Advances in Biological Chemistry
Vol.4 No.1(2014), Article ID:42711,9 pages DOI:10.4236/abc.2014.41005
N-acetylcysteine amide protects against dexamethasone-induced cataract related changes in cultured rat lenses
![]()
1Department of Chemistry, Missouri University of Science and Technology, Rolla, USA
2Department of Ophthalmology, Baskent University, Istanbul, Turkey
Email: *nercal@mst.edu
Copyright © 2014 Shakila Tobwala et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Shakila Tobwala et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received 5 December 2013; revised 10 January 2014; accepted 21 January 2014
KEYWORDS
Dexamethasone; Cataracts; Oxidative Stress; Antioxidant; N-Acetylcysteine Amide
ABSTRACT
Glucocorticoids (GCs) are one of the most widely used immunosuppressive and anti-inflammatory agents. However, their long term and systemic use is associated with adverse drug reactions including posterior subcapsular cataracts as one of its ocular complications. Balanced redox state is crucial for maintenance of lens transparency, and a high content of glutathione (GSH) in the lens is believed to play a key role in doing so. Depletion of GSH is implicated in the etiopathogenesis of dexamethasone-induced cataracts and, therefore, the present study was sought to evaluate the efficacy of a novel thiol antioxidant, N-acetylcysteine amide (NACA), in preventing dexamethasone-induced cataractogenesis. Cataract formation was induced by incubation of rat lenses with 5 µM dexamethasone. To assess whether NACA had a significant impact on dexamethasone-induced cataracts, the rat lenses were divided into four groups: 1) control group (Dulbecco’s Modified Eagle Medium (DMEM), 2) dexamethasone group (DMEM with 5 µM dexamethasone), 3) NACA-only group (50 µM NACA solution), and 4) NACA pretreatment group (50 µM NACA for 6 hours followed by 5 µM dexamethasone only for 18 hours). Lenses were cultured for 7 days at 37˚C under 5% CO2. Lenses were evaluated daily using a dissecting microscope and photographed and graded for the development of opacity. The rat lenses in both the control and the NACA-only groups were clear, whereas all lenses within the dexamethasone-only group developed well-defined cataracts. Overall observations indicated that NACA inhibits cataract formation by limiting lipid peroxidation and increasing the ratio of GSH/GSSG in lens. Therefore, NACA can be developed into a potential adjunctive therapeutic option for patients undergoing therapy with GCs to inhibit glucocorticoid-induced cataracts.
1. INTRODUCTION
Glucocorticoids (GCs) are steroid hormones that play a role in physiological processes and are widely used as immunosuppressive and anti-inflammatory agents in the treatment of many clinical conditions, including rheumatoid arthritis, asthma, autoimmune diseases, and various ocular diseases [1,2]. However, their clinical use is restricted due to a wide range of complications associated with their long-term topical and systemic use. One of the ocular complications of glucocorticoid toxicity is the development of posterior subcapsular cataracts (PSCs) [3-8]. Unfortunately, certain patients cannot avoid longterm steroid therapy and therefore, development of adjunctive therapeutics for the prevention of steroid-induced cataracts is highly desirable.
Oxidative stress and depletion of GSH are implicated in the etiopathogenesis of glucocorticoid-induced cataracts [9-14]. Under physiological conditions, the lens utilizes its various antioxidant defenses to effectively protect itself and maintain the reduced state of thiols, which is essential for retaining clarity of the lens. However, under oxidative stress, depletion of GSH disturbs the thiolredox status. This sets off a cascade of oxidative damage resulting in the oxidation of all major macro-molecules in cells, including lipids, proteins, and nucleic acids, and eventually in the disruption of function and integrity of lens cells [15-19]. Oxidative damage to proteins may lead to structural and functional changes, including conformational changes resulting in inhibition of enzymatic and binding activities, fragmentation, denaturation, aggregation, altered gene expression and regulation, and modulation of cell signaling [20]. In addition, lipid hydroperoxides may cause changes in membrane permeability [21-24] along with uncoupling of the membranebound enzyme Na+/K+-ATPase and oxidative inhibition of Ca2+-ATPase [25,26], and DNA damage [27]. Changes in the redox ratio of GSH lead to crosslinking, aggregation, insolubility, and fragmentation of crystalline proteins, which results in the formation of cataracts [12, 28-31]. Epidemiological studies have documented loss of GSH in various types of cataracts [32-34]. A decrease in GSH levels after glucocorticoid exposure [35,36] along with an increase in the levels of lipid peroxide in the lens [36] has been reported. Furthermore, protection by antioxidants like vitamin E and ascorbic acid [7,37] against damage caused by a soluble corticosteroid suggests the role of oxidative stress in steroid-induced cataract formation.
At present, the treatment for cataracts requires surgery to remove the natural lens that has developed opacification, replacing it with a synthetic lens to provide transparency. Although cataract surgery is considered to be one of the safest procedures available, treatment is relatively expensive and there is a significant rate of postsurgical complications, including the development of a posterior capsular opacification [38]. Since depletion of GSH is hypothesized to be a key initiating event in the development of PSCs, the use of a GSH prodrug as an adjunctive therapeutic agent would be an effective, nonsurgical treatment to prevent and treat cataracts. However, progress in this area has been modest. Although studies have indicated that antioxidants like N-acetyl carnosine and N-acetyl cysteine (NAC) may ameliorate the risk for cataracts [39,40]. NAC is not highly bioavailable and does not readily penetrate into cells, thereby requiring fairly high doses that can increase the side effect profile. A potential candidate that possesses far better characteristics for development as an ophthalmologic agent to address oxidative stress damage is the low molecular weight thiol antioxidant, N-acetylcysteine amide (NACA). NACA’s characteristics as a drug were improved over NAC by neutralizing the carboxylic group of NAC, which makes the NACA molecule more lipophilic and therefore enhances its ability to penetrate cellular membranes. The enhanced ability to penetrate cells allows NACA to be administered at a lower dose than NAC, giving the drug a greater therapeutic index and lowering the risk of side effects that traditionally have been associated with higher doses of NAC [41,42]. NACA is an excellent source of sulfhydryl (SH) groups that can be converted by the cells into metabolites capable of stimulating GSH synthesis. The molecule also can promote intracellular detoxification and act directly as a free radical scavenger. NACA acts as a carrier of NAC and its antioxidant and free radical scavenging abilities are equal to or better than those of NAC [43].
