Open Journal of Molecular and Integrative Physiology
Vol.2 No.3(2012), Article ID:21842,6 pages DOI:10.4236/ojmip.2012.23012
Constant daily hypoxia leads to different degrees of pulmonary hypertension in two different rat strains
![]()
Division of Pulmonary, Critical Care and Sleep Medicine, Louis Stokes Cleveland Department of Veterans Affairs Medical Center, University Hospitals Case Medical Center and Case Western Reserve University, Cleveland, USA
Email: *cserhal@ameritech.net
Received 14 April 2012; revised 29 May 2012; accepted 17 June 2012
Keywords: Pulmonary Hypertension; Daily Hypoxia; Right Ventricular Hypertrophy; Pulmonary Artery; Contractility
ABSTRACT
Pulmonary diseases associated with diurnal hypoxemia are known to be associated with pulmonary hypertension in some patients. In this study we examined the effects of daily hypoxia (10% oxygen; 8h/day for 14 days) on two strains of rats to simulate sleep related hypoxia in pulmonary diseases expecting to find differences in vascular responses, the development of right ventricular hypertrophy and pulmonary hypertension according to genetic background. In response to daily hypoxia, Sprague Dawley rats developed right ventricular hypertrophy while Brown Norway rats did not. Both strains developed pulmonary hypertension (elevated right ventricular pressure) although the increase was significantly greater in the Sprague Dawley strain. Pulmonary artery (first branch) vasoconstrictive responses to potassium chloride were increased equally in both strains and the subsequent vasodilation with acetylcholine were reduced equally with daily hypoxia in both strains. Taken together, these findings suggest that the genetic makeup of the rats contributed significantly to the development of right ventricular hypertrophy and the degree of pulmonary hypertension. Moreover, this response is not secondary to differences in the intralobar pulmonary vascular reactivity. Genetic background could explain why certain patients do worse with hypoxia inducing pulmonary vascular diseases.
1. INTRODUCTION
The incidence of pulmonary hypertension in chronic obstructive and interstitial lung diseases is dependent on the disease, the degree of hypoxia and possibly an individual’s genetic response [1-4]. Although the incidence and degree of pulmonary hypertension in these diseases may be low, it carries important prognostic information in that its presence infers a much worse prognosis than its absence [5]. A genetic contribution to the pulmonary hypertension phenotype in these diseases is not clear. Genetic predisposition is however clearly present in human studies of pulmonary hypertension in populations living at high altitudes, exposed to ambient hypoxia [6].
The development of pulmonary hypertension has been examined in rat strains exposed to continuous steady-state hypoxia. In these animal models, measured physiologic differences included vascular reactivity and right ventricular hypertrophy [7]. Thomas et al. showed that the physiologic response to continuous hypoxia was a graded response that starts with an increase in pulmonary vascular reactivity followed by an increase in right ventricular pressure and finally right ventricular hypertrophy [8]. However, Jin et al. reported that the Sprague Dawley rat strain developed significantly less vascular reactivity than that developed by the Wistar rat strain, indicating a role for genetic predisposition in the response to hypoxia [7].
In contrast to the effect of continuous hypoxia explored in these animal models, humans develop pulmonary hypertension in response to hypoxemia associated with cardiopulmonary conditions. This pathophysiology is recognized by World Health Organization as class 3 pulmonary hypertension. Hypoxemia is either only noticed or significantly worse during sleep in these patients [9,10]. Such nocturnal hypoxemic episodes are associated with acute increases in pulmonary arterial pressure that in turn may lead to the development of wake time pulmonary hypertension [1,11]. These findings have prompted physicians to measure overnight oximetry to determine the presence and severity of hypoxia during the hours of sleep as a risk factor [12]. Such a paradigm of daily hypoxia might be a better mimic of hypoxia secondary to pulmonary diseases [12]. Nattie et al. reported that 8 hours of daily hypoxia over a 3 week period led to sustained increases in right ventricular pressure in Sprague Dawley strain of rats [13]. Whether such a daily exposure would have that same effect in different strains has not been explored. Such a study design might provide insight into why certain patients develop signs of pulmonary hypertension in hypoxia causing diseases while others do not.
In this study we investigated the physiologic effects of daily hypoxia on Sprague Dawley and Brown Norway rats with respect to three parameters: the change in large pulmonary artery contractility right ventricular pressure and right ventricular mass. We hypothesize that there is a difference in the vascular response and the development of right ventricular hypertrophy and pulmonary hypertension between Sprague Dawley and Brown Norway strains of rats when exposed to eight hours of constant daily hypoxia over two weeks.
