPA1. Mfn1 and Mfn2 are large proteins, encoded by nuclear genes, with a GTPase activity and two transmembrane domains in the mitochondrial outer membrane. They also present a large domain facing the cytosol and are required for outer membrane fusion. The third enzyme called OPA1 is implicated in the inner membrane fusion.
To precise the roles of each of these proteins, various animal and cellular models were developed. Mfn1- and Mfn2-deficient mice were designed: they died at midgestation [11]. Embryonic cells lacking Mfn showed a drastic diminution in mitochondrial fusion resulting in fragmented mitochondria. More recently, transgenic mice expressing a pathogenic Mfn2 with a T105M mutation were created [12], they exhibited the clinical features of the Charcot-Marie-Tooth (CMT) disease, axonal type 2. A histologic analysis of the animal tissues showed a improper mitochondrial distribution in axons. Accordingly, mutations in the MFN2 gene were identified in patients with some forms of CMT (OMIM 608506 and 608507).
A mouse carrying a mutation in the Opa1 gene was created, this resulted in a severe reduction in the level of Opa1 protein in all tissues. The homozygous mutation induced premature death in embryons as soon as 13.5 days pc. Histologic studies from adult heterozygotes showed an increase in mitochondrial fission and fragmentation associated with neuronal degeneration [13] that lead to optic nerve atrophy and a visual function defect.
In mammals, mitochondrial fission is driven by Drp1, a dynamin-like protein [14]. Drp1 is a cytosolic enzyme that is recruited to mitochondria when fission occurs. This is a soluble protein with a GTPase activity located on the N-terminal domain. It also has a GTPase effector domain in the C-terminal domain. It is likely that, in mammalian cells, proteins that allow the anchorage of Drp1 onto mitochondria are required. Members of the dynamin subfamily contains a GTPase domain and include dynamin-1, -2 and -3. They have been shown to participate in endocytosis. Members of the dynaminrelated subfamily also contain the GTPase domain. They are implicated in either intracellular trafficking or mitochondrial morphology [15].
Several cell models were created to precise the role of Drp1. In 1998, a mutant form of DRP1 was expressed in COS-7 cells. The introduction of a mutation in the GTPase domain induced severe alterations in mitochondrial morphology and led to the formation of mitochondrial aggregates [16]. On the other hand, it was found that nitric oxide increased mitochondrial fission and induced neuronal damage and it was suggested that this regulation may, at least in part, be involved in the onset of the Alzheimer disease [17].
It was shown that a deletion in the Drp1 gene in mice was lethal at the embryonic stage [18]. Homozygote Drp1-/- embryos have smaller heart and liver and histological studies evidenced the presence of enlarged mitochondria. Peroxisomes were also larger in Drp1-/- cells. The analysis of apoptotic events in Drp1-/- cells suggested that Drp1 may be involved in later apoptotic events: e.g. cytochrome c release and caspase activation.
2.1.2. Mitochondria Transport
Mitochondria can migrate in the cell to special cell compartments. This seems especially true in neurons in which the location of mitochondria may be critical [19,20]. Mitochondrial transport inside mammalian cells is largely dependent on the microtubule network and transport is ensured by kinesin and dynein motors [21].
2.1.3. Mitophagy
Mitophagy is a mechanism that allows the degradation of mitochondria through an autophagic process [22]. Mitochondria will first be included into an autophagosome that will then fuse with lysosomes and its content hydrolyzed. Initially described as a mechanism involved in maintaining the integrity of cells during starvation, mitophagy seems also to participate at controlling mitochondrial number [23].
2.2. Peroxisome
The number of peroxisomes present in one cell seems to be associated with several cellular mechanisms that involved peroxisome generation and pexophagy.
The origin and the generation of peroxisome remains an open question. Whether peroxisomes can replicate autonomously like mitochondria or derive from other cell organelles (e.g. lysosomes or endoplasmic reticulum) has not found a definitive answer yet [24]. The autonomous replication of peroxisome has been proposed a few decades ago. Today, this mechanism seems doubtful and peroxisomes appeared more likely to derive from endoplasmic reticulum (ER) or from the fission of preexisting peroxisomes [25].