Promising results with NACA [44-51] in various oxidative stress-related disorders encouraged us to investigate the protective role of NACA in the prevention of dexamethasone-induced cataracts. Our data showed that NACA inhibits dexamethasone-induced cataract formation by limiting lipid peroxidation and increasing the ratio of GSH/GSSG in lenses. NACA can potentially be developed into a promising adjunctive therapeutic option for patients undergoing therapy with glucocorticoids.
2. MATERIALS AND METHODS
2.1. Materials
Dexamethasone and N-(1-pyrenyl)-maleimide (NPM) were obtained from Sigma-Aldrich (St. Louis, MO). High performance liquid chromatography (HPLC) grade solvents were purchased from Fisher Scientific (Fair Lawn, NJ). NACA was gifted by Dr. Glenn Goldstein (David Pharmaceuticals, New York, NY). All other chemicals were bought from Sigma-Aldrich (St. Louis, MO).
2.2. Ex Vivo Rat Lens Culture and Drug Treatment
All animal procedures conformed to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and by the Animal Care and Use Protocol Review Committee at the Missouri University of Science and Technology. For rat lens culture, eyes from 21-dayold Sprague-Dawley male rats were enucleated to expel the lens using plastic-coated forceps and fine scissors. Eyes were immediately transferred to Dulbecco’s MEM (pH 7.2; Sigma, St. Louis, MO), containing 0.1% bovine serum albumin (BSA; GibcoBRL, Grand Island, NY) and antibiotic solution (GibcoBRL; 100 U/mL penicillin, 100 µg/mL streptomycin, and 0.25 µg/mL amphotericin B) [52]. The culture media was changed daily. Approximately 24 hours after the preparation of organ cultures, clear lenses were selected and were randomly divided into four groups: 1) control group (DMEM), 2) dexamethasone group (DMEM with 5 µM dexamethasone), 3) NACA-only group (50 µM NACA in DMEM), and 4) NACA pretreatment group (pretreatment with 50 µM NACA for 6 hours followed by 5 µM dexamethasone only for 18 hours in DMEM). Lenses in each group were cultured in DMEM media as detailed above for 7 days at 37˚C under 5% CO2. Lenses were evaluated daily using a dissecting microscope and photographed to check for the development of opacity.
2.3. Intracellular Glutathione (GSH) Measurement
Intracellular GSH content was determined by reverse phase HPLC, according to the method developed in our laboratory [53]. Lens samples were homogenized in serine borate buffer (SBB). Twenty microliters of this homogenate were added to 230 µl of HPLC grade water and 750 µl of NPM (1 mM in acetonitrile). The resulting solutions were incubated at room temperature for 5 min. The reaction was stopped by adding 10 µl of 2 N HCl. The samples were then filtered through a 0.45 µm filter (Advantec MFS, Inc. Dulin, CA, USA) and injected onto the HPLC system. 5 µl of the sample were injected for analysis using a Thermo Finnigan TM Spectra SYSTEM SCM1000 Vacuum Membrane Degasser, FinniganTM SpectraSYSTEM P2000 Gradient Pump, FinniganTM SpectraSYSTEM AS3000 Autosampler, and FinniganTM SpectraSYSTEM FL3000 Fluorescence Detector (λex = 330 nm and λem = 376 nm). The HPLC column was a Reliasil ODS-1 C18 column (Column Engineering, Ontario, CA, USA). The mobile phase was 70% acetonitrile and 30% water and was adjusted to a pH of 2.5 through the addition of 1 ml/L of both acetic and o-phosphoric acids. The NPM derivatives were eluted from the column isocratically at a flow rate of 1 ml/min.
2.4. Total Glutathione and Glutathione Disulfide (GSSG) Measurement
Total glutathione content was determined by reverse phase HPLC. Lens samples were homogenized in SBB. Twenty microliters of this homogenate were added to 60 µl of NADPH (2 mg/ml) in nanopure water and 20 μl of 1 unit/ml glutathione reductase were added to reduce GSSG. After 10 min of incubation at room temperature, the treated samples were diluted with 150 μl H2O, and then immediately derivatized with 750 μl of 1.0 mM NPM. The samples were analyzed as detailed for the determination of GSH using reverse phase HPLC. Data from the original GSH levels and the total current GSH levels in each sample were subsequently used to calculate the levels of GSSG present in each sample [54].
2.5. Determination of Glutathione Reductase (GR) Activity
Glutathione reductase is the enzyme responsible for recycling GSSG into GSH via a reduction mechanism, utilizing both GSSG and NADPH as a substrate. The activity of this enzyme was determined using a commercial kit from OxisResearch (Portland, OR, USA). The oxidation of NADPH to NADP+ was accompanied by a decrease in absorbance at 340 nm, providing a spectrophotometric means for monitoring the enzyme activity of GR. The activity of GR in cells was determined by adding homogenate to a solution containing both GSSG and NADPH and then recording the absorbance as a function of time at 340 nm. The rate of decrease in the A340 was directly proportional to the GR activity in each sample.
2.6. Lipid Peroxidation Measurement
Malondialdehyde (MDA) is a thiobarbituric acid reactive substance (TBARS). The extent of lipid peroxidation was determined by measuring concentrations of TBAMDA complex. Lens homogenate (350 μl), 100 μl of 500 ppm butylated hydroxytoluene, and 550 μl of 10% trichloroacetic acid were combined, and the suspension was boiled for 30 min. An aliquot (500 μl) of the supernatant was removed and 500 μl of thiobarbituric acid added. From this solution, 500 μl were removed and added to 1.0 ml of n-butanol. This mixture was vortexed, and centrifuged for 5 min at 110 × g to facilitate phase separation. Fluorescence was then measured (λex = 515 nm and λem = 550 nm) [55].
2.7. Statistical Analysis
All reported values were represented as the mean ± S.D. of quadruplets. Statistical analysis was performed using the GraphPad Prism software (GraphPad, San Diego, CA). Statistical significance was ascertained by one way analysis of variance, followed by Tukey’s multiple comparison tests. Values of p < 0.05 were considered significant. In the figures, “*” represents a significant difference in comparison with the control group, and “#” represents a significant difference in comparison with the Dex-only group.