2. METHODS
All procedures were approved by the local Institutional Animal Care and Use Committee and conformed to animal care guidelines established by the National Institutes of Health. Adult (6 weeks of age) male Sprague Dawley (420 ± 14 g, n = 18) and Brown Norway (318 ± 12 g, n = 18) rats were used for the experiments. Animals had free access to water and food throughout the experiments.
2.1. Exposure Protocols
Rats were housed under one of two experimental conditions: normoxia or daily hypoxia. Daily hypoxia exposure was as follows. Cages were placed in a chamber sealed with Plexiglas doors. The chamber was connected to compressed air (21% O2) and nitrogen (0% O2) tanks via a sensor controller pinch-valve switch and set to deliver either gas at a constant pressure of 5 psi through the inflow to the cage. A fan was used to disperse the gas at the inflow and equivalent oxygen levels were measured in multiple areas of the cage. Animals in the daily hypoxia group were exposed to compressed air or hypoxic gas beginning at 8:00 AM and ending at 4:00 PM each day for 14 days to coincide with sleep cycle. Control animals were placed in the chamber between 8:00 AM and 4:00 PM daily with room air flowing to simulate experimental conditions. When not in the exposure chamber, animals were housed at the animal facility under room air conditions. Inspired PO2, intrachamber pressure and intrachamber PCO2 were closely monitored during the hypoxia exposure. Sensors placed inside the cages of the experimental rat groups showed that flushing cages with compressed normoxic air (21% O2) or nitrogen (to maintain a hypoxia level of 10% O2) resulted in changes in inspired PO2 which were maintained over the eight hour period with no change in PO2 or intrachamber pressure.
2.2. In Vivo Hemodynamic Measurements
Right ventricular systolic pressure (RVP, mmHg) was measured as previously described by Fagan [14]. Briefly, rats were anesthetized with an intraperitoneal injection of ketamine, xylazine and acepromazine anesthesia cocktail with extra doses of the same cocktail given as needed during the experiment. The anesthetized animals were then placed in the supine position while spontaneously breathing room air. A 24-gauge needle was introduced percutaneously into the right ventricle via a subxiphoid approach. RVP was verified in real time and recorded. Animals from the daily hypoxia exposure group were kept hypoxic until immediately before hemodynamic measurement after at least a four hour exposure to hypoxia. After RVP measurements the chest was opened, the aorta was severed and the animal exsanguinated.
2.3. Measurements of RV Mass
Immediately after death, the heart was resected and the atria were removed at the plane of the atrioventricular valves. The RV free wall was then dissected free of the left ventricle (LV) and septum (S). The RV and LV plus S were weighed (g), and the RV-to-(LV + S) ratio was calculated.
2.4. In Vitro Measurement of Contractility of Pulmonary Artery Rings
As described previously, 2 mm long pulmonary artery rings were dissected from the right and left first branch pulmonary arteries and suspended at 17.5 mmHg [15]. Maximum contraction was determined with exposure to 80 mM of KCL and calculated in grams of force per gram of tissue. Maximum dilation was determined with exposure to acetylcholine (300 µM) following a maximum contraction induced by 80 mM KCL [16]. The degree of response to acetylcholine was quantified as percent dilation from maximum contraction.
2.5. Data Analysis
All values presented are means ± SE. Two-way analysis of variance (ANOVA) for repeated measures was performed. The post-hoc Student-Newman-Kuels test was conducted in the event of significant ANOVA results. Statistical comparisons between two groups were performed using unpaired t-tests. The level for statistical significance was set at 0.05 (two-tailed).
3. RESULTS
In-vivo measurements of right ventricular pressure (RVP) showed a significant increase in pressure when Sprague Dawleys were conditioned with 2 weeks of eight hour constant daily hypoxia compared to normoxic controls; this increase of pressure was also present in the Brown Norway strain after hypoxic exposure (Figure 1). A compareson was made across strains to identify whether the degree of rise in right ventricular pressure was different in the two strains. There was a significantly greater increase in the right ventricular pressure of Brown Norway when compared to Sprague Dawley strain of rats as measured by percentage difference (Figure 2).
