Journal of Encapsulation and Adsorption Sciences
Vol.2 No.3(2012), Article ID:23093,12 pages DOI:10.4236/jeas.2012.23008
Complexation of Both Enantiomers of 2-Phenylpropionic Acid with Cyclodextrin: Determination of Binding Constant, Stoichiometry, Bioavailability and Co-Conformation
Pharmaceutical Sciences Research Division, King’s College, London, UK
Email: *ali.aboel_dahab@kcl.ac.uk
Received June 14, 2012; revised July 20, 2012; accepted August 16, 2012
Keywords: β-Cyclodextrin; Binding Constant; Bioavailability; 2-Phenylpropionic Acid; UV and Circular Dichroism
ABSTRACT
Although enantiomers of 2-phenylpropionic acids (2-PPAs), or profens are important group of nonsteroidal anti-inflammatory drugs (NSAIDs) and have been in clinical use for many years, there is no literature covering its binding interaction in particular with cyclodextrins. NSAIDs are marketed as racemates, chiral discrimination and knowledge of enantiomeric bioavailability is essential. Circular dichroism (CD) spectroscopy is the technique of choice for elucidating chirality and monitoring and characterizing molecular recognition phenomena in solution. Methods employing the fundamentals of the simultaneous measurements of absorbance and CD and a novel efficient titration method have been developed to study the binding of β-Cyclodextrin (β-CyD) and the two enantiomers of 2-PPA as a function of pH. The effect on physicochemical properties and bioavailability was investigated. The binding constant, stoichiometry and pKa for both the free and the bound drugs were determined using a Levenburg-Marquadt non-linear equation. The exact nature of the enantiomer discriminating interactions by cyclodextrins (CyDs) is not well understood. In this work, the interactions and co-conformations of both enantiomers of 2-PPA with β-CyD were explained and estimated using spectroscopic variations upon complexation. The results indicated a change in the physicochemical properties of 2-PPAs upon complexation and highlighted the enantioselective binding of β-CyD as a function of pH. The charge on the guest molecule and its stereochemistry are of great importance in regulating the stability of the guest/β-CyD complexes; hence the bioavailability of drugs. This work elucidates 2-PPAs/β-CyD binding interactions and highlights the effect of β-CyD on drugs with an effective novel method for binding titration and the potential of the simultaneous measurements of absorbance and CD in future chiral drug interactions studies.
1. Introduction
The 2-arylpropionic acids (2-APAs), or profens, are an important class of NSAIDs, that have been in clinical use for about 40 years. Widely used members of this drug class include naproxen, ibuprofen, ketoprofen, and flurbiprofen. The most important therapeutic activity of 2- APAs consists in their anti-inflammatory and pain relieving action. Although all NSAIDs cause a full range of gastrointestinal adverse effects, they vary in frequency and severity [1-3]. Deaths from gastrointestinal toxic effects of NSAIDs are assumed to be the 15th most common cause of death in the United States [4]. Among the different methods proposed to overcome the problems associated with NSAIDs, is the molecular encapsulation of these drugs by cyclodextrins (CyDs), which is probably the most widely used method [5,6]. Therefore, full understanding of this type of interaction is of great importance. These carbohydrates are mainly used to increase the aqueous solubility and stability of drugs, but they can also, for example, be used to convert liquid drugs into microcrystalline powders, to prevent drug-drug or drugadditive interactions; reducing gastrointestinal or ocular irritation, and reducing or eliminating unpleasant taste or smell. Labile drug stabilization by cyclodextrins [7] and their ability to ameliorate drug irritation, and thus improve drug contact time at the absorption site in nasal, ocular, rectal and transdermal delivery are some other important factors that contribute to the cyclodextrin improved bioavailability. The cyclodextrins enhance the bioavailability of insoluble drugs by increasing their solubility, dissolution and permeability, which is increased by direct action on mucosal membranes enhancing drug absorption and bioavailability [8]. This is achieved by making the drug available at the surface of the biological barrier, e.g., skin, mucosa, or the eye cornea, from where it partitions into the membrane without disrupting the lipid layers of the barrier [9]. In such cases, it is important to use just enough cyclodextrin to solubilise the drug in the aqueous vehicle since excess cyclodextrin may decrease the drug bioavailability [10] . In the case of water-soluble drugs e.g. antiviral and antisense drugs, which are highly polar and soluble in aqueous medium can be poorly bioavailable due to inefficient transport across the hydrophobic lipid bilayer constituting cellular membranes. In this case, the encapsulation of soluble drugs by cyclodextrins can be useful since the host molecules may act as concentration-regulating agents, decreasing the adverse side effects at the organ or cellular level and improving the optimal dose of the drug and its frequency of administration [11-13] . Moreover, it has been shown that the rates for formation and dissociation of drug/cyclodextrin complexes are very close to diffusion controlled limits with complexes being continually formed and broken down [14]. Consequently, the presence of water-soluble drug/cyclodextrin complexes right at the hydrated epithelial surface will frequently increase the availability of dissolved drug molecules, especially of lipophilic drugs with poor aqueous solubility [15]. However, it was reported that cyclodextrins, because of their ability to remove cholesterol, may increase membrane fluidity and induce membrane invagination through a loss of bending resistance and results in cell lysis. Also, removal of phospholipids, especially phosphatidyl choline and sphingomyelin from the outer half of the membrane bilayer by cyclodextrin causes bilayer imbalance.