3. RESULTS
3.1. Prevention of Dexamethasone-Induced Cataracts by NACA
To examine the effects of dexamethasone on the lens, we used an ex vivo rat lens model. We isolated rat lenses and treated them in organ culture to cause steroid-induced cataracts directly. The lenses were incubated with 5 µM dexamethasone and morphologic changes in the whole lenses were recorded photographically with a dissection microscope. Opacity first appeared at day 3 in the lenses incubated with dexamethasone. All lenses in the dexamethasone group developed cataracts by day 7. In contrast, only 25% of the lenses developed cataracts in the NACA pretreatment group. Untreated control lenses remained transparent until day 7 (Figure 1).
3.2. Effect of NACA on Intracellular GSH Levels in Dexamethasone Treated Cultured Rat Lens
To support our hypothesis that GSH depletion induces cataract formation in the Dex-treated group, we measured the levels of intracellular GSH. Figure 2 shows the effect of Dex on lens GSH levels in the presence and absence of NACA. A 7-day exposure with 5 μM of Dex decreased the GSH level to 21% of that of the control. A pretreatment with 50 μM of NACA increased the GSH level close to control.
3.3. Effect of NACA on Oxidized Glutathione (GSSG) Levels and GSH/GSSG Ratio in Dexamethasone Treated Cultured Rat Lenses
GSSG levels in the lenses of the Dex-only group were found to have significantly increased by approximately three times the GSSG levels in the control group. The amounts of GSSG found in lenses with NACA pretreatment were significantly lower compared to the Dex-only group and were close to that of the control group. A graph with these results is shown in Figure 3. An interesting result was obtained by observing the ratio between the GSH and the GSSG levels in the lenses of each group. As expected, the control group was found to have the highest ratio of GSH to GSSG. The ratio dropped to about 6% of control in the Dex-only group (Figure 4). However, NACA pretreatment increased this ratio significantly to approximately 50% of the control group value.

Figure 1. Appearance of cultured rat lens with dexamethasone and NACA at day 7. Opacity was observed only in the Dex group at day 7 (Figure 1(c)), which was significantly prevented by pretreatment with NACA. (a) Control; (b) NACAonly; (c) 5 µM Dex; (d) 5 µM Dex + 50 µM NACA. All reported values were represented as the mean ± S.D. (n = 8).
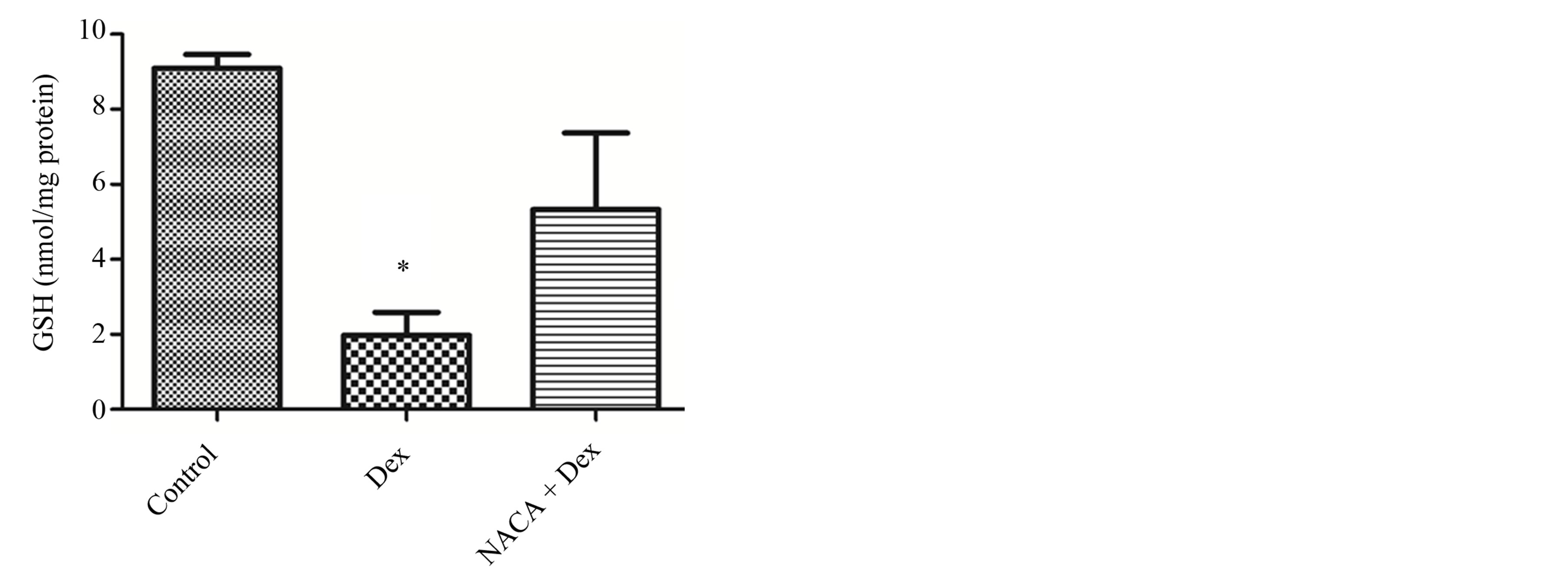
Figure 2. Intracellular GSH levels in lenses after treatment with dexamethasone and NACA. GSH levels were measured after 7 days of treatment for control, NACA, Dex, and Dex + NACA groups. Exposure to Dex (5 μM) significantly decreased intracellular GSH level. Pretreatment with NACA (50 μM), prevented such a dramatic decrease. The NACA-only treated group showed no significant difference when compared to the control. *p ≤ 0.05 compared to the control group. All reported values were represented as the mean ± S.D. of quadruplets.
