Open Journal of Inorganic Chemistry
Vol.05 No.04(2015), Article ID:60345,8 pages
10.4236/ojic.2015.54013
Synthesis, Crystal Structure and Characterization of a New Dihydrogenomonophosphate: (C7H9N2O)H2PO4
Saloua Belghith*, Yahya Bahrouni, Latifa Ben Hamada
Laboratoire d’Energie et de Matériaux (LabEM-LR11ES34), Ecole Supérieure des Sciences et de la Technologie de Hammam Sousse, Tunisia
Email: *saloua.belghith@yahoo.fr
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 18 August 2015; accepted 16 October 2015; published 19 October 2015
ABSTRACT
Chemical preparation, X-ray single-crystal, thermal behavior, and IR spectroscopy investigations are given for a new organic cation dihydrogenomonophosphate (C7H9N2O)H2PO4 (denoted ABHP) in the solid state. This compound crystallizes in the monoclinic space group P21/n. The unit cell dimensions are: a = 11.011(5) Å, b = 5.861(1) Å, c = 15.944(4) Å and β = 100.81(5) with V = 1010.7(6) Å3 and Z = 4. The structure has been solved using direct method and refined to a reliability R factor of 0.048. The atomic arrangement can be described as inorganic clusters [H4P2O8]2− anions between which are located the organic dimmers (C14H18N4O2)2+ through multiple hydrogen bonds (Figure 1).
Keywords:
Chemical Synthesis, X-Ray Diffraction, H-Bonds, Differential Thermal Analysis, Infrared Spectroscopy

1. Introduction
Among the various categories of phosphates, monophosphates are the most numerous not only because they are the first to be investigated, but also because they are the most stable and they have a technological interest in several areas, such as magnetism, electricity, optics and the biomaterials research. The acidic monophosphate

Figure 1. Graphical abstract of (C7H9N2O)H2PO4.
anions like [HPO4]2− and [H2PO4]− exhibit a characteristic geometry to build infinite network via hydrogen bonds with various geometries: chains [1] [2] , ribbons [3] , two-dimensional networks [4] [5] and three-dimen- sional networks [6] . These entities can be associated to organic molecules to produce compounds having a particular interest in some application areas. As a contribution to the study of this monophosphate family, this compound was synthesized within a systematic search on new materials resulting from the association of organic and inorganic entities, which could be of particular interest in non-linear optics [7] . These compounds have also a great interest due to their biological and pharmacological activity as anti-tumor and inhibition of the activity of cholesterol. In addition, it has many applications in the field of agriculture [8] . We report in this work the chemical preparation and the structural investigation of a new organic phosphate (C7H9N2O)H2PO4. The characterization by differential thermal analysis (TG-DTA) and IR spectroscopy is also reported.
2. Experiment
2.1. Chemical Preparation
Crystals of ABHP are easily prepared by slow evaporation at room temperature of an aqueous solution of orthophosphoric acid (85 wt.% H3PO4) and the organic molecule: 2-Aminobenzamide (C7H8N2O); in the molar ratio 1:1. Schematically the synthesis reaction is:

when the most of the solution is evaporated, prismatic crystals appears deep down the vessels. The crystals are stable under normal conditions of temperature and humidity.
2.2. Investigation
The title compound has been studied by various physico-chemical methods: X-ray diffraction, Infrared spectroscopy and Thermal analysis.
2.2.1. X-Ray Structure Determination
The intensity data collection was performed using a CAD4 Enraf-Nonius diffractometer and monochromated Mo Kα radiation. The strategy used for the structure determination and its final results are gathered in Table 1. For the crystal of the title compound, 90 frames were recorded, each being of 2˚ in
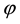 and 120 s duration. Each frame is doubled to eliminate the uncertain electronic impulses. The first 10 frames were used for indexing reflections using the DENZO package and refined to obtain final cell parameters [9] . Preliminary photographs indicated monoclinic symmetry and systematically absent reflections showed the space group to be P21/n. The structure was solved with a direct method, from the SHELXS-97 programs, which permitted the location of the
and 120 s duration. Each frame is doubled to eliminate the uncertain electronic impulses. The first 10 frames were used for indexing reflections using the DENZO package and refined to obtain final cell parameters [9] . Preliminary photographs indicated monoclinic symmetry and systematically absent reflections showed the space group to be P21/n. The structure was solved with a direct method, from the SHELXS-97 programs, which permitted the location of the