2.2.1. Origin and Turnover
The ER origin of peroxisome has been demonstrated on cells lacking peroxisomes but still able to generate new peroxisomes. This observation led to the conclusion that peroxisome may derive from the internal system of the ER [26]. However, the precise mechanisms involved in this de novo formation remain to be identified and furthermore the mechanisms identified in yeast are not likely to occur in mammalian cells [24].
Besides the de novo synthesis, peroxisome can also arise from the division of already existing peroxisomes. Part of this process is known, at least three regulatory elements have characterized: Pex11p, Pex25p and Pex27p positively control this phenomenon. The elongation of peroxisome is clearly under the control of Pex11p [27]. In mammalian cells, however, the other steps have not been fully identified.
Peroxisome is capable of adapting its size, its number and its content depending on the needs of the cell and its environment [24]. Peroxisome proliferation is under a transcriptional control exerted by fatty acids. Fission also participates in regulating peroxisome number.
It was reported that DRP1 was essential for the peroxisome division. DRP1a and PEX11 proteins did not seem to interact directly but the isoform a of DRP1 and PEX11- beta are overexpressed during this process [28].
2.2.2. Pexophagy
The number of peroxisomes present in one cell is also controlled by an autophagic process called pexophagy [29]. Several mechanisms seem to allow such degradation of peroxisomes and under the control of physiological conditions. It is likely that pexophagic events are balanced with fission and de novo synthesis.
2.3. Protein Import
If the mitochondrial genome only encodes for a few proteins, more than 1500 proteins are present in the mitochondria and derive from nuclear genes and are imported into the mitochondria. In peroxisome, the lack of DNA makes that all proteins present in this organelle are encoded by nuclear genes and transported into the peroxisome.
2.3.1. Import of Mitochondrial Proteins
Despite the presence of a genome in mitochondria, most of the proteins present in this organelle are derived from nuclear genes and then imported into mitochondrial compartments. A recent paper extensively reviewed the mechanisms involved in the import of almost 1500 proteins into the mitochondria [30]. Nucleus-encoded mitochondrial proteins are recognized on the mitochondrial surface by receptors. To be recognized, these proteins have to exhibit a targeting signal and depending on the nature of this signal, the proteins are directed to their specific mitochondrial subcompartments (outer membrane, intermembrane space, inner membrane or matrix). Two main groups of targeting signals are distinguished: one group is characterized by the presence of a targeting signal at the amino-terminal extension of the precursor protein, this presequence is usually removed by a proteolytical attack. In the second group signals are not cleavable and remain present in the mature mitochondrialocated proteins [30].
2.3.2. Import of Peroxisomal Proteins
The knowledge on peroxisomal import of proteins is not as well defined as the one regarding mitochondrial proteins. Import of proteins inside the peroxisome involved an ATP-dependent process. Proteins to be imported into the peroxisome are recognized by receptors located at the surface of the peroxisome that interact with specific protein sequences. Two types of targeting signals have been identified: peroxisomal targeting signal 1 (PTS1) and PTS2. PTS2 is the less common signal. Proteins harboring this signal are taken in charge by the Pex7p receptor. PTS1 harboring proteins are much common and are imported by the Pex5p. The import process involves 3 different steps that have been reviewed in [31]. But contrarily to what happen in mitochondria, all the peroxisomal enzymes are encoded by nuclear genes.
3. THE METABOLIC CONNEXION
Fatty acids may undergo several metabolic pathways but most fatty acids are catabolized by the beta oxidative pathways. Mitochondria and peroxisomes are both able to beta oxidize fatty acids. However in mammals, both systems exhibit different specificities in terms of substrate and products and also different functions.
Describing all the functions of mitochondria will take much than a single review. To be very schematic and simplistic, one may consider that the major role of mitochondrion is to control energy production and produce ATP. Ultimately, ATP is produced by the citric acid cycle also known as the Krebs cycle, but many pathways can provide substrate to the Krebs cycle and allow for ATP production. Among those is the beta oxidation of fatty acids. Besides fatty acid oxidation, mitochondria are implicated in the biosynthesis of haem, in the citric acid cycle and as cell physiology regulator as they participate in the apoptosis processes and in ageing.