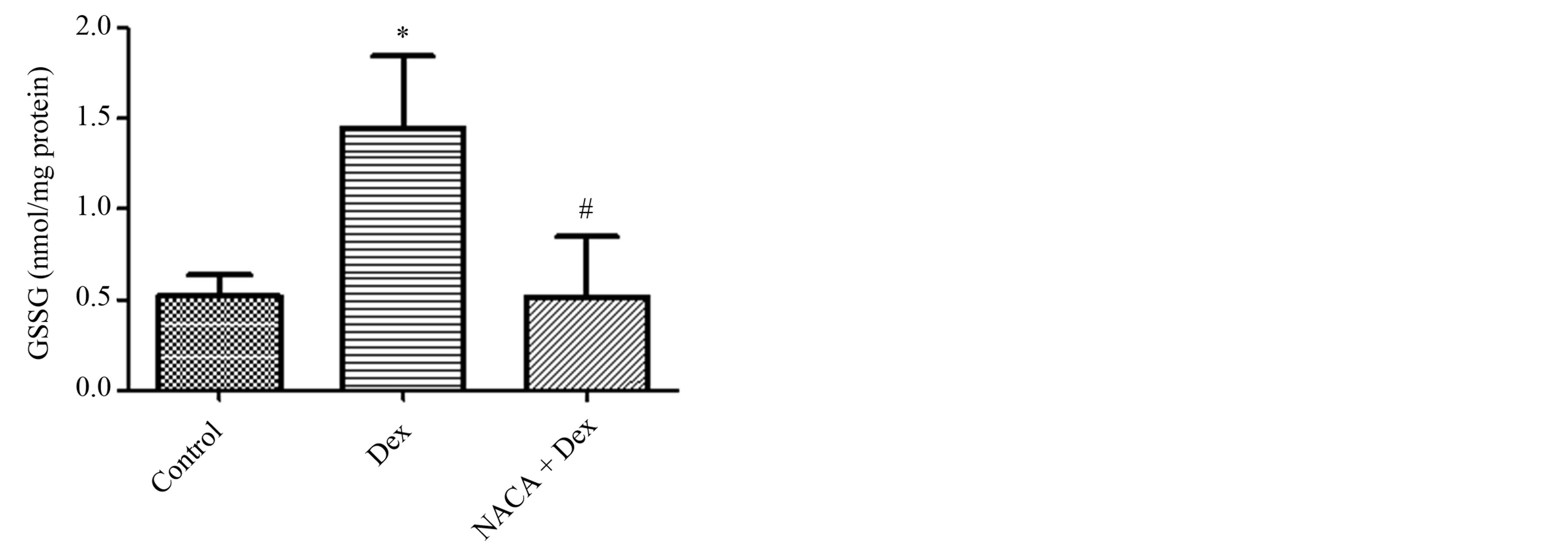
Figure 3. Intracellular GSSG levels in lenses after treatment with dexamethasone and NACA. The GSSG level was significantly higher in the Dex-only group than in the control. This GSSG level was significantly reduced upon pretreatment with NACA. The NACA-only treated group showed no significant difference when compared to the control. *p ≤ 0.05 compared to the control group, and #p ≤ 0.05 compared to the Dex group. All reported values were represented as the mean ± S.D. of quadruplets.
3.4. Effect of NACA on Glutathione Reductase (GR) Activity in Dexamethasone Treated Cultured Rat Lenses
GR is a key antioxidant enzyme involved in maintenance of cellular GSH homeostasis. It reduces GSSG back to the reduced form, GSH. A significant reduction in the activity of GR was observed upon Dex treatment. However, NACA pretreatment increased the activity of GR (Figure 5).
3.5. Effect of NACA on Lipid Peroxidation Byproduct: MDA
MDA was used as an index of lipid peroxidation (LPO). Dex-treated lens had significantly higher levels of MDA, as compared to those of the control (Figure 6). Pretreatment with 50 µM of NACA completely reduced this increase, with MDA levels becoming nearly the same as those of the control, and with a p value of <0.05, as compared to that of the control. The NACA-only treated group showed no significant difference when compared to the control.
4. DISCUSSION
Posterior sub-capsular cataract is one of the ocular complications of glucocorticoid toxicity. Despite a well-established link between the use of GC’s and cataracts, treatment with glucocorticoids cannot be avoided in some cases. GC-induced cataract formation is directly attributed to oxidative stress that occurs within the lens. Oxidation, which can be caused by an overabundance of oxidative stress generators, such as molecular oxygen, hydrogen peroxide, and free radicals, produces a major insult upon the lens, which can lead to the loss of GSH, LPO, and a decrease in antioxidant enzyme activity

Figure 4. GSH/GSSG ratio in lenses after treatment with dexamethasone and NACA. The GSH/ GSSG ratio in the Dex only group was significantly lower than in the control group. This GSH/ GSSG ratio was significantly increased upon pretreatment with 50 μM NACA. The NACA-only treated group showed no significant difference when compared to the control. *p ≤ 0.05 compared to the control group, and #p ≤ 0.05 compared to the Dex group. All reported values were represented as the mean ± S.D. of quadruplets.
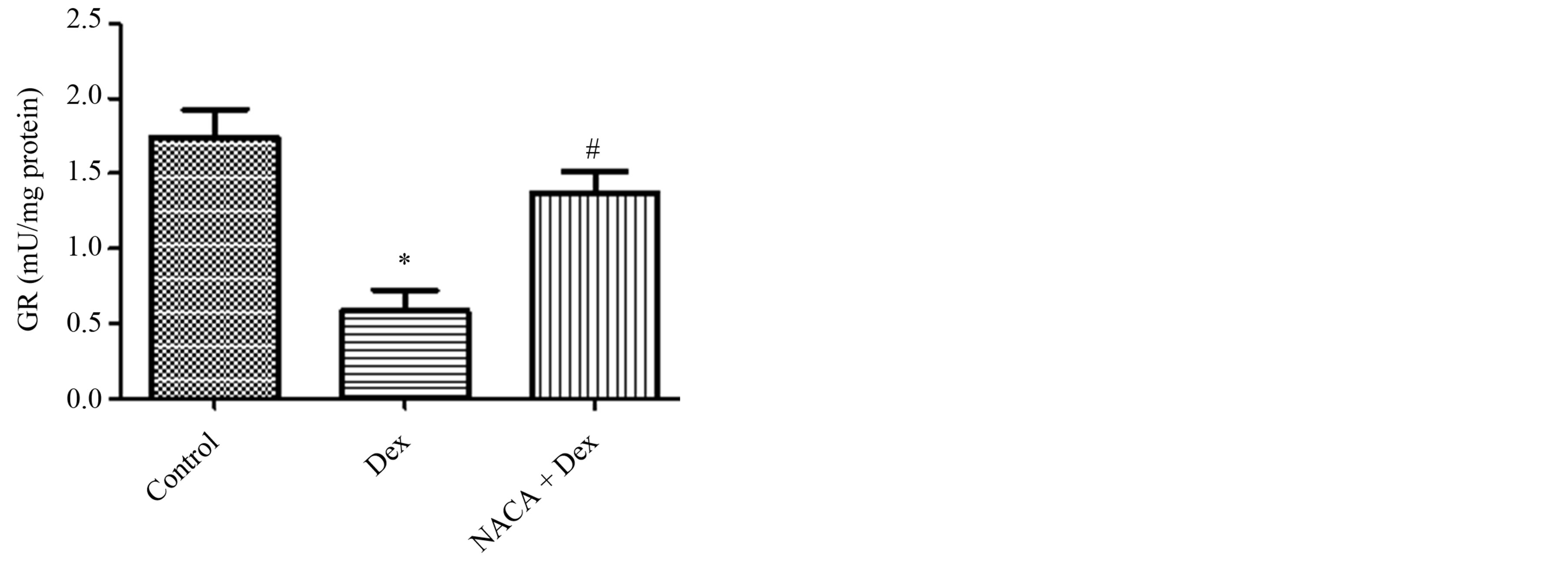
Figure 5. Glutathione reductase activity in lenses after treatment with dexamethasone and NACA. GR activity was significantly lower in the Dexonly group than in the control group, while NACA pretreatment increased its activity. The NACAonly treated group showed no significant difference when compared to the control. *p ≤ 0.05 compared to the control group, and #p ≤ 0.05 compared to the Dex group. All reported values were represented as the mean ± S.D. of quadruplets.