Chirality often plays a fundamental role in determining the pharmacodynamics and pharmacokinetics of a drug. Stereoselectivity occurs in pharmacodynamic processes and the differences between enantiomers can be either qualitative or quantitative [16]. All 2-APAs contain one asymmetric carbon atom in their molecular structure and hence, they can appear in the two enantiomeric forms, as the S-(+) and the R-(−) species which differ in their potency as therapeutic drugs. These are chiral compounds marketed as racemic mixtures. In recent years, it has become clear that the biological effects of many drugs are produced by specific stereoisomers and other stereoisomers may in fact be harmful [17,18] . In fact, quite a number of properties pivotal to drug development and related pharmacokinetics, side effects, toxicology, pharmacyodynamics, and galenics (that is, formulation of pharmaceutical preparations) have been firmly shown to exhibit a marked enantioselectivity [19]. CyDs are well known chiral selectors; they offer the potential discrimination of enantiomeric substances. It is thought that chiral discrimination is brought about by differential interaction of each enantiomer with the seconddary hydroxyl groups (at the 2- and 3-positions of the glucose monomers) which line the wider rim of the cyclodextrin cavity. While both hydrophobic and electrostatic forces are thought to be responsible for complex formation on the basis of theoretical studies, the exact nature of the enantiomer discriminating interactions is not well understood [20]. Hydrogen bonding has been proposed from molecular modelling studies of the propranolol-β-cyclodextrin complex [21] and electrostatic forces from an examination of solid-state complex structures [22]. Thus, it is difficult to say precisely which compounds will be resolved and such predictions as are available are based on empirical observations. The extent of complexation (binding) is an important factor, especially in therapeutics, where the pharmacological effect of a drug is directly related to its nature (meaning in free or complexed form) [23] . Complexation of indomethacin with β-cyclodextrin, for example, does not eliminate the side effect of the drug because 40% of it is in free form [24]. Some non-linear least squares methods in the determination of association constants have been mentioned in the literature [25]. Nevertheless some problems may arise because of the choice of data, initial estimates, convergence or multiple local minima, all typical of nonlinear regression analysis [26]. The binding of a ligand (L) with a drug (D) to form a complex (DL) in which the total ligand is varied at fixed drug concentration constitutes a characteristic example of a 1:1 weak complex model [27].
Interaction of an electron donor (ED) with an electron acceptor (EA), often leads, to the formation of what is known as a charge-transfer complex, a phenomenon described by the equilibrium ED + EA 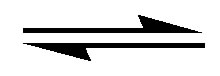 EDEA [28]. The UV-visible spectrum of a charge-transfer complex generally reveals a bathochromic shift relative to the spectra of the starting materials, frequently permitting isolated analysis of the complex itself. The equilibrium B + C
EDEA [28]. The UV-visible spectrum of a charge-transfer complex generally reveals a bathochromic shift relative to the spectra of the starting materials, frequently permitting isolated analysis of the complex itself. The equilibrium B + C  BC may also serve as a useful model for investigating the interaction between an anion and an enzyme, drugs or dyes with polynucleotides, or the entrapment of an organic entity within the hydrophobic cavity of a macrocyclic host. The progress of such reactions can be monitored by means of the experimentally determined value of some spectroscopic variable, e.g., fluorescence quantum yield [26,29], absorption coefficient [30,31], induced circular dichroism (ICD) [30] and chemical shift [26,32] . Moreover, questions connected to the influence of pH of the solutions on the thermodynamic parameters of the (drug/CyD) complexation process have not been investigated in details yet. Only a few articles are devoted to this problem, where indirect methods have been used, such as potentiometry [33] (where Ibuprofen, Ketoprofen and Flurbiprofen were used as guest molecules) and phase-solubility studies with Naproxen [34]. The pH of the media influences in particular the ionisation state of the guest molecule and hence the extent of complexation to CyD molecules. Such molecular encapsulation will affect many of the physicochemical properties of the drugs, such as their aqueous solubility and rate of dissolution. In general, no covalent bonds are formed or broken during the drug-CyD complex formation and under physiological conditions the complexes are readily dissociated and the free drug molecules are in a rapid equilibrium with the drug molecules bound within the CyD cavity. In this work, the interaction of β-CyD with both enantiomers of 2-PPA in the ionised and non-ionised form is investigated using simultaneous measurement of UV and Circular Dichroism. The extent of binding, stoichiometry and the change of physicochemical properties are estimated with accuracy. The mathematical analysis of the spectroscopic variations in binding titrations and the equations used to estimate the binding constant are explained. Also, the coconformation of β- CyD and both enantiomers is explained according to spectroscopic change upon complexation.
