Open Journal of Pediatrics
Vol.06 No.02(2016), Article ID:67210,10 pages
10.4236/ojped.2016.62026
Apert Syndrome: A Case Report and Review of Literature
Simon Pius1*, Halima Abubakar Ibrahim1, Mustapha Bello1, Kefas Mbaya2, Jose Pwavimbo Ambe1
1Dapartment of Paediatrics, University of Maiduguri Teaching Hospital, Maiduguri, Nigeria
2Department of Surgery, University of Maiduguri Teaching Hospital, Maiduguri, Nigeria

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 29 April 2016; accepted 5 June 2016; published 9 June 2016
ABSTRACT
Apert syndrome, also known as acrocephalosyndactyly, is one of the causes of craniofacial syndrome or deformity. It is a rare congenital disorder characterized by premature fusion of cranial sutures (craniosynostosis), malformation of skull, hands, face and feet. This congenital deformity has incidence of 1/50,000 to 1/80,000 live births and is an autosomal dominant in inheritance. Apert syndrome, fibroblast growth factor receptor 2 (FGFR2) and the missense substitution mutations occur at adjacent amino acids (i.e. Ser252Trp, Ser 252Phe, Pro253Arg) between the second and third extra cellular immunoglobulin domain of FGFR2, which maps to chromosome bands 10q26. Increased paternal age has been implicated in the development of Apert syndrome. The syndrome has to be thoroughly evaluated as early definitive diagnosis is important in order to distinguish Apert syndrome from other forms of craniosynostosis like Carpenter syndrome, Crouzon disease, Pfeiffer and Saethre-Chotzen syndrome. It is generally accepted that management of Apert syndrome is multidisciplinary in approach, which should compose of neonatologists, neurosurgeons, craniofacial surgeons, plastic surgeons, otolaryngologists, orthodontists, orthopaedic surgeons, ophthalmologists, radiologists, geneticists, clinical psychologists and speech and language pathologists for the effective management of this condition. Early diagnosis and treatment is important because Apert syndrome when treated early has good prognosis in adult life.
Keywords:
Apert Syndrome, Congenital Malformations, Autosomal Dominant, Premature Fusion of Sutures, Hoof/Rosebud Hand

1. Introduction
Apert syndrome, also known as Acrocephalosyndactyly, is a rarecongenital malformation characterized by premature fusion of the cranial sutures (craniosynostosis), malformations of the skull, hands, face and feet [1] - [3] .
Apert syndrome was described by Dr Eugene Charles Apert, a French Physician in 1906, in nine people who shared similar attributes and characteristics, and was classified into three types depending on the extent and area overwhelmingly involved [1] . It has been described as a branchial arch syndrome, affecting the first branchial (orpharyngeal) arch, the precursor of the maxilla and mandible [4] [5] .
Linguistically “acro” is a Greek word for “peak” referring to a “peaked” head that is common in the syndrome. “Cephalo”, also a Greek, is a combining form meaning “head”. “Syndactyly” refers to webbing of fingers and toes [4] . Embryologically, the hands and feet have selective cell that die, called selective cell death or apoptosis, causing separation of digits, however, in acrocephalosyndactyly selective cell death fail to occur and the skin and rarely bone between the fingers and toes fuses [6] .
The inheritance of Apert syndrome is usually autosomal dominant, but sporadic cases have been reported and probably represent new mutations. Sporadic transmission indicates that a family may have a child with Apert syndrome when no other member of the family is affected. The recurrence risk of having another child with Apert syndrome for two unaffected parents is negligible. However, there is a higher mutation rate in males because the germ-cell divisions in males are greater than those in females. Hence the mutation rates increase with advanced paternal age [7] . Apert syndrome is caused by mutations in a gene called fibroblast growth factor receptor two which is located on chromosome 10 [8] [9] .
The incidence of infants born with Apert syndrome ranges between 1 in 50,000 to 80,000 live births in the over world [7] . This rare clinical abnormality has to be differentiated from other craniofacial syndromes such as Carpenter syndrome (cloverleaf skull deformity), Crouzon disease (craniofacial dysostosis), Pfeiffer syndrome and Seathre-Chotzen syndrome [1] .