Figure 6. MDA levels in rat lenses after treatment with Dex and NACA. It was found that, after 7 days of Dex treatment, the MDA levels significantly increased. Dex (5 μM) induced a significant increase in the MDA level. Pretreatment with 50 µM of NACA decreased lipid peroxidation significantly. The NACAonly treated group showed no significant difference when compared to the control. *p ≤ 0.05 compared to the control group, and #p ≤ 0.05 compared to the Dex group. All reported values were represented as the mean ± S.D. of quadruplets.
[56-58]. GSH is an indispensable and primary lenticular antioxidant [12]. A wide body of evidence indicates loss of GSH because of its oxidation to GSSG, since its levels increase drastically once cataracts develop. Therefore, an alternative method for treating or preventing the occurrence of GC-induced cataracts would be through the use of a GSH prodrug. With this background, we evaluated the effects of a novel antioxidant and a potent GSH prodrug, N-acetylcysteine amide (NACA), in the prevention of cataracts induced by Dex in rat lenses. Results from morphological observations indicated that NACA was able to reduce the opacification of the lens within this exvivo dexamethasone-induced cataract model (Figure 1).
As discussed earlier, GSH is an essential lenticular antioxidant and is present in high concentrations in the lens, providing a first line of defense against oxidative damage [59], as well as playing an important role in antioxidant defense and redox regulation [60]. Results from this study indicate that treatment with dexamethasone decreased lenticular GSH and the GSH/GSSG ratio significantly. In addition, it increased GSSG levels significantly. However, pretreatment with NACA significantly increased the ratio of GSH/GSSG by decreasing the levels of GSSG. The ratio of reduced to oxidized glutathione (GSH/GSSG) serves as a representative marker of the antioxidative capacity of a cell. Depletion of GSH could be due to several possible mechanisms, including but not limited to the efflux of GSH from the lens, sequestration of GSH by incorporation into mixed disulfide aggregates, enhanced consumption of GSH in the process of detoxification of ROS, and downregulation of enzymes involved in GSH biosynthesis. Another possible explanation for the decrease in GSH levels under oxidative stress is the reduced GR activity. Some studies have indicated that loss of GSH will directly affect the activity of the GSH-dependent enzyme GR. This enzyme plays an important role in GSH homeostasis. It has been reported that, under oxidative stress, the protein sulfhydryl (protein-SH) groups are lost [61], which are essential for enzyme activity [62]. Under such circumstances, GSH is not regenerated, so depletion of GSH indicates that the tissue is undergoing oxidative stress. The decreased activity of GR seen in our experiments with Dex treatment supports our hypothesis of GSH depletion upon treatment with Dex. A decrease in the lenticular GSH levels has been observed upon treatment with glucocorticoids [13,35,63]. In vivo treatment of rabbits with the Dex eye drops, as used for cataract surgery, reduced GSH levels in the lens [63]. In addition, free radical scavengers have also been reported to prevent GC-induced cataract formation [64-66] by increasing GSH levels and decreasing LPO. Decreasing the levels of GSH, when reactive oxygen species are present, can trigger a cascade of further oxidative damage, such as LPO, which has been associated with the formation of cataracts in patients [67-70].
The extent of LPO was determined in this study by measuring the amount of MDA, a by-product of LPO, within the lens. Unavailability of GSH as a substrate for glutathione peroxidase stalled the process of lipid peroxide decomposition and, thus, increased the levels of MDA in the Dex-treated group. NACA supplied an adequate amount of GSH as a substrate for glutathione peroxidase to effectively decompose lipid peroxides in the rats, reducing MDA levels (Figure 6). The multiple roles of NACA in preventing cataract formation include direct scavenging of free radicals, providing cysteine for GSH synthesis, and nonenzymatic reduction of the preexisting toxic GSSG into GSH.
Our results suggest that NACA can prevent the formation of dexamethasone-induced cataracts by directly and indirectly maintaining the GSH/GSSG ratio in healthy lenses, allowing the lens to better cope with oxidative stress. NACA could confer a protective effect by providing a substrate for the generation of GSH, maintaining antioxidant levels within the lens and, possibly, through disulfide-exchange mechanisms. Treatment with NACA may prove to have a major therapeutic role in dexamethasone-induced cataracts. In future studies, we will focus on the prophylactic role of NACA in GC-induced cataract formation and investigate the development of a topical formulation for the application of this antioxidant in an in vivo model.
ACKNOWLEDGEMENTS
Dr. Ercal is supported by 1R15EY022218-01A1 from the National Eye Institute (NEI), National Institutes of Health (NIH). The contents of this paper are solely the responsibility of the authors and do not represent official views of the NIH. The authors appreciate the efforts of Barbara Harris in editing the manuscript.
REFERENCES
- Geley, S., Fiegl, M., Hartmann, B.L. and Kofler, R. (1996) Genes mediating glucocorticoid effects and mechanisms of their regulation. Reviews of Physiology, Biochemistry and Pharmacology, 128, 1-97.
- Reichardt, H.M., Tronche, F., Berger, S., Kellendonk, C. and Schutz, G. (2001) New insights into glucocorticoid and mineralocorticoid signaling: Lessons from gene targeting. Advances in Pharmacology, 47, 1-21.