Right ventricular hypertrophy occurred in Sprague Dawley rat strain as manifested by the significant increase of 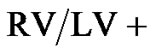 Septum ratio in hypoxia conditioned rats (Figures 3 and 4). In contrast, there was no statistically significant difference in RV hypertrophy in Brown Norway strain of rats after exposure to hypoxia although there
Septum ratio in hypoxia conditioned rats (Figures 3 and 4). In contrast, there was no statistically significant difference in RV hypertrophy in Brown Norway strain of rats after exposure to hypoxia although there
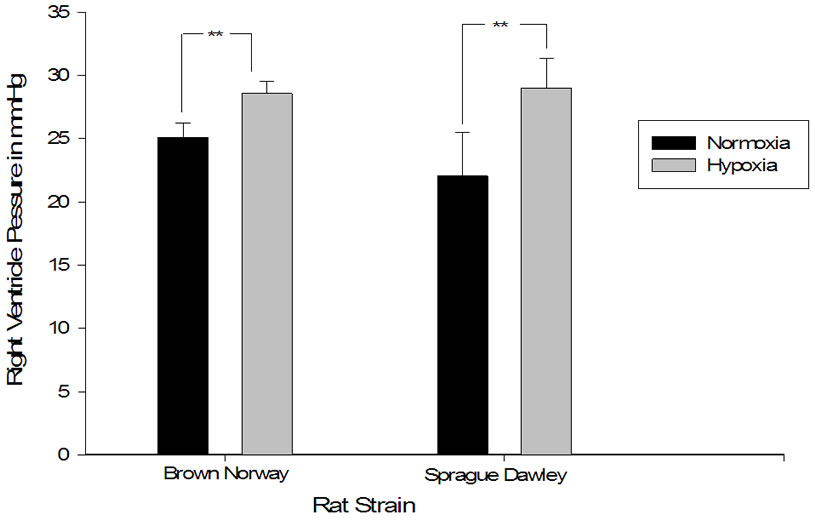
Figure 1. Right ventricular pressure increased in both Sprague Dawley and Brown Norway rat strains in response to daily hypoxia. Right ventricular pressure (RVP; mmHg) as an estimate of pulmonary artery pressure in normoxic Sprague Dawley (n = 9), normoxic Brown Norway (n = 9), and Sprague Dawley (n = 8) and Brown Norway (n = 9) exposed to daily hypoxia for 2 weeks. Asterisks indicate significance of differences (p < 0.05).
Figure 2. Right ventricular pressure increased significantly more in the Sprague Dawley rat strain when compared to Brown Norway in response to daily hypoxia. Right ventricular pressure expressed as a percentage of increase from baseline (RVP; percent) as a measure of pressure increase after hypoxic exposure in Sprague Dawley exposed to daily hypoxia for 2 weeks (n = 8) and Brown Norway exposed to daily hypoxia for 2 weeks (n = 9). Asterisks indicate significance of differences (p = 0.035).
was a trend towards higher 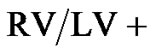 S ratio with hypoxia (p = 0.089).
S ratio with hypoxia (p = 0.089).
The degree of vascular reactivity was higher in both rat strains after hypoxic exposure as determined by two surrogates of reactivity: 1) force generated per gram of
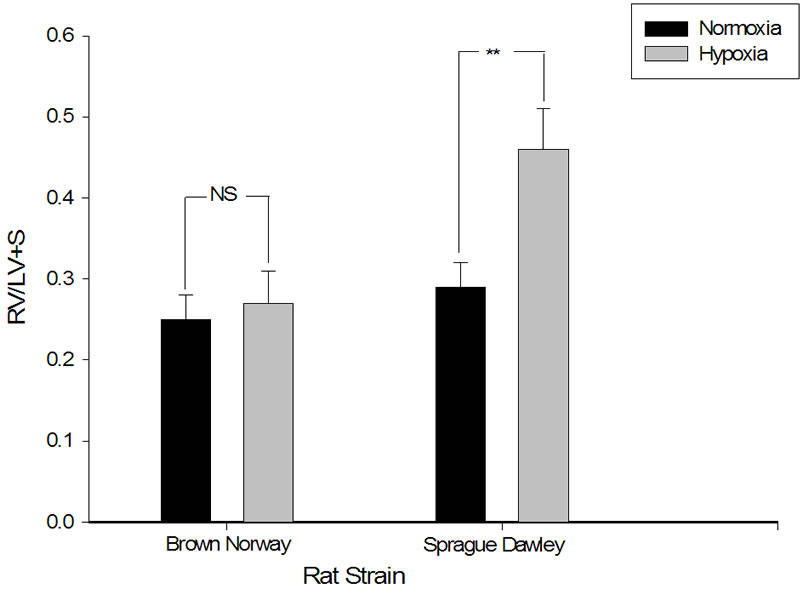
Figure 3. Right ventricular hypertrophy in response to daily hypoxia seen in Sprague Dawley but not Brown Norway rat strains. Right ventricular (RV) mass [RV-to-left ventricle + septum ratio (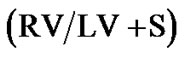 )] in normoxic Sprague Dawley and Brown Norway strain rats (n = 9 and 9 respectively), and Sprague Dawley and Brown Norway strains exposed to eight hours constant daily hypoxia for 2 weeks (n = 9 and 9 respectively). Asterisks indicate significance of differences (p = 0.027), while NS indicates non statistical significance.