This case study is reported here in order to remind clinician especially the neonatologists who are first confronted with such congenital abnormality that, this inherited condition, even though is rare as it thought to be, whenever it is observed at birth, it should be thoroughly evaluated so as to differentiate it from other causes of craniosynostosis mentioned above because Apert syndrome when identified early and treated its, outcome is good in adulthood life.
2. Case Report
BHM, an hour old newborn referred from a secondary health facility with complains of multiple congenital malformation and difficulty in breathing notice at birth.
Patient is products of term gestation, delivered via spontaneous vaginal delivery at the referral hospital and child cry immediately after birth with APGAR score of 7 at one minute and 8 at five minutes respectively. Had a normal and adequate post delivery care, however while being attended to, patient was noticed to have abnormal features and difficulty in breathing with bluish discoloration of the extremities. No history of congenital malformation in the siblings and no family history of foetal wastage or maternal exposure to radiation and use of unprescribed medications. Nomaternal febrile episodes in pregnancy and had tetanus toxoid (TT), intermittent presumptive therapy (IPT) and prescribed haemetinics. Mother is 30 yrs old secondary school leaver from a non-consanguineous marriage in polygamy of two wives, while father is 40 yrs old businessman who deals in traditional fragrance.
Examination findings were: child had slightly dusky trunk with acrocyanosis, was anecteric, he weighed 3.95 kg, his length was 47 cm, was noticed to have multiple congenital malformations
Head: Brachycephally with head circumference of 30 cm, flat occiput, had prominent forehead, anterior fontanelle was widely patent measuring 5 by 6 cm, and posterior fontanelle was also widely patent measuring 4 by 3 cm with sutural diasthesis involving saggital and mitopic sutures. Coronal suture was fussed with ridging. There was also pinched nose, downward slanting of pelpabrel fissures and trapezoid mouth. He was conscious with good activity and normally presents primitive reflexes; he had increased tone in all limbs.
Musculoskeletal system (MSS): had syndactilly, hands appear spoon-shaped, characterized by fusion of all the fingers. Both feet are in equine partae with all toes fused giving it a clubb-like feature. RR of 54CPM, breath sound was bronchovesicular. HR was 152 BPM, heart sound were 1 and 2 only no murmur heard.
Child had unilateral left choanal atresia. Abdomen was flat, moved with respiratory effort, umbilical stump was secured and clean and had well formed male external genitalia.
At admission a working diagnosis of type I Apert (acrocephalosyndactyly) syndrome with choanal atresia was made. Second consideration of at risk for sepsis was also entertained. Results of investigations include venous packed cell volume 62%, random blood sugar of 6.6 mmol/L, blood chemistry revealed Na+ = 134 mmol/L, K+ = 8.4 mmol/L, CL− = 94 mmol/L, 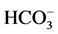 = 15 mmol/L, Urea = 5.9 mmol/L. X-ray of the hands and feet revealed fused bones of the fingers and toes. Choanography showed membranous blockage of left nostril. ENT surgery was done and had a course of antibiotics parenteral cefuroxime and Gentamycin for 14 days. He was discharged and was seen at follow up on two occasions by our team and the ENT team 5th week and 10th week and the baby is thriving well at least for now (Figures 1-6).
= 15 mmol/L, Urea = 5.9 mmol/L. X-ray of the hands and feet revealed fused bones of the fingers and toes. Choanography showed membranous blockage of left nostril. ENT surgery was done and had a course of antibiotics parenteral cefuroxime and Gentamycin for 14 days. He was discharged and was seen at follow up on two occasions by our team and the ENT team 5th week and 10th week and the baby is thriving well at least for now (Figures 1-6).
3. Discussion
The inheritance of Apert syndrome as an autosomal dominant is a small fraction, while sporadic cases are majority and frequent. Sporadic transmission indicates that a family may have a child with Apert when no other member of the family is affected. The recurrence risk of having another child Apert syndrome for two unaffected parents is negligible. However, there is a higher mutation rate in males because the germ-cell divisions in males are greater than those in females. Hence, the mutation rate increases with increased paternal age [6] [7] .

Figure 1. Photograph of the neonate showing the spade like hands.

Figure 2. Picture of the hand showing single nail over the matted finger both left and right.

Figure 3. Picture of the neonate showing hoof/ rosebud feet.

Figure 4. Picture of upper part of the neonate showing depressed dorsum of the nose and trapezoid mouth.