3.1. Beta Oxidation in Mitochondria and Peroxisomes
Beta oxidation is the major pathway for the degradation of fatty acids. In eukaryotic microorganisms and in plant cell, peroxisomes are the unique site for the oxidation of fatty acids [32]. In mammalian cells, mitochondria and peroxisomes are able to oxidize fatty acids through a beta oxidation process sharing many similarities in terms of enzymatic reactions. However, the substrates, the enzymes involved, the final products and the overall goal of these beta oxidations are different [33,34].
In both the mitochondria and the peroxisome, the general schema of the beta oxidation is as follow: the activated fatty acid (acylCoA) is first dehygrogenated, then the double bound is hydrated. This intermediary compound is then dehydrogenated and finally cleaved. Starting with a C:n acyl this series of reactions will lead to a C:n-2 acyl (Figure 1).
Peroxisomes oxidize more complex fatty acids than mitochondria. Among the substrates for the peroxisomal beta oxidation are the very long chain fatty acids (>C22), the branched fatty acids, some prostaglandins and leukotrienes. On the other hand, mitochondria mainly oxidize long chain fatty acid provided by the food and supply acetyl-CoA for subsequent ATP synthesis. Mitochondrial metabolism is strictly dependent upon the en-
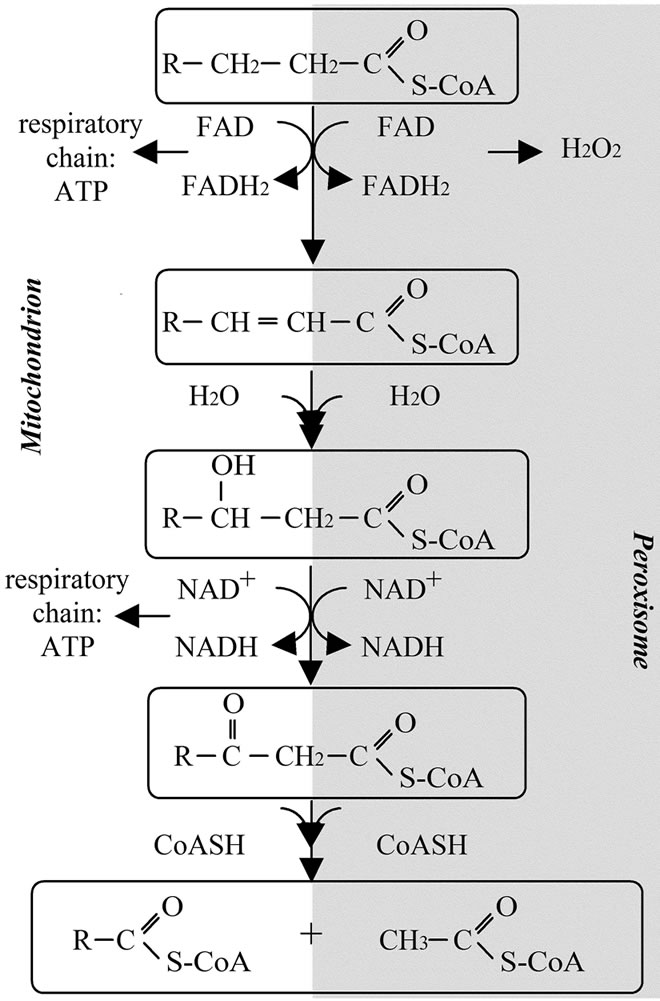
Figure 1. Schematic representation of mitochondrial and peroxisomal beta oxidations.
ergy need whereas peroxisomal metabolism is not. Mitochondrial beta oxidation is directly associated with the production of ATP whereas peroxisomal metabolism of fatty acid leads to the production of H2O2 (Figure 1).
While the reactions are roughly the same in both the mitochondrial and the peroxisomal beta oxidations, the precise nature of the enzymes is different.
3.1.1. Mitochondrial Fatty Acid Oxidation
In the mitochondria, fatty acid beta oxidation requires four reactions and involves prior to oxidation the esterification of fatty acids into acyl-CoA and the transfer of the acyls from the cytosol to the mitochondrial matrix. The formation of acyl-CoA takes place in the cytosol, it consists in an ATP-dependent reaction catalyzed by acylCoA synthases. Several acyl-CoA synthases exist with different types of substrates (either short-chain acyl-CoA or medium-chain acyl-CoA or long-chain acyl-CoA). Once activated acyl-CoA can cross the mitochondrial membrane: long chain acyl-CoA are taken in charge by a multiproteic system known as the carnitine system. This system is composed of two acyl-transferases: the carnitine palmitoyltransferase 1 and 2 and the carnitine acylcarnitine translocase. This system allows the transfer of acyl-CoA from the cytosol to the mitochondrial matrix.