- Jobling, A.I. and Augusteyn, R.C. (2002) What causes steroid cataracts? A review of steroid-induced posterior subcapsular cataracts. Clinical and Experimental Optometry, 85, 61-75. http://dx.doi.org/10.1111/j.1444-0938.2002.tb03011.x
- Carnahan, M.C. and Goldstein, D.A. (2000) Ocular complications of topical, periocular, and systemic corticosteroids. Current Opinion in Ophthalmology, 11, 478-483.
- Wang, C., Dawes, L.J., Liu, Y., Wen, L., Lovicu, F.J. and McAvoy, J.W. (2013) Dexamethasone influences FGFinduced responses in lens epithelial explants and promotes the posterior capsule coverage that is a feature of glucocorticoid-induced cataract. Experimental Eye Research, 111, 79-87. http://dx.doi.org/10.1016/j.exer.2013.03.006
- Urban, R.C. and Cotlier, E. (1986) Corticosteroid-induced cataracts. Survey of Ophthalmology, 31, 102-110. http://dx.doi.org/10.1016/0039-6257(86)90077-9
- Praveen, M.R., Shah, G.D., Vasavada, A.R., Shah, A.R., Johar K., Gami Y., et al. (2011) Posterior capsule opacification in eyes with steroid-induced cataracts: Comparison of early results. Journal of Cataract & Refractive Surgery, 37, 88-96. http://dx.doi.org/10.1016/j.jcrs.2010.08.035
- Liu, A. and Manche, E.E. (2011) Bilateral posterior subcapsular cataracts associated with long-term intranasal steroid use. Journal of Cataract & Refractive Surgery, 37, 1555-1558. http://dx.doi.org/10.1016/j.jcrs.2011.05.020
- Nishigori, H., Lee, J.W., Yamauchi, Y. and Iwatsuru, M. (1986) The alteration of lipid peroxide in glucocorticoidinduced cataract of developing chick embryos and the effect of ascorbic acid. Current Eye Research, 5, 37-40. http://dx.doi.org/10.3109/02713688608995163
- Anderson, E.I., Wright, D.D. and Spector, A. (1979) The state of sulfhydryl groups in normal and cataractous human lens proteins. II. Cortical and nuclear regions. Experimental Eye Research, 29, 233-243. http://dx.doi.org/10.1016/0014-4835(79)90004-6
- Lou, M.F., Dickerson, J.E. Jr., Garadi, R. and York, B.M., Jr. (1988) Glutathione depletion in the lens of galactosemic and diabetic rats. Experimental Eye Research, 46, 517-530. http://dx.doi.org/10.1016/S0014-4835(88)80009-5
- Reddy, V.N. (1990) Glutathione and its function in the lens—An overview. Experimental Eye Research, 50, 771- 778. http://dx.doi.org/10.1016/0014-4835(90)90127-G
- Spector, A. (1995) Oxidative stress-induced cataract: Mechanism of action. The FASEB Journal, 9, 1173-1182.
- Taylor, A. and Davies, K.J. (1987) Protein oxidation and loss of protease activity may lead to cataract formation in the aged lens. Free Radical Biology & Medicine, 3, 371- 377. http://dx.doi.org/10.1016/0891-5849(87)90015-3
- Babizhayev M.A. and Costa E.B. (1994) Lipid peroxide and reactive oxygen species generating systems of the crystalline lens. Biochimica et Biophysica Acta, 1225, 326-337. http://dx.doi.org/10.1016/0925-4439(94)90014-0
- Dische, Z. and Zil, H. (1951) Studies on the oxidation of cysteine to cystine in lens proteins during cataract formation. American Journal of Ophthalmology, 34,104-113.
- Kleiman, N.J. and Spector, A. (1993) DNA single strand breaks in human lens epithelial cells from patients with cataract. Current Eye Research, 12, 423-431. http://dx.doi.org/10.3109/02713689309024624
- Spector, A. and Roy, D. (1978) Disulfide-linked high molecular weight protein associated with human cataract. Proceedings of the National Academy of Sciences of the United States of America, 75, 3244-3248. http://dx.doi.org/10.1073/pnas.75.7.3244
- Zigler, J.S. Jr., Huang, Q.L. and Du, X.Y. (1989) Oxidative modification of lens crystallins by H2O2 and chelated iron. Free Radical Biology & Medicine, 7, 499-505. http://dx.doi.org/10.1016/0891-5849(89)90025-7
- Simpanya, M.F., Ansari, R.R., Suh, K.I., Leverenz, V.R. and Giblin, F.J. (2005) Aggregation of lens crystallins in an in vivo hyperbaric oxygen guinea pig model of nuclear cataract: Dynamic light-scattering and HPLC analysis. Investigative Ophthalmology & Visual Science, 46, 4641- 4651. http://dx.doi.org/10.1167/iovs.05-0843
- Zigler, J.S. Jr., Bodaness, R.S., Gery, I. and Kinoshita, J.H. (1983) Effects of lipid peroxidation products on the rat lens in organ culture: A possible mechanism of cataract initiation in retinal degenerative disease. Archives of Biochemistry and Biophysics, 225, 149-156. http://dx.doi.org/10.1167/iovs.05-0843
- Zigler, J.S. Jr., Gery, I., Kessler, D. and Kinoshita, J.H. (1983) Macrophage mediated damage to rat lenses in culture: A possible model for uveitis-associated cataract. Investigative Ophthalmology & Visual Science, 24, 651- 654.
- Goosey, J.D., Tuan, W.M. and Garcia, C.A. (1984) A lipid peroxidative mechanism for posterior subcapsular cataract formation in the rabbit: A possible model for cataract formation in tapetoretinal diseases. Investigative Ophthalmology & Visual Science, 25, 608-612.
- Zigler, J.S. Jr. and Hess, H.H. (1985) Cataracts in the Royal College of Surgeons rat: Evidence for initiation by lipid peroxidation products. Experimental Eye Research, 41, 67-76. http://dx.doi.org/10.1016/0014-4835(85)90095-8
- Borchman, D., Paterson, C.A. and Delamere, N.A. (1989) Oxidative inhibition of Ca2+-ATPase in the rabbit lens. Investigative Ophthalmology & Visual Science, 30, 1633- 1637.
- Kagan V.E. (1988) Lipid peroxidation in biomembranes. CRC Press, Boca Raton, 181.