Figure 5. Radiograph of the hands and feet showing fusion of the bones of the digits.

Figure 6. Photograph of the upper part of the trunk low set ears, beaked nose.
In contrast, Glaser and colleagues reported a significantly greater mutation rate in a group of young men who had children with Apert syndrome and suggested that there are many other relevant environmental factors in addition to increase in paternal age [10] . In the absence of a family history, diagnosis is difficult before birth of the baby especially in developing country like our where efficient antenatal care and possible prenatal diagnosis is lacking [6] .
Holten and colleagues concluded that there is a genetic anomaly causing variable and uncoordinated differentiation of the mesenchyme at the time of embryologic separation into various skeletal components, particularly in the distal limb bud and craniofacial skeleton. And the disease process continue postnatally, especially in endochondral bone growth [6] [11] . In 1995, A.O.M. Wilkie published a paper showing evidence that acrocephalosyndactyly was caused by a defect on the fibroblast growth factor receptor 2 gene, on chromosome 10 [8] . More than 98% of cases of Apert syndrome are caused by specific missense substitution mutations, involving adjacent amino acids (i.e. Ser 252Trp, Ser252Phe, Pro 253Arg) in the linker between the second and third extra cellular immunoglobulin domains of FGFR2, which maps to chromosome bands 10q26 [4] .
The remaining cases are due to Alu-element insertion mutations in or near exon 9 of FGFR2. Most of the sporadic cases resulting from new mutations are related to the increased paternal age effect. The incidence of FGFR2 mutations increases exponentially with increased paternal age, probably due to an increase in the frequency of these mutations and a selective advantage in the male germ-cell line [10] [12] . Park and colleagues concluded that the two missense mutations in exon IIIa of theFGFR2 gene resulting in Sre252Trp or Pro 253Arg, there no differences between the 2 mutations with respect to their phenotypic features, Yet there are overlap between Apert, Crouzon, Seathre-Chotzen, Carpenter and Pfeiffer syndromes [13] [14] .
Most new mutations, estimated at 1/65,000 live births imply that germ line transversion rates at these two positions are currently the highest known in human genome. The rarity of familial cases can be explained by reduced genetic fitness of individuals because of severe malformations and the presence of mental retardation in many cases [4] . The FGFR2 mutations lead to an increase in the number of precursor cells that enter the osteogenic pathway. Ultimately, this leads to increased subperiosteal bone matrix formation and premature calvaria ossification during foetal development. The order and rate of suture fusion determine the degree of deformity and disability [1] [4] . Once a suture becomes fused, growth perpendicular to that suture becomes restricted, and the fused bones act as a single bony structure.
Compensatory growth occurs at the remaining open sutures to allow continued brain growth; however, complex multiple sutural synostosis frequently extends to premature fusion of the sutures at the base of the skull, causing midfacial hypoplesia, shallow orbits, a foreshortened nasal dorsum, maxillary hypoplesia and occasional upper airway obstruction [15] - [17] .
The first genetic evidence that syndactyly in Apert syndrome is a keratinocyte growth factor receptor (KGFR)-mediated effect was provided by the observation of the correlation between KGFR expression in fibroblasts and severity of syndactyly. Patients with Ser252Trp and those with Pro 253Arg have different phenotypic expression. The syndactyly is more severe with Pro253Arg mutation for both hands and feet, like in this patients who has type III (“hoof or rosebud”) syndactyly in both the hands and feet, in contrast cleft palate is significantly more common with Ser252Trp mutation though our patient don’t have [18] .
Amblyopia and strabismus is more common in patients with the FGFR2 Ser252Trp mutation, and optic disc pallor is more frequent in patients with the FGFR2 Pro253Arg mutation. Patients with FGFR2 Ser252Trp mutation have a significantly greater prevalence of visual impairment compared with patients with the FGFR2 Pro253Arg mutation [19] .
The spectrum of abnormalities in Apert syndrome as described recently along with the recommendations for orthodontic treatment is wide [20] . Many of the characteristic facial features of Apert syndrome results from the premature fusion of the skull bones. The head is unable to grow normally, which leads to a sunken appearance in the middle of the face, bulging and wide-set eyes, a beaked nose, and an underdeveloped upper jaw (maxillary hypoplesia) leading to crowded teeth and other dental problems. This case is a neonate but will be monitored for evolution of these complications.