The beta oxidation itself consists in several cycles each consisting in 4 enzymatic reactions that would remove 2C from a C:n acyl-CoA giving rise to a C:n-2 acyl-CoA. The four enzymes involved are successively the acyl-CoA dehydrogenase, the 2-enoyl-CoA hydratase, the 3-hydroxyacyl-CoA dehydrogenase and finally the 3-oxoacyl-CoA thiolase. For each enzyme several isoforms may exist. The mitochondrial beta oxidation is complete which means that the final product of this series of reactions is acetyl-CoA. The mitochondrial beta oxidation is coupled with the respiratory chain and the Krebs cycle and in fine led to the production of ATP [35].
3.1.2. Peroxisomal Fatty Acid Oxidation
The peroxisomal beta oxidation starts also with the conversion of the fatty acid (especially very long chain fatty acids or branched fatty acids) into acyl-CoA. The acylCoA may then enter into the peroxisome using a different system than in mitochondria. The entry of acyl-CoA, in the peroxisome, involves an ABC transporter (ABCD1) [36]. The first reaction is catalyzed by an acyl-CoA oxidase (ACOX) often considered as a key step in this pathway. Several isoforms of ACOX have been described, and all of them are dimeric FAD-containing proteins. In ACOX catalyzed reaction, the electrons from FAD are directly transferred to oxygen generating H2O2 while, in the mitochondrial reaction, the electrons are directed to the respiratory chain.
The following step in this pathway involves the reaction catalyzed by the multifunctional enzyme (MFE) that catalyzes the second and the third reactions of the pathway. Two MFE are present in the peroxisome: MFE-1 (also known as the L-bifunctional protein, LBP) and MFE-2 (also called the D-bifunctional protein, DBP). They catalyzed the same reaction with mirror image stereochemistry. Intriguingly, these two enzymes do not share many properties neither structural nor functional. The last reaction is catalyzed by the peroxisomal thiolase. It consists in the thiolytic cleavage of 3-ketoacyl-CoA to acetyl-CoA and acyl-CoA shortened by two carbon atoms [32].
Another major difference between mitochondrial and peroxisomal beta oxidation is the nature of the final products of the pathways. In mitochondria, acyl-CoA is fully degraded to acetyl-CoA whereas in the peroxisome, the reaction is incomplete giving rise to medium chain acyl-CoA [37]. These compounds are then taken in charge by the carnitine octanoyl transferase and converted into acyl-carnitine that can leave the peroxisome [38] and, at least for some of them, may be fully oxidized in the mitochondria. However, the part of peroxisome in energy production remains limited.
3.2. Reactive Oxygen Species: Mitochondria vs Peroxisomes
Mitochondria and peroxisomes are both directly implicated in the production and the scavenging of reactive oxygen species (ROS).
In mitochondria, the electron transport chain is the major pathway producing ROS. Superoxide ions are produced at the complex 1 level and superoxide can then be converted into more potent ROS. An uncontrolled production of ROS will lead to damage to all macromolecules present in the cells including lipids, proteins and DNA. In the mitochondria, the hypothesis of a vicious cycle involving ROS has been many times proposed. In this hypothesis, overproduction of ROS induced damage to mitochondrial enzymes and DNA, leading to alteration in the respiratory chain. Inadequate function in the respiratory chain will then increase the production of ROS. ROS overproduction in mitochondria leads to the release of various compounds including cytochrome c that trigger apoptosis [39,40]. Antioxidant enzymes such as MnSOD, glutathione reductase and antioxidant molecules such as thioredoxin, glutathione and NADH are present in the mitochondria and may limit oxidative damages [41].
Peroxisome metabolism strongly accounts for the ROS production. The peroxisome contains many oxidases that produce various types of ROS including nitric oxide, hydroxyl radicals, superoxide radicals and hydrogen peroxide [42]. Peroxisome also contains many antioxidant systems. Catalase, Cu/ZnSOD and glutathione peroxidase are among the enzymes present in the peroxisomal matrix that participate at the defense against oxidative damages.