)] in normoxic Sprague Dawley and Brown Norway strain rats (n = 9 and 9 respectively), and Sprague Dawley and Brown Norway strains exposed to eight hours constant daily hypoxia for 2 weeks (n = 9 and 9 respectively). Asterisks indicate significance of differences (p = 0.027), while NS indicates non statistical significance.
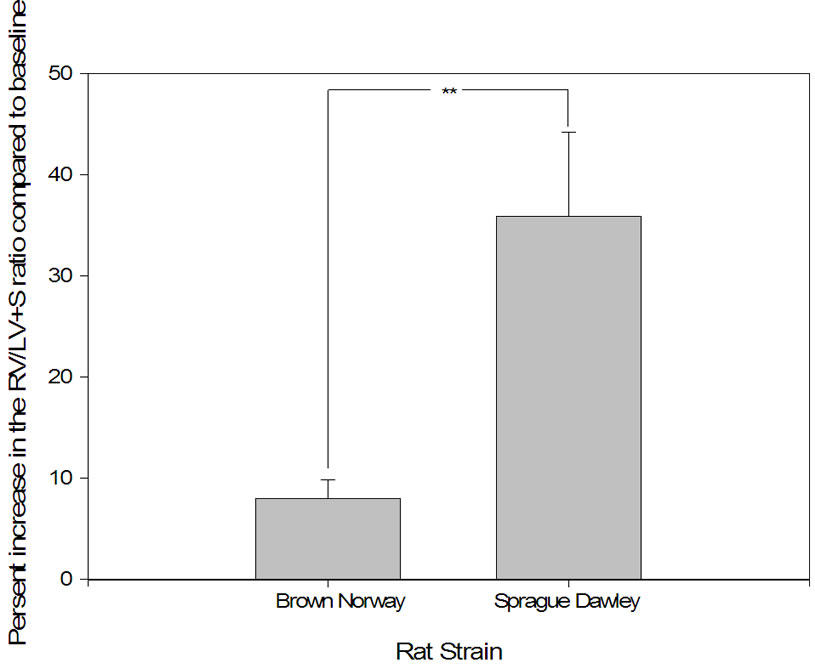
Figure 4. Right ventricular hypertrophy developed significantly more in Sprague Dawley versus Brown Norway rat strains in response to daily hypoxia. Right ventricular (RV) mass [RV-toleft ventricle + septum ratio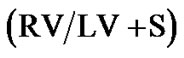 ] as a percentage of increase from baseline (
] as a percentage of increase from baseline (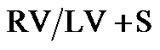 ; percent) as a measure of pressure increase after hypoxic exposure in Sprague Dawley exposed to daily hypoxia for 2 weeks (n = 9) and Brown Norway exposed to daily hypoxia for 2 weeks (n = 9). Asterisks indicate significance of differences (p < 0.001).
; percent) as a measure of pressure increase after hypoxic exposure in Sprague Dawley exposed to daily hypoxia for 2 weeks (n = 9) and Brown Norway exposed to daily hypoxia for 2 weeks (n = 9). Asterisks indicate significance of differences (p < 0.001).
tissue in 2 mm pulmonary vascular rings when exposed to a maximal dose of potassium chloride and 2) the percentage of dilation in response to acetylcholine after such contraction. The degree of contraction was higher in pulmonary artery rings from both strains after exposure to hypoxia and the response to acetylcholine was blunted in both (Figures 5 and 6). Interestingly, the increase in the force of contraction and the blunting of the acetylcholine were similar in both strains when compared to each other (p = 0.54).