BC may also serve as a useful model for investigating the interaction between an anion and an enzyme, drugs or dyes with polynucleotides, or the entrapment of an organic entity within the hydrophobic cavity of a macrocyclic host. The progress of such reactions can be monitored by means of the experimentally determined value of some spectroscopic variable, e.g., fluorescence quantum yield [26,29], absorption coefficient [30,31], induced circular dichroism (ICD) [30] and chemical shift [26,32] . Moreover, questions connected to the influence of pH of the solutions on the thermodynamic parameters of the (drug/CyD) complexation process have not been investigated in details yet. Only a few articles are devoted to this problem, where indirect methods have been used, such as potentiometry [33] (where Ibuprofen, Ketoprofen and Flurbiprofen were used as guest molecules) and phase-solubility studies with Naproxen [34]. The pH of the media influences in particular the ionisation state of the guest molecule and hence the extent of complexation to CyD molecules. Such molecular encapsulation will affect many of the physicochemical properties of the drugs, such as their aqueous solubility and rate of dissolution. In general, no covalent bonds are formed or broken during the drug-CyD complex formation and under physiological conditions the complexes are readily dissociated and the free drug molecules are in a rapid equilibrium with the drug molecules bound within the CyD cavity. In this work, the interaction of β-CyD with both enantiomers of 2-PPA in the ionised and non-ionised form is investigated using simultaneous measurement of UV and Circular Dichroism. The extent of binding, stoichiometry and the change of physicochemical properties are estimated with accuracy. The mathematical analysis of the spectroscopic variations in binding titrations and the equations used to estimate the binding constant are explained. Also, the coconformation of β- CyD and both enantiomers is explained according to spectroscopic change upon complexation.
2. Experimental
2.1. Materials
β-Cyclodextrin, R and S enantiomers of 2-phenyl propionic acid were purchased from Sigma-Aldrich, UK. NaOH and HCl, were purchased from BDH, UK. Water was distilled in-house at KCL biophysics laboratory. Measurements of pH were made using a ThermoElectron Russell pH combination electrode (KCMAW11) connected to a Mettler-Toledo meter (Corning pH 105). Weighing was carried out using a Mettler Toledo MT5 microbalance.
• 2.2. Methods
Spectroscopic measurements and data analysis
CD and UV spectra were measured simultaneously using a Jasco J720 spectrometer (Jasco Inc., Tokyo, Japan) as described by Aboel Dahab et al. [35] where absorbance spectra were obtained from the photomultiplier HV measurements. Data was analysed using the relevant equation with both visual and mathematical fitting in the MathSoft MathCad computer program and the Levenburg-Marquadt non-linear equation fitting routine in the Microcal Origin 6 computer program.