4. CONCLUSIONS
Mitochondria and peroxisomes share various properties, they are both able to degrade fatty acids, but their objectives are different and the way to succeed also differs. Their functions are not redundant but cooperative. The biogenesis and the turnover of mitochondria and peroxisomes may show a few similarities but again the mechanisms involved are specific for each organelle as well as the control exerted on these mechanisms.
A few years ago, mitochondrial and peroxisomal metabolisms of fatty acids were regarded as closely linked. One hypothesis was then, than very long chain fatty acids were first oxidized in the peroxisome leading to the formation of shortened fatty acids as the peroxisomal beta oxidation is not complete. Products from the peroxisomal beta oxidation were then taken in charge by the mitochondria and fully degraded. This has to be tempered. One of the major aspects to be remembered is that mitochondrial metabolism is closely linked to energy production while peroxisomal metabolism is not. Mitochondrial beta oxidation is associated with respiratory chain and the tricarboxylic cycle and leads to the production of ATP. On the other hand peroxisomal metabolism is not, the function of peroxisome is more likely associated with mechanisms of detoxification and mechanisms of biosynthesis of bioactive molecules, i.e. specific fatty acids.
Another point that is distinct between mitochondrial and peroxisomal beta oxidations is the fact that peroxisomal beta oxidation is regarded as incomplete as the final product is not acetyl moieties but medium length acyls. One explanation may be that, in the peroxisome, the presence of the carnitine octanoyl transferase induces a metabolic competition for shortened acyls between the beta oxidation and the transesterification of acyls-CoA into acyl-carnitine and a subsequent metabolism other than the beta oxidation [43].
In conclusion, these two organelles may have similar metabolic pathways but their physiologic functions are obviously different. It is clear that these two organelles communicate with each other, however, the language they use has still to be determined.
REFERENCES
- Montoya, J., Lopez-Gallardo, E., Diez-Sanchez, C., Lopez-Perez, M.J. and Ruiz-Pesini, E. (2009) 20 years of human mtDNA pathologic point mutations: carefully reading the pathogenicity criteria. Biochimica et Biophysica Acta, 1787, 476-483. doi:10.1016/j.bbabio.2008.09.003
- Howell, N., Elson, J.L., Chinnery, P.F. and Turnbull, D.M. (2005) MtDNA mutations and common neurodegenerative disorders. Trends in Genetics, 21, 583-586. doi:10.1016/j.tig.2005.08.012
- Schmidt, O., Pfanner, N. and Meisinger, C. (2010) Mitochondrial protein import: From proteomics to functional mechanisms. Nature Reviews Molecular Cell Biology, 11, 655-667. doi:10.1038/nrm2959
- Kukat, A. and Trifunovic, A. (2009) Somatic mtDNA mutations and aging—Facts and fancies. Experimental Gerontology, 44, 101-105. doi:10.1016/j.exger.2008.05.006
- Pang, C.Y., Ma, Y.S. and Wei, Y.U. (2008) MtDNA mutations, functional decline and turnover of mitochondria in aging. Frontiers in Bioscience, 13, 3661-3675. doi:10.2741/2957
- Okamoto, K. and Shaw, J.M. (2005) Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annual Reviews Genetics, 39, 503-536. doi:10.1146/annurev.genet.38.072902.093019
- Chen, H. and Chan, D.C. (2009) Mitochondrial dynamics—fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Human Molecular Genetics, 18, R169-R176. doi:10.1093/hmg/ddp326
- Chan, D.C. (2006) Mitochondria: Dynamic organelles in disease, aging, and development. Cell, 125, 1241-1252. doi:10.1016/j.cell.2006.06.010