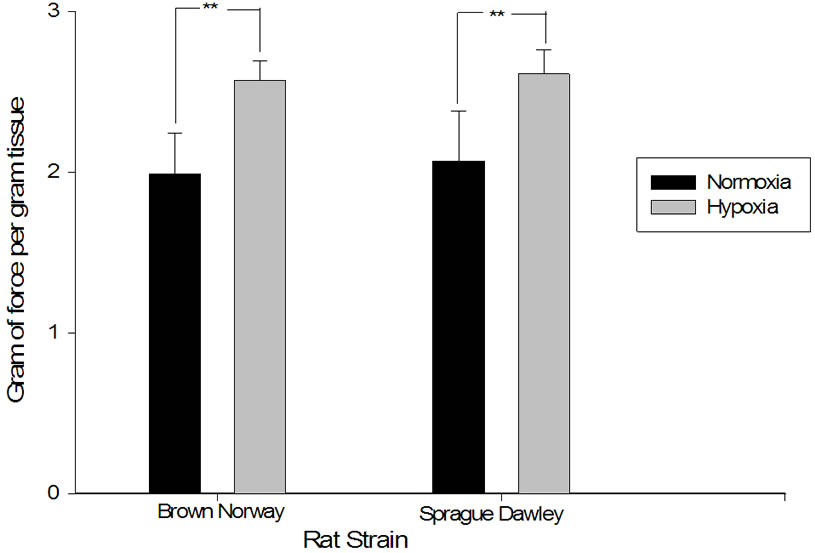
Figure 5. Contractility increased after hypoxic exposure in both Sprague Dawley and Brown Norway rat strains. Maximal contraction of 2 mM pulmonary artery rings plotted in grams of tension per gram of tissue in response to 80 mM potassium chloride. The figure compares normoxia in Sprague Dawley and Brown Norway (n = 8 and 7 respectively) to daily hypoxia for two weeks in Sprague Dawley and Brown Norway strain of rats (n = 8 and 9 respectively). Asterisks indicate significance of differences (p < 0.05).
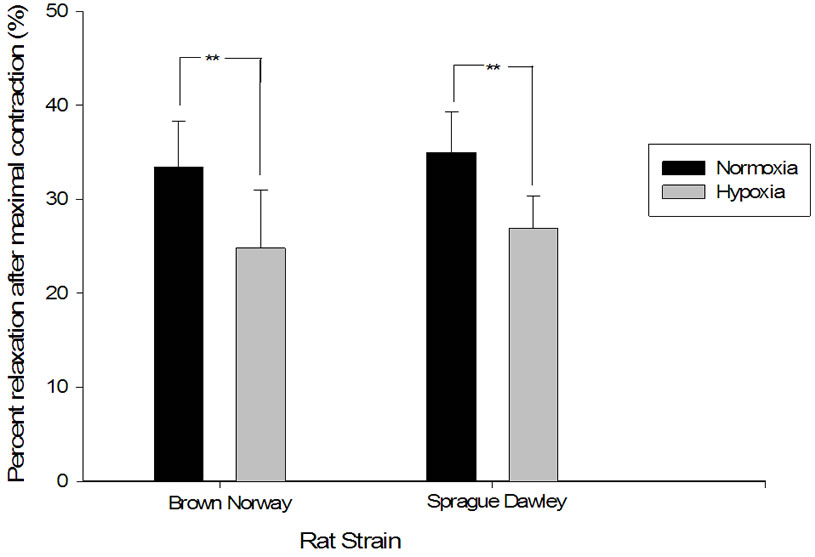
Figure 6. Relaxation in response to acetylcholine was blunted in both Brown Norway and Sprague Dawley rat strains after exposure to daily hypoxia. Maximal relaxation in response to 300 µM of acetylcholine after maximal contraction with 80 mM potassium chloride. The figure compares normoxia in Sprague Dawley and Brown Norway (n = 8 and 7 respectively) to daily hypoxia for two weeks in Sprague Dawley and Brown Norway strain of rats (n = 8 and 9 respectively). Asterisks indicate significance of differences (p < 0.05).
4. DISCUSSION
Our results confirm the previous reports that daily hypoxic exposures over time leads to increased pulmonary artery pressure and right ventricular hypertrophy and that these changes do occur in the setting of eight hours of constant daily hypoxia [13]. The hypothesis that the degree of pulmonary artery pressure differs between these two strains of rat when exposed to eight hours of constant daily hypoxia over two weeks is confirmed. However, at this time point pulmonary vascular responsiveness is no different and more importantly the Brown Norway did not respond with an increase in right ventricular hypertrophy despite an increase in pulmonary artery pressure.