Table 1. Crystal data and structure refinement.
PO4 groups. The remaining non-hydrogen atoms were located by the successive difference Fourier maps using the SHELXL-97 programs [10] . In the final least-squares refinement of atomic parameters with isotropic thermal factors of the H atoms, R decreased to 4.8% (Rw = 12,79%) for ABHP. The final atomic coordinates are given in Table 2. Main geometrical features, bond distances and angles are reported in Table 3.
2.2.2. Thermal Analysis
Setaram TG-DTA92 thermoanalyzer was used to perform thermal treatment on samples of ABHP. The TG-DTA thermograms were obtained with 15.20 mg. Samples were placed in an open platinum crucible and heated in air with 3˚C/min heating rate; an empty crucible was used as reference.
Table 2. The final atomic coordinates and equivalent temperature factors for (C7H9N2O)H2PO4.
Table 3. Main interatomic distances (Å) and bond angles (˚) for (C7H9N2O)H2PO4.
2.2.3. Infrared Spectroscopy
IR spectrum of ABHP was recorded at room temperature with a Biored FTS 6000 FTIR spectrometer over the wave number range of 4000 - 400 cm−1 with a resolution of about 4 cm−1. Thin, transparent pellet was made by compacting an intimate mixture obtained by shaking 2 mg of the samples in 100 mg of KBr.
3. Results and Discussion
3.1. ABHP Structure Description
A view of the structure projected along the b direction (Figure 2) shows that The
 inorganic entities have a layered organisation parallel to (a, c) plane, with an interplane period of c/2 = 7.972 Å. The organic cations are trapped in the interlayer spacing, and neutralize the negative charge of the inorganic layers. The asymmetric unit of the crystal structure consists of one phosphate anion and one organic cation.
inorganic entities have a layered organisation parallel to (a, c) plane, with an interplane period of c/2 = 7.972 Å. The organic cations are trapped in the interlayer spacing, and neutralize the negative charge of the inorganic layers. The asymmetric unit of the crystal structure consists of one phosphate anion and one organic cation.
Within the inorganic layer (Figure 3), The
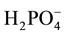 tetrahedron are associated in pairs, forming centrosymmetric finite clusters [H4P2O8]2−. The P-P distance between two
tetrahedron are associated in pairs, forming centrosymmetric finite clusters [H4P2O8]2−. The P-P distance between two
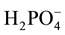 groups linked by four hydrogen bonds is 4.148 Å. The P-O distances range from 1.496(2) to 1.568(3) Å. These values are comparable to the reported data [11] -[14] . The longest P-O distances, 1.563(3) and 1.568(3) Å, are due to the presence of the acidic hydrogen atoms on the PO4 tetrahedron. The average values of P-O distances and O-P-O angles are 1.534(3) Å and 109.39(15)˚. They are in good agreement with that generally observed in such anions in other phosphates [15] .
groups linked by four hydrogen bonds is 4.148 Å. The P-O distances range from 1.496(2) to 1.568(3) Å. These values are comparable to the reported data [11] -[14] . The longest P-O distances, 1.563(3) and 1.568(3) Å, are due to the presence of the acidic hydrogen atoms on the PO4 tetrahedron. The average values of P-O distances and O-P-O angles are 1.534(3) Å and 109.39(15)˚. They are in good agreement with that generally observed in such anions in other phosphates [15] .

Figure 2. Projection along the b axis of the atomic arrangement in (C7H9N2O)H2PO4.