Early fusion of the skull bones also affects the development of the brain, which leads to central nervous system (CNS) abnormalities including malformation of the corpus callosum and limbic system structures, gyral abnormalities, hypoplastic white matter, and heterotopic gray matter. Most patients have ventriculomegaly, resulting from distortion imposed upon a large brain within a misshapen skull leading to (cone-shaped) head with flat occiput, short anterior-posterior diameter, prominent elongated forehead and a short broad nose and reduced nasolabial angle. Progressive hydrocephalus occurred only in 10% of cases [4] [21] . Cardiovascular and genitourinary anomalies occurred in 10% and 9.6% of cases, respectively. Intelligence varied from normal to mental deficiency, even though fewer numbers were reported to have good integration into the society with normal social life [22] [23] . In patient with Apert syndrome, webbed or fused fingers and toes (syndactyly) are characteristic, and the severity of fusion varies from three digits on each hand and foot is fused together. In the most severe form, all of the fingers and toes are fused; this is the case of this patient we are reporting, because our patient has type III (hoof or rosebud) syndactyly which was well corroborated by the X-ray of the hands and feet showing fusion of the bones of the digits in Figure 5 above.
In addition, signs and symptoms of Apert syndrome may include hearing impairment resulting from persistent middle ear effusion, unusually heavy sweating (hyperhidrosis), oily skin with severe acne, patches of missing hair in the eyebrows, fusion of spinal bones in the (cervical vertebrae), may be associated with opening in the roof of the mouth (a cleft palate), this case did not have cleft palate. Apert syndrome patients also have characteristic abnormalities in other bones including the shoulders, elbows, hips, knees, and ribs [24] - [26] .
Diagnosis of Apert syndrome is mainly dependent upon the clinical and radiological findings, because molecular analysis for the detection of the specific mutation is expensive and is farfetched in this part of the world. Skull radiography reveals craniosynostosis which usually involves coronal sutures and the radiography of the hands and feet shows the various fusions in the hands and feet bones [1] [27] . This case had coronal sutural fusion and complete bone fusion in digits hands and feet.
Computerized tomography (CT) scan with 3-dimensional reconstruction analysis of the calvaria and cranial bases has become the most useful radiological examination in identifying skull shape and presence or absence of involved sutures. CT scan precisely reveals the pathological anatomy and permits specific operative planning. Magnetic resonance imaging (MRI) is the imaging modality of choice for detecting intracranial abnormalities. The common intracranial malformations reported on neuroimaging included ventriculomegaly, corpus callosum hypoplesia, septumpellucidum hypoplesia, cavum verga and arachnoid cyst [28] [29] . We could not afford to do CT-scan and MRI, due to financial constraints.
Treatment of Apert syndrome lest we forget, involve the formation of a strong multidisciplinary team comprising of neonatologists, neurosurgeons, craniofacial surgeons, plastic surgeons, otolaryngologists, orthodontists, orthopedic surgeons, ophthalmologists, radiologists, clinical psychologists and speech and language pathologists for the care of these patients to improve their quality of life. The modality is generally divided into two major approaches, the first one which include the medical management and the second one is the surgical management which is the main modality of the treatment. The management involves protection of the cornea, by instillation of lubricating ointment in the eyes at bedtime to protect corneas from desiccation and also artificial tear drops during the day [4] .
Upper airway obstructions during the neonatal period involve the removal of excessive nasal secretions, treat the upper airway infections. Also humidification with added oxygen and judicious use of topical nasal decongestants is helpful. Sleep apnoea monitoring (a sleep recording of multiple physiologic variables) currently is the most reliable method for determining the presence of sleep apnoea in Apert syndrome [1] [4] . There may be a need to employ continuous positive pressure ventilation. In the presence of chronic middle ear effusion associated with bilateral conductive hearing deficit, antibiotic therapy is essential.
In addition evaluation of psychosocial, speech and hearing is important so as to undertake rehabilitative measures to prevent cognitive impairment and delay in speech and language development [6] [30] [31] .
The Main aim of surgery in treatment of Apert syndrome includes the following, to release the cranial sutures in order to permit a proper brain development, to prevent hydrocephalus due to raised intracranial pressure, repair the cleft in order to prevent nasal regurgitation as well as facilitate proper speech. Correction of midfacial hypoplesia to facilitate proper breathing improves body image and self-esteem. Surgical release of the digits (syndactyly) is to provide better grasp.