The hypoxia paradigms currently popular for simulating pathophysiologic responses to hypoxia in humans fall into two categories: steady-state exposures of 8 - 24 hours and intermittent exposure of varying times with hypoxia/ reoxygenation cycles no greater than 10 minutes. The latter approach is proposed for the study of the hypoxic consequences of sleep apnea, and such various short-cycle hypoxia reoxygenation paradigms are accompanied by metabolic effects on lipid metabolism [17] and glucose intolerance [18]. These effects are in part secondary to the development of free oxygen radicals formed in the hypoxia reoxygenation process [19]. The common approach to model the hypoxia of chronic lung disease is a 23 - 24 hour exposure to hypoxia for 2 - 3 weeks. One recent study compared steady state constant to intermittent exposures, there is a differential effect on pulmonary vasculature which is at least partially modulated by the presence or absence of hypercapnia [20]. Neither of these paradigms however, simulate hypoxia in pulmonary disorders that results from the normal physiology of sleep [21]. The paradigm we use in this study is designed to mimic nocturnal hypoxia secondary to ventilation/perfusion mismatch and the hypoventilation of sleep present in patients with hypoxia inducing-pulmonary disorders [8,14]. We chose eight hours of hypoxia during the light period to coincide with the major sleep period of the rat. This paradigm has been shown by others to result in prototypic hypoxic responses, increases in hematocrit, pulmonary artery pressure, and right ventricular hypertrophy—a finding we found and confirmed in one strain [13-15,22].
The choice of strains was based on two considerations. First, we have previously shown that a comparison of these two strains in regard to the ventilatory chemoresponsivness shows that to the same environmental challenges the Brown Norway strain has a reduced hypoxic and hypercapnic response compared to the Sprague Dawley, and the response to reoxygenation is also different with the Brown Norway showing post-hypoxic frequency and ventilatory decline while the Sprague Dawley shows a sustained post-hypoxic frequency and ventilation [23,24]. Second, the difference in terms of genetic distance between the two strains, the Brown Norway is very different (63% difference in synonymous single nucleotide polymorphisms) from the Sprague Dawley [25]. Therefore, the genetic differences between the strains, while not inclusive of all known polymorphisms among rat strains, is sufficient to test the hypothesis that genetic background might play a role in the response to chronic, daily hypoxia.
When Thomas et al. investigated the development of pulmonary hypertension in rats exposed to constant hypoxia for 3, 5 and 20 days, the response was found to be graded and time dependent [8]. Thus, our findings can be explained by two possible hypotheses. Brown Norway rats are resistant to the development of pulmonary hypertension by virtue of their genetic make-up and this would be the maximum effect seen after hypoxic exposure. Alternatively, a more plausible explanation is that there is an exposure length effect in which pulmonary hypertension is a stepwise disease starting with increased pulmonary vascular reactivity followed by increased right ventricular pressure which eventually leads to right ventricular hypertrophy. This theory would explain why both strains developed increased contractility and right ventricular pressure but not right ventricular hypertrophy at two weeks of exposure. Assuming such a mechanism is true; the finding of no difference in the pulmonary reactivity between the two strains might imply that the pulmonary arteries from both strains have reached maximal heightened reactivity. This kinetic effect is supported by the previous findings that confirm the development of increased vascular reactivity and increased right ventricular pressure over a twenty day period [8].
The increased pulmonary vascular contractility is probably secondary to a combination of increased reactivity and a fixed vascular component (remodeling). In humans, acute increases in pulmonary arterial pressure are associated with nocturnal hypoxemic episodes and may lead to the development of pulmonary hypertension [1,14]. It is also documented that patients with sleep apnea and resting daytime pulmonary hypertension have increased hypoxic pulmonary vasoconstriction as well as increased resistance to pulmonary blood flow, suggesting both a reactive and fixed (remodeled) vascular component of the pulmonary hypertension [22,26]. Another important consideration in diseases associated with hypoxia is the fact that the presence of hypercapnea leads to more severe pulmonary hypertension [27]. In the present report, the rats were not made hypercapnic, and, although we did not measure arterial blood gasses, the animals were likely hypocapnic. The action of carbon dioxide in the pulmonary circulation may depend on the pulmonary arterial pressure with hypercapnia causing vasoconstriction at low pulmonary arterial pressure and vasodilatation at high pressure [28]. How a hypercapnic environment could have affected our results is not entirely clear. In summary, the present study reports the development of different degrees of pulmonary hypertension in two strains of rats in response to constant daily hypoxia, simulating the hypoxia-reoxygenation cycle seen with hypoxia in diseases such as chronic obstructive pulmonary disease. Although the episodic nature of the hypoxia may be similar to that seen in diseases like chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis, further studies are needed to determine the mechanism of resistance of certain strains and the genetic factors contributing to differences in right ventricular remodeling in this exposure paradigm.