- Park, J.W. and Floyd, R.A. (1992) Lipid peroxidation products mediate the formation of 8-hydroxydeoxyguanosine in DNA. Free Radical Biology & Medicine, 12, 245-250. http://dx.doi.org/10.1016/0891-5849(92)90111-S
- Harding, J.J. (1970) Free and protein-bound glutathione in normal and cataractous human lenses. Biochemical Journal, 117, 957-960.
- Linetsky, M., James, H.L. and Ortwerth, B.J. (1999) Spontaneous generation of superoxide anion by human lens proteins and by calf lens proteins ascorbylated in vitro. Experimental Eye Research, 69, 239-248. http://dx.doi.org/10.1006/exer.1999.0710
- Fu, S., Dean, R., Southan, M. and Truscott, R. (1998) The hydroxyl radical in lens nuclear cataractogenesis. The Journal of Biological Chemistry, 273, 28603-28609. http://dx.doi.org/10.1074/jbc.273.44.28603
- Rathbun, W.B. and Bovis, M.G. (1986) Activity of glutathione peroxidase and glutathione reductase in the human lens related to age. Current Eye Research, 5, 381- 385. http://dx.doi.org/10.3109/02713688609025177
- David, L.L. and Shearer, T.R. (1984) State of sulfhydryl in selenite cataract. Toxicology and Applied Pharmacology, 74, 109-115. http://dx.doi.org/10.1016/0041-008X(84)90276-X
- Calvin, H.I., Medvedovsky, C. and Worgul, B.V. (1986) Near-total glutathione depletion and age-specific cataracts induced by buthionine sulfoximine in mice. Science, 233, 553-555. http://dx.doi.org/10.1126/science.3726547
- Padgaonkar, V., Giblin, F.J. and Reddy, V.N. (1989) Disulfide cross-linking of urea-insoluble proteins in rabbit lenses treated with hyperbaric oxygen. Experimental Eye Research, 49, 887-899. http://dx.doi.org/10.1016/S0014-4835(89)80047-8
- Dickerson, J.E. Jr., Dotzel, E. and Clark, A.F. (1997) Steroid-induced cataract: New perspective from in vitro and lens culture studies. Experimental Eye Research, 65, 507-516. http://dx.doi.org/10.1006/exer.1997.0359
- Nishigori, H., Kosano, H. and Umeda, I.O. (2004) Inhibition of glucocorticoid-induced cataracts in chick embryos by RU486: A model for studies on the role of glucocorticoids in development. Life Sciences, 75, 3027-3033. http://dx.doi.org/10.1016/j.lfs.2004.04.051
- Creighton, M.O., Sanwal, M., Stewart-DeHaan, P.J. and Trevithick, J.R. (1983) Modeling cortical cataractogenesis. V. Steroid cataracts induced by solumedrol partially prevented by vitamin E in vitro. Experimental Eye Research, 37, 65-76. http://dx.doi.org/10.1016/0014-4835(83)90150-1
- Hirsch, R.P. and Schwartz, B. (1983) Increased mortality among elderly patients undergoing cataract extraction. JAMA Ophthalmology, 101, 1034-1037. http://dx.doi.org/10.1001/archopht.1983.01040020036004
- Babizhayev, M.A., Burke, L., Micans, P. and Richer, S.P. (2009) N-Acetylcarnosine sustained drug delivery eye drops to control the signs of ageless vision: Glare sensitivity, cataract amelioration and quality of vision currently available treatment for the challenging 50,000-patient population. Clinical Interventions in Aging, 4, 31-50.
- Zhang, S., Chai, F., Yan, H, Guo, Y. and Harding, J.J. (2008) Effects of N-acetylcysteine and glutathione ethyl ester drops on streptozotocin-induced diabetic cataract in rats. Molecular Vision, 14, 862-870.
- Cotgreave, I.A. (1997) N-acetylcysteine: Pharmacological considerations and experimental and clinical applications. Advances in Pharmacology, 38, 205-227. http://dx.doi.org/10.1016/S1054-3589(08)60985-0
- Atlas, D., Melamed, E. and Offen, D. (1999) Brain targeted low molecular weight hydrophobic antioxidant compounds. US Patent No. 5874468.
- Ates, B., Abraham, L. and Ercal, N. (2008) Antioxidant and free radical scavenging properties of N-acetylcysteine amide (NACA) and comparison with N-acetylcysteine (NAC). Free Radical Research, 42, 372-377. http://dx.doi.org/10.1080/10715760801998638
- Tobwala, S., Zhang, X., Zheng, Y., Wang, H.J., Banks, W.A. and Ercal, N. (2013) Disruption of the integrity and function of brain microvascular endothelial cells in culture by exposure to diesel engine exhaust particles. Toxicology Letters, 220, 1-7. http://dx.doi.org/10.1016/j.toxlet.2013.03.023
- Banerjee, A., Trueblood, M.B., Zhang, X., Manda, K.R., Lobo, P., Whitefield, P.D., Hagen, D.E. and Ercal, N. (2009) N-acetylcysteineamide (NACA) prevents inflammation and oxidative stress in animals exposed to diesel engine exhaust. Toxicology Letters, 187, 187-193. http://dx.doi.org/10.1016/j.toxlet.2009.02.022
- Carey, J.W., Pinarci, E.Y., Penugonda, S., Karacal, H. and Ercal, N. (2011) In vivo inhibition of l-buthionine- (S,R)-sulfoximine-induced cataracts by a novel antioxidant, N-acetylcysteine amide. Free Radical Biology and Medicine, 50, 722-729. http://dx.doi.org/10.1016/j.freeradbiomed.2010.12.017
- Grinberg, L., Fibach, E., Amer, J. and Atlas, D. (2005) Nacetylcysteine amide, a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free Radical Biology and Medicine, 38, 136-145. http://dx.doi.org/10.1016/j.freeradbiomed.2004.09.025
- Offen, D., Gilgun-Sherki, Y., Barhum, Y., Benhar, M., Grinberg, L., Reich, R., Melamed, E. and Atlas, D. (2004) A low molecular weight copper chelator crosses the blood-brain barrier and attenuates experimental autoimmune encephalomyelitis. Journal of Neurochemistry, 89, 1241-1251. http://dx.doi.org/10.1111/j.1471-4159.2004.02428.x
- Carey, J.W., Tobwala, S., Zhang, X., Banerjee, A., Ercal, N., Pinarci, E. and Karacal, H. (2012) N-acetyl L-cysteine amide protects retinal pigment epithelium against methamphetamine-induced oxidative stress. Journal of Biophysical Chemistry, 3, 101-110. http://dx.doi.org/10.4236/jbpc.2012.32012
- Tobwala, S., Fan, W., Stoeger, T. and Ercal, N. (2013) N-acetylcysteine amide, a thiol antioxidant, prevents bleomycin-induced toxicity in human alveolar basal epithelial cells (A549). Free Radical Research, 47, 740-749. http://dx.doi.org/10.3109/10715762.2013.819974
- Zhang, X., Tobwala, S. and Ercal, N. (2012) N-acetylcysteine amide protects against methamphetamine-induced tissue damage in CD-1 mice. Human & Experimental Toxicology, 31, 931-944. http://dx.doi.org/10.1177/0960327112438287
- Xie, G.L., Yan, H. and Lu, Z.F. (2011) Inhibition of glucocorti-coid-induced alteration of vimentin by a glucocorticoid receptor antagonist RU486 in the organ-cultured rat lens. Molecular Vision, 17, 32-40.