Starting with the cranial surgery, it includes removal of the synostosis sutures so as to relieve the increased intracranial pressure, reshaping of the calvaria and this allows more normal cranial development to proceed with respect to shape, volume and bone quality. Surgical operations such lateral and medial tarsorrhaphy is performed in severe cases to narrow the palpebral fissures cosmetically and to protect the corneas and maintain vision. In case of upperairway obstruction compromising respiration may require orotracheal intubation [4] . Tracheostomy is usually indicated in sleep apnoea that is severely affecting the neonate, so also bilateral myringotomy and placement of ventilation tubes in cases of chronic middle ear effusion associated with bilateral conductive hearing deficit [1] [4] [32] .
Orbital surgery is to correct ocular proptosis, reduction of intraorbital distance thereby correcting the interior malrotation. As the child grows, nasal reconstruction focuses on correction of the excessively obtuse nasofrontal angle, flat nasal dorsum, and ptotic nasal tip [33] [34] . In addition, some form of midfacial surgery involve normalization of midface appearance, expansion of the inferior orbit, volumetric expansion of the nasal and nasopharyngeal airways and establishment of a normal dentoskeletal relationship [4] [35] [36] . Also mandibular osteotomies are usually performed to improve dentoskeletal relations for masticatory and aesthetic benefit.
Recently, some advancement has been made in other surgical approaches such as surgical care involving, early release of the coronal suture and fronto-orbital advancement and reshaping to reduce dysmorphic and unwanted skull growth changes. Craniosynostosis requires multistaged operative surgeries which usually proffer significant cosmetic improvement. Ideally initial surgery is often performed as early as age 3 months [4] .
Most often, the procedural options being applied; that is the Le Fort II which is typically used if forehead retrusion is not present, while the Le Fort III osteotomy or the monobloc advancement is by distraction osteogenesis [37] [38] . Distraction osteogenesis involves methodical lengthening of bone by gradual mechanical displacement of a surgically created fracture. Application of this technique in the correction of midfacial structure deformity may produce more durable results in Paediatric population and appears to improve the outcome of the midface advancement compared with the traditional techniques [39] .
In the presence of ventriculomegaly, hydrocephalus do occur in 10%, so as part of surgical manipulation, shunting procedure is performed in order to prevent development of hydrocephalus and consequent development of raised intra cranial pressure [4] [40] .
Syndactyly generally speaking, there is no standard treatment for the hand/foot malformations in Apert syndrome, due to the differences and severity in clinical manifestations in different patients. Every patient is therefore individualized in approach to treatment, aiming at an adequate balance between hand functionality and cosmetics. However some guidelines can be given depending on the severity of the deformities.
In general it is initially recommended to release the first and the fourth interdigital spaces, thus releasing the border rays [41] . This makes it possible for the child to grasp things by hand, a very important function for the child’s development. Later the second and third interdigital spaces have to be released. Because there three hand types in Apert, all with their different deformities, they all need a different approach regarding their treatment. However, the case being reported has type III syndactyly (“hoof” or “rosebud” hand) e.g. complex syndactyly of the first web space, tight fusion of all digits with one conjoint nail of the middle three fingers and complex syndactyly of the fourth web space.
Type III hands are the most challenging to treat because of their complexity. First of all, it is advised to release the first and the fourth web space, thus converting it to type I hand. In order to increase the first web space, lengthening of the thumb can be done. It is also considered that in severe cases an amputation of the index finger can be done [42] [43] . However, before making this decision, it is important to weigh the potential improvement to be achieved against the possible psychological problems of the child later due to aesthetics of the hand. Later, the second and/or third inter digital web space should be released, bearing in mind with the growth of the child and respectively the hands, secondary revisions are usually needed to treat the contractures and to improve the cosmetic appearance of the hands.