5. ACKNOWLEDGEMENTS
These studies were supported by NIH grant HL-70697 and Merit review grants from the Department of Veterans Affairs Research Service. We would also like to thank Michelle Moyer for help with statistical analysis and setup of pulmonary vascular ring apparatus and Jesse Dostal for help with statistical analysis and hemodynamic measurements.
REFERENCES
- Bradley, T.D., Rutherford, R., Grossman, R.F., Lue, F., Zamel, N., Moldofsky, H. and Phillipson, E.A. (1985) Role of daytime hypoxemia in the pathogenesis of right heart failure in the obstructive sleep apnea syndrome. American Review of Respiratory Disease, 131, 835-839.
- Hedner, J. (1996) Vascular function in OSA. Sleep, 19, S213-S217.
- Shahar, E., Whitney, C.W., Redline, S., Lee, E.T., Newman, A.B., Javier, Nieto, F., O’Connor, G.T., Boland, L.L., Schwartz, J.E. and Samet, J.M. (2001) Sleep-disordered breathing and cardiovascular disease: Cross-sectional results of the sleep heart health study. American Journal of Respiratory and Critical Care Medicine, 163, 19-25.
- Weitzenblum, E., Krieger, J., Apprill, M., Vallee, E., Herhart, M., Ratomaharo, J., Oswald, M. and Kurtz, D. (1988) Daytime pulmonary hypertension in patients with obstructive sleep apnea syndrome. American Review of Respiratory Disease, 138, 345-349.
- Tobin, M.J. (2001) Chronic obstructive pulmonary disease, pollution, pulmonary vascular disease, transplantation, pleural disease, and lung cancer in AJRCCM 2000. American Journal of Respiratory and Critical Care Medicine, 164, 1789-1804.
- Niermeyer, S., Yang, P., Shanmina, Drolkar, Zhuang, J., and Moore, L.G. (1995) Arterial oxygen saturation in Tibetan and Han infants born in Lhasa, Tibet. New England Journal of Medicine, 333, 1248-1252. doi:10.1056/NEJM199511093331903
- Jin, X.R., Fan, M., Wang, Z.Q., Su, Y.C., Yang, G.T., Hu, H.B., Zhang, Y.P., Feng, C.J., Ding, Y.X. and Wang, D.X. (1990) Strain difference in pulmonary vascular responsiveness to hypoxia in rats. Journal of Tongji Medical University, 10, 134-140. doi:10.1007/BF02986450
- Thomas, B.J. and Wanstall, J.C. (2003) Alterations in pulmonary vascular function in rats exposed to intermittent hypoxia. European Journal of Pharmacology, 477, 153-161. doi:10.1016/j.ejphar.2003.08.015
- Rafanan, A.L., Golish, J.A., Dinner, D.S., Hague, L.K. and Arroliga, A.C. (2001) Nocturnal hypoxemia is common in primary pulmonary hypertension. Chest, 120, 894-899. doi:10.1378/chest.120.3.894
- Simonneau, G., Galie, N., Rubin, L.J., Langleben, D., Seeger, W., Domenighetti, G., Gibbs, S., Lebrec, D., Speich, R., Beghetti, M., Rich, S. and Fishman, A. (2004) Clinical classification of pulmonary hypertension. Journal of the American College of Cardiology, 43, 5S-12S. doi:10.1016/j.jacc.2004.02.037
- Fagan, K.A. and Weil, J.V. (2001) Potential genetic contributions to control of the pulmonary circulation and ventilation at high altitude. High Altitude Medicine & Biology, 2, 165-171. doi:10.1089/152702901750265279
- Mortola, J.P. (2004) Breathing around the clock: An overview of the circadian pattern of respiration. European Journal of Applied Physiology, 91, 119-129. doi:10.1007/s00421-003-0978-0
- Nattie, E.E., Bartlett, D. Jr. and Johnson, K. (1978) Pulmonary hypertension and right ventricular hypertrophy caused by intermittent hypoxia and hypercapnia in the rat. American Review of Respiratory Disease, 118, 653-658.
- Fagan, K.A. (2001) Selected contribution: Pulmonary hypertension in mice following intermittent hypoxia. Journal of Applied Physiology, 90, 2502-2507.