- Winters, R.A., Zukowski, J., Ercal, N., Matthews, R.H. and Spitz, D.R. (1995) Analysis of glutathione, glutathione disulfide, cysteine, homocysteine, and other biological thiols by high-performance liquid chromatography following derivatization by n-(1-pyrenyl) maleimide. Analytical Biochemistry, 227, 14-21. http://dx.doi.org/10.1006/abio.1995.1246
- Ates, B., Ercal, B.C., Manda, K., Abraham, L. and Ercal, N. (2009) Determination of glutathione disulfide levels in biological samples using thiol-disulfide exchanging agent, dithiothreitol. Biomedical Chromatography, 23, 119-123. http://dx.doi.org/10.1002/bmc.1083
- Draper, H.H., Squires, E.J., Mahmoodi, H., Wu, J., Agarwal, S. and Hadley, M. (1993) A comparative evaluation of thiobarbituric acid methods for the determination of malondialdehyde in biological materials. Free Radical Biology and Medicine, 15, 353-363. http://dx.doi.org/10.1016/0891-5849(93)90035-S
- Bhuyan, K.C. and Bhuyan, D.K. (1984) Molecular mechanism of cataractogenesis: III. Toxic metabolites of oxygen as initiators of lipid peroxidation and cataract. Current Eye Research, 3, 67-81. http://dx.doi.org/10.3109/02713688408997188
- Meister, A. (1991) Glutathione deficiency produced by inhibition of its synthesis, and its reversal; Applications in research and therapy. Pharmacology & Therapeutics, 51, 155-194. http://dx.doi.org/10.1016/0163-7258(91)90076-X
- Mitton, K.P., Dean, P.A.W., Dzialoszynski, T., Xiong, H., Sanford, S.E. and Trevithick, J.R. (1993) Modelling cortical cataractogenesis. 13. Early effects on lens ATP/ADP and glutathione in the strepto-zotocin rat model of the diabetic cataract. Experimental Eye Research, 56, 187-198. http://dx.doi.org/10.1006/exer.1993.1026
- Truscott, R.J. (2005) Age-related nuclear cataract-oxidation is the key. Experimental Eye Research, 80, 709-725. http://dx.doi.org/10.1016/j.exer.2004.12.007
- Lou, M.F. (2003) Redox regulation in the lens. Progress in Retinal and Eye Research, 22, 657-682. http://dx.doi.org/10.1016/S1350-9462(03)00050-8
- Wu, G., Fang, Y.Z., Yang, S., Lupton, J.R. and Turner, N.D. (2004) Glutathione metabolism and its implications for health. The Journal of Nutrition, 134, 489-492.
- Agarwal, R. and Shukla, G.S. (1999) Potential role of cerebral glutathione in the maintenance of blood-brain barrier integrity in rat. Neurochemical Research, 24, 1507-1514. http://dx.doi.org/10.1023/A:1021191729865
- Pescosolido, N., Miccheli, A., Manetti, C., Iannetti, G.D., Feher, J. and Cavallotti, C. (2001) Metabolic changes in rabbit lens induced by treatment with dexamethasone. Ophthalmic Research, 33, 68-74. http://dx.doi.org/10.1159/000055646
- Nishigori, H., Hayashi, R., Lee, J.W., Maruyama, K. and Iwatsuru, M. (1985) Preventive effect of ascorbic acid against glucocorticoid-induced cataract formation of developing chick embryos. Experimental Eye Research, 40, 445-451. http://dx.doi.org/10.1016/0014-4835(85)90157-5
- Nishigori, H., Yasunaga, M., Mizumura, M., Lee, J.W. and Iwatsuru, M. (1989) Preventive effects of pyrroloquinoline quinone on formation of cataract and decline of lenticular and hepatic glutathione of developing chick embryo after glucocorticoid treatment. Life Sciences, 45, 593-598. http://dx.doi.org/10.1016/0024-3205(89)90044-1
- Setogawa, T., Kosano, H., Ogihara-Umeda, I., Kayanuma, T. and Nishigori, H. (1994) Preventive effect of SA3443, a novel cyclic disulfide, on glucocorticoid-induced cataract formation of developing chick embryo. Experimental Eye Research, 58, 689-695. http://dx.doi.org/10.1006/exer.1994.1066
- Rikans, L.E. and Hornbrook, K.R. (1997) Lipid peroxidation, antioxidant protection and aging. Biochimica et Biophysica Acta, 1362, 116-127. http://dx.doi.org/10.1016/S0925-4439(97)00067-7
- Bhuyan, K.C., Bhuyan, D.K. and Podos, S.M. (1986) Lipid peroxidation in cataract of the human. Life Sciences, 38, 1463-1471. http://dx.doi.org/10.1016/0024-3205(86)90559-X
- Micelli-Ferrari, T., Vendemiale, G., Grattagliano, I., Boscia, F., Arnese, L., Altomare, E. and Cardia, L. (1996) Role of lipid peroxidation in the pathogenesis of myopic and senile cataract. British Journal of Ophthalmology, 80, 840- 843. http://dx.doi.org/10.1136/bjo.80.9.840
- Simonelli, F., Nesti, A., Pensa, M., Romano, L., Savastano, S., Rinaldi, E. and Auricchio, G. (1989) Lipid peroxidation and human cataractogenesis in diabetes and severe myopia. Experimental Eye Research, 49, 181-187. http://dx.doi.org/10.1016/0014-4835(89)90088-2
NOTES
*Corresponding author.