4. Conclusion
Apert syndrome (acrocephalosyndactyly) is an autosomal dominant condition in which increased paternal age has been implicated in its development. It is an uncommon condition that causes craniosynostosis and limb deformities. Other syndromes that are associated with craniosynostosis include Carpenter syndrome, Crouzons disease, Pfeiffer syndrome, Saethre Chotzen syndrome. These syndromic conditions should be evaluated in detail so that each syndrome should be differentiated with precision from each other. Apert syndrome, fibroblast growth factor receptor 2 (FGFR2) and the missense substitution mutations occur at adjacent amino acids (i.e. Ser252Trp, Ser 252Phe, Pro253Arg) between the second and third extra cellular immunoglobulin domain of FGFR2, which maps to chromosome bands 10q26. While that of syndactyly is a keratinocyte growth factor receptor, and syndactyly is more severe with Pro253Arg mutations. When a neonatologist is confronted with this condition, effort should be made to evaluate the patient thoroughly so as to make a definitive diagnosis because when properly diagnosed and treated early in life, Apert syndrome has good outcome in both intelligence and aesthetic appearance in adult life.
Cite this paper
Simon Pius,Halima Abubakar Ibrahim,Mustapha Bello,Kefas Mbaya,Jose Pwavimbo Ambe, (2016) Apert Syndrome: A Case Report and Review of Literature. Open Journal of Pediatrics,06,175-184. doi: 10.4236/ojped.2016.62026
References
- 1. Gazi, S.R., Manu, P.S. and Vijayalakshmi, S. (2014) Apert’s Syndrome—A Rare Craniofacial Anomaly. South-East Asian Journal of Case Report and Review, 3, 645-667.
- 2. Apert, M. (1906) De l’acrocephalosyndactylie. Bulletins et mémoires de la société des hopitaux de Paris, 23, 1310-1313.
- 3. Cohen, M.M.J. (1986) Craniosynostosis: Diagnosis, Evaluation and Management. Raven Press, New York.
- 4. Satyanand, T., Sachin, K. and Mohit, S. (2010) Etiology, Symptoms and Treatment of Apert Syndrome, A Congenital Disorder: An Overview. International Journal of Pharma and Bio Sciences, 1, 1-7.
- 5. Blank, C.E. (1960) Apert’s Syndrome (A Type of Acrocephalosyndactyly)—Observations on a British Series of 39 Cases. Annals of Human Genetics, 24, 151-164.
http://dx.doi.org/10.1111/j.1469-1809.1959.tb01728.x - 6. Premalatha, Kannan, V.P. and Madhu. (2010) Apert Syndrome. Case Report. The Journal of Indian Society of Pedodontics and Preventive Dentistry, 28, 322-325.
- 7. Kilic, I., Baykara, Y., Semerci, C.N., Ergin, H., Lale, N. and Tufan, S. (2004) Apert Syndrome. Turkish Journal of Medical Sciences, 34, 405-448.
- 8. Wilkie, A.O., Slaney, S.F., Oldridge, M., Poole, M.D., Ashworth, G.J., Hockley, A.D., et al. (1995) Apert Syndrome Results from Mutations of FGFR2 and Is Allelic with Crouzon Syndrome. Nature Genetics, 9, 165-172.
http://dx.doi.org/10.1038/ng0295-165 - 9. Ibrahimi, O.A., Eliseenkova, A.V., Plotnikov, A.N., Yu, K., Ornitz, D.M. and Mohammadi, M. (2001) Structural Basis for Fibroblast Growth Factor Receptor 2 Activation in Apert Syndrome. Proceedings of the National Academy of Sciences of the United States of America, 98, 7182-7187.
http://dx.doi.org/10.1073/pnas.121183798 - 10. Glaser, R.L., Broman, K.W. and Schulman, R.L. (2003) The Paternal Age Effect in Apert Syndrome Is Due, in Part to the Increased Frequency of Mutation in Sperm. The American Journal of Human Genetics, 73, 939-947.
http://dx.doi.org/10.1086/378419 - 11. Holten, I.W., Smith, A.W., Bourne, A.J. and David, D.J. (1997) The Apert Syndrome Hand: Pathologic Anatomy and Clinical Manifestation. Plastic and Reconstructive Surgery, 99, 1681-1687.
- 12. Goriely, A., Mcvean, G.A., Rojmyr, M., Ingemarsson, B. and Wilkie, A.O. (2003) Evidence for Selective Advantage of Pathologic FGFR2 Mutations in the Male Germ Line. Science, 301, 643-646.
http://dx.doi.org/10.1126/science.1085710 - 13. Lejeunle, E., Cameron, R. and El-Ghouzzi, V. (1990) Clinical Variability in Patients with Apert Syndrome. Journal of Neurosurgery, 90, 443-447.