Figure 3. Projection of one anionic layer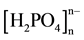 , viewed down the crystallographic c axis.
, viewed down the crystallographic c axis.
The calculated average values of distortion indices corresponding to the different angles and distances in the PO4 tetrahedral [DI(OPO) = 0.019; DI(PO) = 0.021 and DI(OO) = 0.007], exhibit a pronounced distortion of the PO distances and OPO angles if compared to OO distances; so the phosphate group can be considered as a rigid regular arrangement of oxygen atoms, with the P atom displaced from their centroid [16] .
The interaction of the orthophosphoric acid with the organic molecule (C7H8N2O) leads to the protonation of nitrogen grafted on the benzene ring and the formation of the (C7H9N2O)+ cation. With regards to the organic cations arrangement, the (C7H9N2O)+ groups are organized in opposition by creating, thus, a local inversion centre. Note that each two cations are associated with a hydrogen bonds N2-H2N2∙∙∙O5 to form a dimmer located in planes z = (n + 1)/2. Interatomic distances and angles in these groups spread within the respective ranges: 1.234(4) - 1.504(5) Å and 117.7(3) - 121.7(3)˚ (Table 3).
Hydrogen bonding play a significant role in the linking the organic molecules with the anionic sheets made by H2PO4 moieties. This interaction contributes to the cohesion of the structure. All the D (donor)-H∙∙∙A (acceptor) hydrogen bonds are listed in Table 4, with an upper limit of 2.149(4) Å for H∙∙∙A distance and a lower limit of 164(4)˚ for the D-H∙∙∙A bond angles. Thus, this atomic arrangement exhibits two types of intermolecular interaction: O-H∙∙∙O and N-H∙∙∙O, classified respectively as strong and weak hydrogen bonds. The structure studied in this work, contains a single hydrogen bond of the first type and the second type five. The only link O3-HO3∙∙∙O4 considered high [O3-HO3∙∙∙O4 = 2.554(3) Å], brings the two anionic species as a cluster (H4P2O8)2−; the second type of hydrogen bonds connecting the inorganic clusters to organic dimmers. A three-dimensional frame work is then created.
3.2. Thermal Behavior
The two curves corresponding to differential thermal analysis (DTA) and thermogravimetric analysis (TGA) in open air are given in Figure 4. The DTA curve shows an important endothermic melting peak at about 197˚C followed by a course of weak endothermic peaks in a wide temperature range [200˚C - 400˚C]. The TGA curve shows an important weight loss corresponding to the progressive pyrolysis of the organic molecule in this temperature range. The stability of the investigated anhydrous (C7H9N2O)H2PO4 below this melting temperature (197˚C) can be explained by the different strong bonds observed by the X-ray diffraction.
Table 4. Bond lengths (Å) and angles (˚) in the Hydrogen-bonding schemea.
aSymmetry operators: (i): −x + 3/2, Y − 1/2, −Z + 3/2; (ii): X − 1/2, −Y + 1/2, Z + 1/2; (iii): −X + 1, −Y + 1, −Z + 2; (iv): −X + 1, −Y + 1, −Z + 1.

Figure 4. TG-DTA thermo grams of (C7H9N2O)H2PO4.
3.3. IR Absorption Spectroscopy
The normal modes of vibration in an isolated PO4 tetrahedron with an ideal Td symmetry have been widely studied [17] . The n1(A1) and n3(F2) symmetric and asymmetric stretching modes are observed in 1200 - 900 cm−1 region, whereas the n2(E) and n4(F2) symmetric and asymmetric bending modes are distinguished in the 600 - 400 cm−1 domain [18] [19] . The group-theoretical analysis shows that the number of normal modes of PO4 tetrahedron is given by the representation: Gint = A1 + E + 2F2. The localisation of two protons on two of the oxygen atoms reduces the ideal symmetry from Td to C2v. The correlation of group to subgroup shows that these modes can be divided into 2A1 + B1 + B2 stretching and 2A1 + A2 + B1 + B2 bending vibration in the C2v symmetry of H2PO4 group. However, in the crystal, The H2PO4 tetrahedron has the lower local symmetry C1, and therefore anisotropic crystal fields may lift degeneracy and allow inactive modes to be active.
Infrared absorption spectrum (Figure 5) of the title compound shows vibration bands characteristic of (C7H9N2O)+ cations and
 anions. Its interpretation is made in terms of internal modes of two atomic groups, PO2 and P(OH)2, included in
anions. Its interpretation is made in terms of internal modes of two atomic groups, PO2 and P(OH)2, included in
 anion. The two stretching vibrations, asymmetric and symmetric of PO2 group, are observed respectively at 1150 and 1074 cm−1; while those related to P(OH)2 occur, as two intense bands, at 940 and 874 cm−1 [20] . Then we attribute the two very intense bands at 1094 cm−1 and 958 cm−1 and the two intense bands at, 880 cm−1 and 853 cm−1 to these four vibrations. The splitting of F2 stretching
anion. The two stretching vibrations, asymmetric and symmetric of PO2 group, are observed respectively at 1150 and 1074 cm−1; while those related to P(OH)2 occur, as two intense bands, at 940 and 874 cm−1 [20] . Then we attribute the two very intense bands at 1094 cm−1 and 958 cm−1 and the two intense bands at, 880 cm−1 and 853 cm−1 to these four vibrations. The splitting of F2 stretching