- Olichon, A., Guillou, E., Delettre, C., Landes, T., Arnaune-Pelloquin, L., Emorine, L.J., Mils, V., Daloyau, M., Hamel, C., Amati-Bonneau, P., et al. (2006) Mitochondrial dynamics and disease, OPA1. Biochimica et Biophysica Acta, 1763, 500-509. doi:10.1016/j.bbamcr.2006.04.003
- Detmer, S.A. and Chan, D.C. (2007) Functions and dysfunctions of mitochondrial dynamics. Nature Reviews Molecular Cell Biology, 8, 870-879. doi:10.1038/nrm2275
- Chen, H., Detmer, S.A., Ewald, A.J., Griffin, E.E., Fraser, S.E. and Chan, D.C. (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. The Journal of Cell Biology, 160, 189-200. doi:10.1083/jcb.200211046
- Detmer, S.A., Van de Velde, C., Cleveland, D.W. and Chan, D.C. (2008) Hindlimb gait defects due to motor axon loss and reduced distal muscles in a transgenic mouse model of Charcot-Marie-Tooth type 2A. Human Molecular Genetics, 17, 367-375. doi:10.1093/hmg/ddm314
- Davies, V.J., Hollins, A.J., Piechota, M.J., Yip, W., Davies, J.R., White, K.E., Nicols, P.P., Boulton, M.E. and Votruba, M. (2007) Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Human Molecular Genetics, 16, 1307-1318. doi:10.1093/hmg/ddm079
- Wakabayashi, J., Zhang, Z., Wakabayashi, N., Tamura, Y., Fukaya, M., Kensler, T.W., Iijima, M. and Sesaki, H. (2009) The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. The Journal of Cell Biology, 186, 805-816. doi:10.1083/jcb.200903065
- Otsuga, D., Keegan, B.R., Brisch, E., Thatcher, J.W., Hermann, G.J., Bleazard, W. and Shaw, J.M. (1998) The dynamin-related GTPase, Dnm1p, controls mitochondrial morphology in yeast. The Journal of Cell Biology, 143, 333-349. doi:10.1083/jcb.143.2.333
- Smirnova, E., Shurland, D.L., Ryazantsev, S.N. and Van de Bliek, A.M. (1998) A human dynamin-related protein controls the distribution of mitochondria. The Journal of Cell Biology, 143, 351-358. doi:10.1083/jcb.143.2.351
- Cho, D.H., Nakamura, T., Fang, J., Cieplak, P., Godzik, A., Gu, Z. and Lipton, S.A. (2009) S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science, 324, 102-105. doi:10.1126/science.1171091
- Ishihara, N., Nomura, M., Jofuku, A., Kato, H., Suzuki, S.O., Masuda, K., Otera, H., Nakanishi, Y., Nonaka, I., Goto, Y., et al. (2009) Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nature Cell Biology, 11, 958-966. doi:10.1038/ncb1907
- MacAskill, A.F. and Kittler, J.T. (2010) Control of mitochondrial transport and localization in neurons. Trends in Cell Biology, 20, 102-112. doi:10.1016/j.tcb.2009.11.002
- Zinsmaier, K.E., Babic, M. and Russo, G.J. (2009) Mitochondrial transport dynamics in axons and dendrites. Results Problems in Cell Differentiation, 48, 107-139. doi:10.1007/400_2009_20
- Pathak, D., Sepp, K.J. and Hollenbeck, P.J. (2010) Evidence that myosin activity opposes microtubule-based axonal transport of mitochondria. The Journal of Neuroscience, 30, 8984-8992. doi:10.1523/JNEUROSCI.1621-10.2010
- Burman, C. and Ktistakis, N.T. (2010) Autophagosome formation in mammalian cells. Seminars in Immunopathology, 32, 397-413. doi:10.1007/s00281-010-0222-z
- Kanki, T. and Klionsky, D.J. (2008) Mitophagy in yeast occurs through a selective mechanism. The Journal of Biological Chemistry, 283, 32386-32393. doi:10.1074/jbc.M802403200
- Smith, J.J. and Aitchison, J.D. (2009) Regulation of peroxisome dynamics. Current Opinion in Cell Biology, 21, 119-126. doi:10.1016/j.ceb.2009.01.009
- Titorenko, V.I. and Mullen, R.T. (2006) Peroxisome biogenesis: The peroxisomal endomembrane system and the role of the ER. The Journal of Cell Biology, 174, 11-17. doi:10.1083/jcb.200604036