- 14. Park, W.J., Theda, C. and Maestri, N.E. (1995) Analysis of Phenotypic Features and FGFR2 Mutations in Apert Syndrome. The American Journal of Human Genetics, 57, 321-328.
- 15. Fanganiello, R.D., Sertie, A.L., Reis, E.M., Yeh, E., Oliveira, N.A., Bueno, D.F., et al. (2007) Apert pSer252Trp Mutation in FGFR2 Alters Osteogenic Potential and Gene Expression of Cranial Periosteal Cells. Molecular Medicine, 13, 422-442.
http://dx.doi.org/10.2119/2007-00027.Fanganiello - 16. Ibrahimi, O.A., Chiu, E.S., McCarthy, J.G. and Mohammadi, M. (2005) Understanding the Molecular Basis of Apert Syndrome. Plastic & Reconstructive Surgery, 115, 264-270.
- 17. Khong, J.J., Anderson, P. and Gray, T.L. (2006) Ophthalmic Findings in Apert Syndrome Prior to Craniofacial Surgery. American Journal of Ophthalmology, 142, 328-330.
http://dx.doi.org/10.1016/j.ajo.2006.02.046 - 18. Slaney, S.F., Oldridge, M. and Hurst, J.A. (1996) Differential Effects of FGFR2 Mutations on Syndactyly and Cleft Palate in Apert Syndrome. American Journal of Human Genetics, 58, 923-932.
- 19. Khong, J.J., Anderson, P.J., Hammerton, M., Roscioli, T., Selva, D. and David, D.J. (2007) Differential Effects of FGFR2 Mutation in Ophthalmic Findings in Apert Syndrome. Journal of Craniofacial Surgery, 18, 39-42.
http://dx.doi.org/10.1097/01.scs.0000249358.74343.70 - 20. Hohoff, A., Joos, U., Meyer, U., Ehmer, U. and Stamm, T. (2007) The Spectrum of Apert Syndrome Phenotype, Particularities in Orthodontic Treatment and Characteristics of Orthodontic Surgery. Head & Face Medicine, 8, 10.
http://dx.doi.org/10.1186/1746-160X-3-10 - 21. Stal, S., Hollier, L.H. and Cole, P. (2011) Apert Syndrome, Uptodate Version 19.3.
- 22. Renier, D., Arnauld, E. and Cinalli, G. (1995) Prognosis for Mental Function in Apert Syndrome. Journal of Neurosurgery, 85, 66-72.
http://dx.doi.org/10.3171/jns.1996.85.1.0066 - 23. Allam, K.A., Wan, D.C. and Khwanngern, K. (2011) Treatment for Apert Syndrome: A Long-Term Follow-Up Study. Plastic & Reconstructive Surgery, 127, 1601-1611.
http://dx.doi.org/10.1097/PRS.0b013e31820a64b6 - 24. Cohen Jr., M.M. and Kreiborg, S. (1993) Skeletal Abnormalities in Apert Syndrome. American Journal of Medical Genetics, 47, 624-632.
http://dx.doi.org/10.1002/ajmg.1320470509 - 25. Rajenderkumar, D., Bamiou, D.E. and Sirimanna, T. (2005) Audiological Profile in Apert Syndrome. Archives of Disease in Childhood, 90, 592-593.
http://dx.doi.org/10.1136/adc.2004.067298 - 26. de Jong, T., Toll, M.S., de Gier, H.H. and Mathijssen, I.M. (2011) Audiological Profile of Children and Young Adults with Syndromic and Complex Craniosynostosis. Archives of Otolaryngology—Head & Neck Surgery, 137, 775-778.
- 27. Cohen Jr., M.M. and Kreiborg, S. (1996) Suture Formation, Premature Sutural Fusion, and Suture Default Zones in Apert Syndrome. American Journal of Medical Genetics, 62, 339-344.
http://dx.doi.org/10.1002/(SICI)1096-8628(19960424)62:4<339::AID-AJMG3>3.0.CO;2-M - 28. Yacubian-Fernandes, A., Palhares, A., Giglio, A., Gabarra, R.C., Zanint, S., Portela, L., et al. (2004) Apert Syndrome: Analysis of Associated Brain Malformations and Conformational Changes Determined by Surgical Treatment. Journal of Neuroradiology, 31, 116-122.
http://dx.doi.org/10.1016/s0150-9861(04)96978-7 - 29. Renier, D., Arnaud, E., Cinalli, G., Sebag, G., Zerah, M. and Marchac, D. (1996) Prognosis for Mental Function in Apert Syndrome. Journal of Neurosurgery, 85, 66-72.
http://dx.doi.org/10.3171/jns.1996.85.1.0066 - 30. Hen, H. Ed. (2006) Apert Syndrome In: Chen, H., Ed., Atlas of Genetic Diagnosis and Counseling, Springer-Verlag, New Jersey, 61-69.