Figure 5. IR spectrum of (C7H9N2O)H2PO4.
mode of PO4 into two very intense components at 1094, and 958 cm−1 corroborate the symmetry lowering of H2PO4 in the solid state. Bending modes of H2PO4 group are observed at lower frequencies. The two medium bands at 748 cm−1 and 552 cm−1 correspond respectively to the rocking ρ(PO2) and to the bending δ(OHPOH) vibrations; whereas the strong band at 665 cm−1 is attributed to the wagging ω(PO2) vibration. The two bands, strong at 495 cm−1 and medium at 434 cm−1 correspond to the torsion τ(PO2) and to the bending δ(O-P-O) vibrations. The two strong bands observed at 1239 cm−1 and 853 cm−1 are assigned to δ(P-O-H) in plane bending and γ(P-O-H) out of plane bending modes [21] .
The presence of a strong band at 1689 cm−1 is assigned to the stretching vibration modes of C=O groups. Abroad band extending from 3300 cm−1 to 1966 cm−1 is observed in the IR spectrum. This broad band must be due the symmetric and asymmetric stretching modes of NH3, NH2, CH and OH. The stretching and bending modes of
 groups appears via the medium band at 3127 cm−1 and the strong band at 1554 cm−1 respectively. The presence of the strong band at 1619 cm−1 correspond to the stretching vibration modes of C=C groups.
groups appears via the medium band at 3127 cm−1 and the strong band at 1554 cm−1 respectively. The presence of the strong band at 1619 cm−1 correspond to the stretching vibration modes of C=C groups.
3.4. Supplementary Material
Crystallographic data for the structural analysis have been deposited at the Cambridge Crystallographic Data Centre, CCDC No. 1419172. Copies of this information may be obtained free of charge from The Director, CCDC, 12 Union Road, Cambridge, CB2 IEZ, UK (fax: +44-1226-336033; e-mail: deposit@ccdc.cam).
Acknowledgements
The authors gratefully acknowledge financial support from the Ministry of Higher Education, Scientific Research and Technology of Tunisia.
Cite this paper
SalouaBelghith,YahyaBahrouni,Latifa BenHamada, (2015) Synthesis, Crystal Structure and Characterization of a New Dihydrogenomonophosphate: (C7H9N2O)H2PO4. Open Journal of Inorganic Chemistry,05,122-130. doi: 10.4236/ojic.2015.54013
References
- 1. Baouab, L., Guerfel, T. and Soussi, M. (2000) Structure, Thermal Behavior, and IR Investigation of Bis(3-amino-1,2,4-triazolium)monohydrogenmonophosphate. Journal of Chemical Crystallography, 30, 805-809.
http://dx.doi:10.1023/A:1013280426251 - 2. Averbuch-Pouchot, M.T., Durif, A. and Guitel, J.C. (1988) Structures of -Alanine, DL-Alanine and Sarcosine Monophosphates. Acta Crystallographica C, 44, 1986-1872.
http://dx.doi.org/10.1107/S0108270188000502 - 3. Baouab, L. and Jouini, A. (1998) Crystal Structures and Thermal Behavior of Two New Organic Monophosphate. Journal of Solid State Chemistry, 141, 343-351.
http://dx.doi.org/10.1006/jssc.1998.7933 - 4. Averbuch-pouchot, M.T. Durif, A. and Guitel, J.C. (1988) Structures of Glycine Monophosphate and Glycine Cyclo-Triphosphate. Acta Crystallographica C, 44, 99-102.
http://dx.doi.org/10.1107/S0108270187008539 - 5. Blessing, R.H. (1986) Hydrogen Bonding and Thermal Vibrations in Crystalline Phosphate Salts of Histidine and Imidazole. Acta Crystallographica B, 42, 613-621.