- South, S.T. and Gould, S.J. (1999) Peroxisome synthesis in the absence of preexisting peroxisomes. The Journal of Cell Biology, 144, 255-266. doi:10.1083/jcb.144.2.255
- Koch, A., Schneider, G., Luers, G.H. and Schrader, M. (2004) Peroxisome elongation and constriction but not fission can occur independently of dynamin-like protein 1. Journal of Cell Science, 117, 3995-4006. doi:10.1242/jcs.01268
- Li, X. and Gould, S.J. (2003) The dynamin-like GTPase DLP1 is essential for peroxisome division and is recruited to peroxisomes in part by PEX11. The Journal of Biological Chemistry, 278, 17012-17020. doi:10.1074/jbc.M212031200
- Sakai, Y., Oku, M., Van der Klei, I.J. and Kiel, J.A. (2006) Pexophagy: Autophagic degradation of peroxisomes. Biochimica et Biophysica Acta, 1763, 1767-1775. doi:10.1016/j.bbamcr.2006.08.023
- Schmidt, O., Pfanner, N. and Meisinger, C. (2010) Mitochondrial protein import: From proteomics to functional mechanisms. Nature Review Molecular Cell Biology, 11, 655-667. doi:10.1038/nrm2959
- Brown, L.A. and Baker, A. (2008) Shuttles and cycles: Transport of proteins into the peroxisome matrix (review). Molecular Membrane Biology, 25, 363-375. doi:10.1080/09687680802130583
- Poirier, Y., Antonenkov, V.D., Glumoff, T. and Hiltunen, J.K. (2006) Peroxisomal β-oxidation—A metabolic pathway with multiple functions. Biochimica et Biophysica Acta, 1763, 1413-1426. doi:10.1016/j.bbamcr.2006.08.034
- Wanders, R.J., Ferdinandusse, S., Brites, P. and Kemp, S. (2010) Peroxisomes, lipid metabolism and lipotoxicity. Biochimica Biophysica Acta, 1801, 272-280.
- Goepfert, S. and Poirier, Y. (2007) Beta-oxidation in fatty acid degradation and beyond. Current Opinion Plant Biology, 10, 245-251. doi:10.1016/j.pbi.2007.04.007
- Eaton, S., Bartlett, K. and Pourfarzam, M. (1996) Mammalian mitochondrial beta-oxidation. Biochemical Journal, 320, 345-357.
- Van Roermund, C.W., Visser, W.F., Ijlst, L., Waterham, H.R. and Wanders, R.J. (2011) Differential substrate specificities of human ABCD1 and ABCD2 in peroxisomal fatty acid beta-oxidation. Biochimica et Biophysica Acta, 1811, 148-152.
- Mannaerts, G.P., Van Veldhoven, P.P. and Casteels, M. (2000) Peroxisomal lipid degradation via β- and α-oxidation in mammals. Cell Biochemistry and Biophysics, 32, 73-87. doi:10.1385/CBB:32:1-3:73
- Le Borgne, F., Ben Mohamed, A., Logerot, M., Garnier, E. and Demarquoy, J. (2011) Changes in carnitine octanoyltransferase activity induce alteration in fatty acid metabolism. Biochemical and Biophysical Research Communications, 409, 699-704. doi:10.1016/j.bbrc.2011.05.068
- Caroppi, P., Sinibaldi, F., Fiorucci, L. and Santucci, R. (2009) Apoptosis and human diseases: Mitochondrion damage and lethal role of released cytochrome c as proapoptotic protein. Current Medicinal Chemistry, 16, 4058-4065.
- Gogvadze, V., Orrenius, S. and Zhivotovsky, B. (2006) Multiple pathways of cytochrome c release from mitochondria in apoptosis. Biochimica et Biophysica Acta, 1757, 639-647. doi:10.1016/j.bbabio.2006.03.016
- Jezek, P. and Hlavata, L. (2005) Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. The International Journal of Biochemistry and Cell Biology, 37, 2478-2503. doi:10.1016/j.biocel.2005.05.013
- Angermuller, S. (1989) Peroxisomal oxidases: Cytochemical localization and biological relevance. Progress Histochemistry and Cytochemistry, 20, 1-65.
- Le Borgne, F., B. Mohamed, A., Logerot, M., Garnier, E. and Demarquoy, J. (2011) Changes in carnitine octanoyltransferase activity induces alteration in fatty acid metabolism. Biochemical and Biophysical Research Communications, 409, 669-704. doi:10.1016/j.bbrc.2011.05.068