- 31. Rajenderkumar, D., Bamiou, D. and Sirimanna, T. (2005) Management of Hearing Loss in Apert Syndrome. Journal of Laryngology & Otology, 119, 385-390.
http://dx.doi.org/10.1258/0022215053945714 - 32. Cunniff, C. (2004) Prenatal Screening and Diagnosis for Paediatricians. Pediatrics, 114, 889-894.
http://dx.doi.org/10.1542/peds.2004-1368 - 33. Tay, T., Martin, F. and Rowe, N. (2006) Prevalence and Causes of Visual Impairment in Craniosynostosis. Clinical & Experimental Ophthalmology, 34, 34-40.
http://dx.doi.org/10.1111/j.1442-9071.2006.01242.x - 34. Von Gemet, S., Golla, A., Ehrenfels, Y., Schuffenhauer, S. and Fairley, J.D. (2000) Genotype-Phenotype Analysis in Apert Syndrome Suggests Opposite Effects of the Two Recurrent Mutations on Syndactyly and Outcome of Craniofacial Surgery. Clinical Genetics, 57, 137-139.
http://dx.doi.org/10.1034/j.1399-0004.2000.570208.x - 35. McCarthy, J.G., Grayson, B. and Booksrein, F. (1984) Le Fort III Advancement Osteotomy in the Growing of Child. Plastic & Reconstructive Surgery, 74, 343-354.
http://dx.doi.org/10.1097/00006534-198409000-00003 - 36. Shetye, P.R., Kapadia, H., Grayson, B.H. and MacCarthy, J.G. (2010) A 10 Year Study of Skeletal Stability and Growth of Midface Following Le Fort III Advancement in a Syndromic Craniosynostosis. Plastic & Reconstructive Surgery, 126, 973-981.
http://dx.doi.org/10.1097/PRS.0b013e3181e60502 - 37. Cedars, M.G., Linck, D.L., Chin, M. and Toth, B.A. (1999) Advancement of Midface Using Distraction Techniques. Plastic & Reconstructive Surgery, 103, 429-441.
http://dx.doi.org/10.1097/00006534-199902000-00010 - 38. Wong, G.B., Kakulis, E.G. and Mulliken, J.B. (2000) Analysis of Fronto-Orbital Advancement for Apert, Crouzzon, Pfeiffer, and Seathre-Chotzen Syndromes. Plastic & Reconstructive Surgery, 105, 2314-2323.
http://dx.doi.org/10.1097/00006534-200006000-00002 - 39. Tessier, P. (1971) The Definitive Plastic Surgical Treatment of the Severe Facial Deformities of Craniofacial Dysostosis. Crouzon’s and Apert’s Diseases. Plastic & Reconstructive Surgery, 48, 419-442.
http://dx.doi.org/10.1097/00006534-197111000-00002 - 40. Do Amaral, C.M.R., Di Domizio, G. and Buzzo, C.L. (2009) Surgical Treatment of Apert Syndrome and Crouzon Anomaly by Gradual Bone Distraction. Plastic & Reconstructive Surgery, 124, 634-638.
- 41. Zucker, R.M. (1991) Syndactyly Correction of the Hands in Apert Syndrome. Clinics in Plastic Surgery, 18, 357-364.
- 42. Fearon, J.A. (2003) Treatment of the Hands and Feet in Apert Syndrome: An Evolution in Management. Plastic and Reconstructive Surgery (Dallas, Texas), 112, 1-12.
http://dx.doi.org/10.1097/01.PRS.0000065908.60382.17 - 43. Upton, J. (1991) Apert Syndrome. Classification and Pathologic Anatomy of Limb Anomalies. Clinics in Plastic Surgery, 18, 321-355.