http://dx.doi.org/10.1107/S0108768186097641 - 6. Kamoun, S., Jouini, A. and Daoud, A. (1990) Structure du Aza-3 Pentanediyle-l,5 Diammonium Monohydrogenomonophosphate Dihydrate. Acta Crystallographica Section C, 64, 1481-1483.
http://dx.doi:10.1107/S0108270189012552 - 7. Masse, R., Bagieu-Beucher, M., Pecault, J., Levy, J.P. and Zyss, J. (1993) Design of Organic-Inorganic Polar Crystals for Quadratic Non Linear Optics. Non-linear Optical, 5, 413-423.
- 8. Allouch, F., Zouari, F., Chabchoub, F. and Salem, M. (2008) 5-Amino-3-methyl-1-phenyl-1H-1,2,4-triazole. Acta Crystallographica E, 64, 684.
http://dx.doi.org/10.1107/S1600536808005801 - 9. Otwinowski, Z. and Minor, W. (1997) In Methods in Enzymology. Academic Press, New York.
- 10. Sheldrick, G.M. (1997) SHELXS97 and SHELXL97. Program for Crystal Structure Solution and Refinement. University of Göttingen, Göttingen.
- 11. Kamoun, S., Kamoun, M., Jouini, A. and Daoud, A. (1989) Structure of Ethylenediammonium Bis(Dihydrogenmonophosphate). Acta Crystallographica C, 45, 481-482.
http://dx.doi.org/10.1107/S0108270188012405 - 12. Bagieu-Beucher, M., Durif, A. and Guitel, J.C. (1989) Structure of Ethylenediammonium Dihydrogentetraoxophosphate(V) Pentahydrogenbis[Tetraoxophosphate(V)]. Acta Crystallographica C, 45, 421-423.
http://dx.doi.org/10.1107/S0108270188011977 - 13. Averbuch-Pouchot, M.T. and Durif, A. (1987) Structures of Ethylenediammonium Monohydrogentetraoxophosphate(V) and Ethylenediammonium Monohydrogentetraoxoarsenate (V). Acta Crystallographica C, 43, 1894-1896.
http://dx.doi.org/10.1107/S0108270187089716 - 14. Kamoun, S., Jouini, A. and Daoud, A. (1991) Structure du propanediammonium-1,3 monohydrogénomonophosphate monohydrate. Acta Crystallographica C, 47, 117-119.
http://dx.doi.org/10.1107/S0108270190003122 - 15. Kaabi, K., Rayes, A., Ben Nasr, C., Rzaigui, M. and Lefevre, F. (2003) Synthesis and Crystal Structure of a New Dihydrogenomonophosphate (4-C2H5C6H4NH3)H2PO4. Materials Research Bulletin, 38, 741-747.
http://dx.doi.org/10.1016/S0025-5408(03)00072-2 - 16. Baur, W.H. (1974) The Geometry of Polyhedral Distortions. Predictive Relationships for the Phosphate Group. Acta Crystallographica B, 30, 1195-1215.
http://dx.doi.org/10.1107/S0567740874004560 - 17. Steger, E. and Herzog, K. (1964) Zum Schwingungsspektrum der Phosphorsäure. I. Infrarotund RAMAN-Spektren von Phosphatlösungen. Zeitschrift für Anorganische und Allgemeine Chemie, 331, 169-182.
http://dx.doi.org/10.1002/zaac.19643310308 - 18. Nakamoto, K. (1986) Infrared and Raman Spectre of Inorganic and Coordination Compounds. Wiley-Interscience, Hoboken.
- 19. Steger, E. and Schmid, W. (1964) Infrarotspektren von Sulfaten und Phosphaten. Berichte der Bunsengesellschaft für physikalische Chemie, 68, 102-109.
http://dx.doi.org/10.1002/bbpc.19640680118 - 20. Haile, S.M., Calkins, P.M. and Broysen, D. (1998) Structure and Vibrational Spectrum of β-Cs3(HSO4)2[H2-x(P1-x, Sx)O4] (x~0.5), a New Superprotonic Conductor, and a Comparison with α-Cs3(HSO4)2(H2PO4). Journal of Solid State Chemistry, 139, 373-387.
http://dx.doi.org/10.1006/jssc.1998.7861 - 21. Chapman, A.C. and Thirlwell, L.E. (1964) Spectra of Phosphorus Compounds—I the Infra-Red Spectra of Orthophosphates. Spectrochimica Acta, 20, 937-947.
http://dx.doi.org/10.1016/0371-1951(64)80094-1
NOTES

*Corresponding author.