- Rogers, T.K., Stewart, A.G. and Morice, A.H. (1992) Effect of chronic hypoxia on rat pulmonary resistance vessels: Vasodilatation by atrial natriuretic peptide. Clinical Science, 83, 723-729.
- McIntyre, C.A., Buckley, C.H., Jones, G.C., Sandeep, T.C., Andrews, R.C., Elliott, A.I., Gray, G.A., Williams, B.C., McKnight, J.A., Walker, B.R. and Hadoke, P.W. (2001) Endothelium-derived hyperpolarizing factor and potassium use different mechanisms to induce relaxation of human subcutaneous resistance arteries. British Journal of Pharmacology, 133, 902-908. doi:10.1038/sj.bjp.0704143
- Li, J., Grigoryev, D.N., Ye, S.Q., Thorne, L., Schwartz, A.R., Smith, P.L., O’Donnell, C.P. and Polotsky, V.Y. (2005) Chronic intermittent hypoxia upregulates genes of lipid biosynthesis in obese mice. Journal of Applied Physiology, 99, 1643-1648. doi:10.1152/japplphysiol.00522.2005
- Polotsky, V.Y., Li, J., Punjabi, N.M., Rubin, A.E., Smith, P.L., Schwartz, A.R. and O’Donnell, C.P. (2003) Intermittent hypoxia increases insulin resistance in genetically obese mice. Journal of Physiology, 552, 253-264. doi:10.1113/jphysiol.2003.048173
- Brindeiro, T.C.M., da Silva, A.Q., Allahdadi, K.J., Youngblood, V. and Kanagy, N.L. (2007) Reactive oxygen species contribute to sleep apnea-induced hypertension in rats. American Journal of Physiology Heart and Circulatory Physiology, 293, H2971-H2976. doi:10.1152/ajpheart.00219.2007
- Snow, J.B., Kitzis, V., Norton, C.E., Torres, S.N., Johnson, K.D., Kanagy, N.L., Walker, B.R. and Resta, T.C. (2007) Differential effects of chronic hypoxia and intermittent hypocapnic and eucapnic hypoxia on pulmonary vasoreactivity. Journal of Applied Physiology, 101, 110- 118. doi:10.1152/japplphysiol.00698.2005
- Tagaito, Y., Polotsky, V.Y., Campen, M.J., Wilson, J.A., Balbir, A., Smith, P.L., Schwartz, A.R. and O’Donnell, C.P. (2001) A model of sleep-disordered breathing in the C57BL/6J mouse. Journal of Applied Physiology, 91, 2758-2766.
- Bao, G., Metreveli, N., Li, R., Taylor, A. and Fletcher, E.C. (1997) Blood pressure response to chronic episodic hypoxia: Role of the sympathetic nervous system. Journal of Applied Physiology, 83, 95-101.
- Subramanian, S., Dostal, J., Erokwu, B., Han, F., Dick, T.E. and Strohl, K.P. (2007) Domperidone and ventilatory behavior: Sprague-Dawley versus Brown Norway rats. Respiratory Physiology & Neurobiology, 155, 22-28. doi:10.1016/j.resp.2006.04.002
- Subramanian, S., Erokwu, B., Han, F., Dick, T.E. and Strohl, K.P. (2002) L-NAME differentially alters ventilatory behavior in Sprague-Dawley and Brown Norway rats. Journal of Applied Physiology, 93, 984-989.
- Kitada, K., Voigt, B., Kondo, Y. and Serikawa, T. (2000) An integrated rat genome map based on genetic and cytogenetic data. Experimental Animals, 49, 119-126. doi:10.1538/expanim.49.119
- Sajkov, D., Wang, T., Saunders, N.A., Bune, A.J., Neill, A.M. and Mcevoy, D.R. (1999) Daytime pulmonary hemodynamics in patients with obstructive sleep apnea without lung disease. American Journal of Respiratory and Critical Care Medicine, 159, 1518-1526.
- Bao, G., Randhawa, P.M. and Fletcher, E.C. (1997) Acute blood pressure elevation during repetitive hypocapnic and eucapnic hypoxia in rats. Journal of Applied Physiology, 82, 1071-1078.
- Baudouin, S.V. and Evans, T.W. (1993) Action of carbon dioxide on hypoxic pulmonary vasoconstriction in the rat lung: Evidence against specific endothelium-derived relaxing factor-mediated vasodilation. Critical Care Medicine, 21, 740-746. doi:10.1097/00003246-199305000-00018
NOTES
*Corresponding author.