Titrations
A study of compounds binding to cyclodextrin as a function of pH potentially requires the consideration of four equilibria:
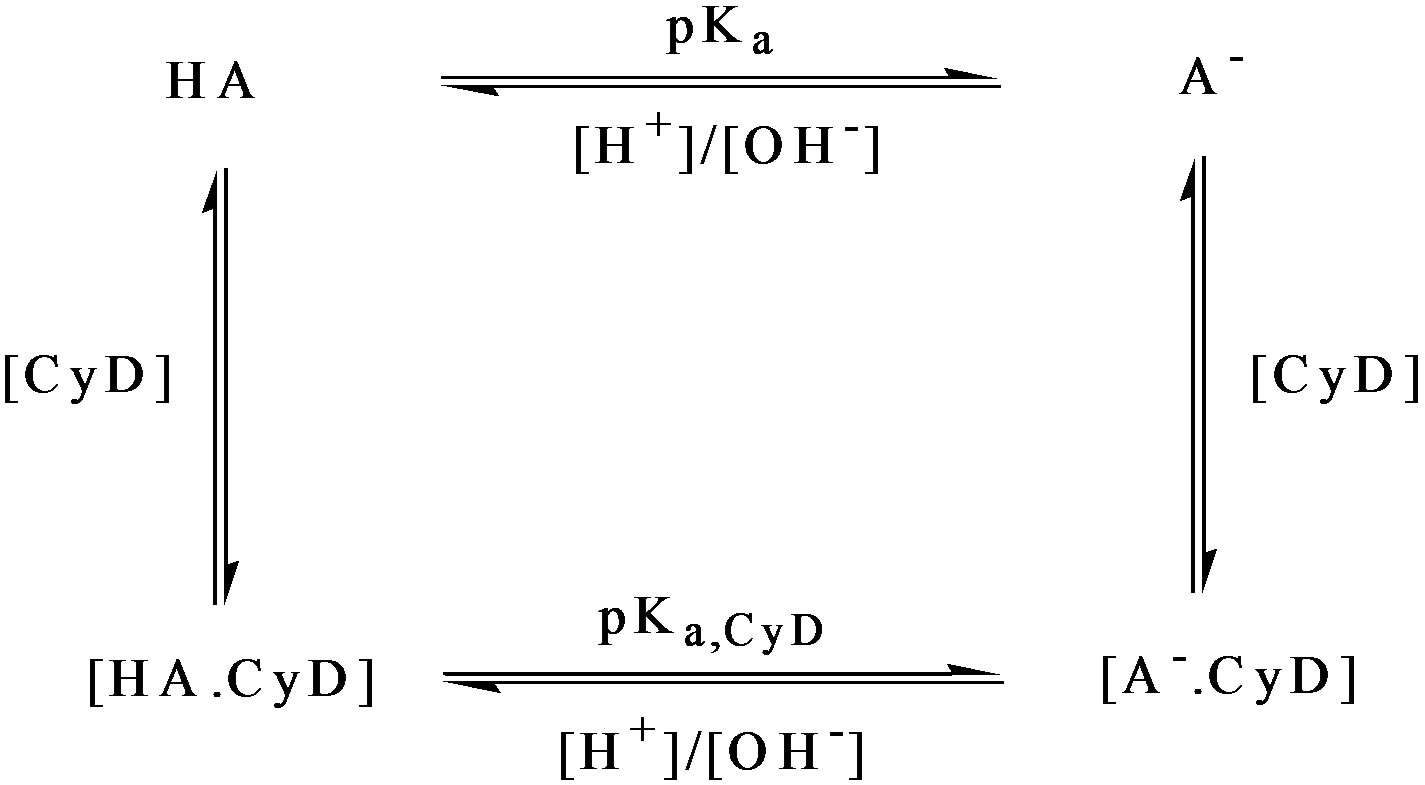
Firstly, cyclodextrin-free pH titrations need to be undertaken for the determination of the free-drug pKa and to establish the pH extremes for the cyclodextrin titration. Cyclodextrin titrations were in practice performed at pH ~ 2 and pH 7. Titrations were not performed below pH 2 in order to avoid cyclodextrin hydrolysis at lower pHs. The pathlengths used in this work were 0.05 cm and 1.0 cm with a wavelength range from 300 to 190 nm. An important feature of the pH titrations described here is the constant analyte concentration. A drug dissolved in a buffer is naturally resistant to pH change. Dissolved in distilled water alone, the solution is likely to be approximately at the value of the solute pKa. Suitable volumes of drug solutions of appropriate concentrations were placed in a small flask and titrated with the addition of 5 - 10 μl aliquots of NaOH/HCl leaving the drug concentration effectively constant. For pH values < pH 3, 5 - 10 μl aliquots of 3M HCl needed to be added; between pH 3 and pH 10, 5 - 10 μl aliquots of 0.1M NaOH/HCl sufficed; for pH > 10, 3M NaOH was required. At each pH interval, solution samples were transferred to the appropriate cell for spectral measurement using a Pasteur pipette and returned to the titration flask for further pH changes.
An important feature to simplify the understanding of drug binding and the mechanics of data analysis is to keep the concentration of the monitored species constant. The most efficacious scheme is presented in Figure 1. In this scheme two initial stock solutions are prepared, solution A containing the guest compound at an appropriate concentration but no host and solution B containing the guest compound at the same concentration as solution A (in order to keep the concentration of 2-PPA fixed at all stages of the titration) but with a host concentration at a reasonable molar excess. The titration can start from zero host concentration (solution A) and proceed via the judicial withdrawal and additions as illustrated in Figure 1.
Alternatively, the titration can start from the fully bound state solution B and progressively reducing the host concentration.
The choice of concentration in equilibrium studies is critical depending upon: 1) binding constants; 2) extinc tion coefficients/pathlength and 3) solubility.
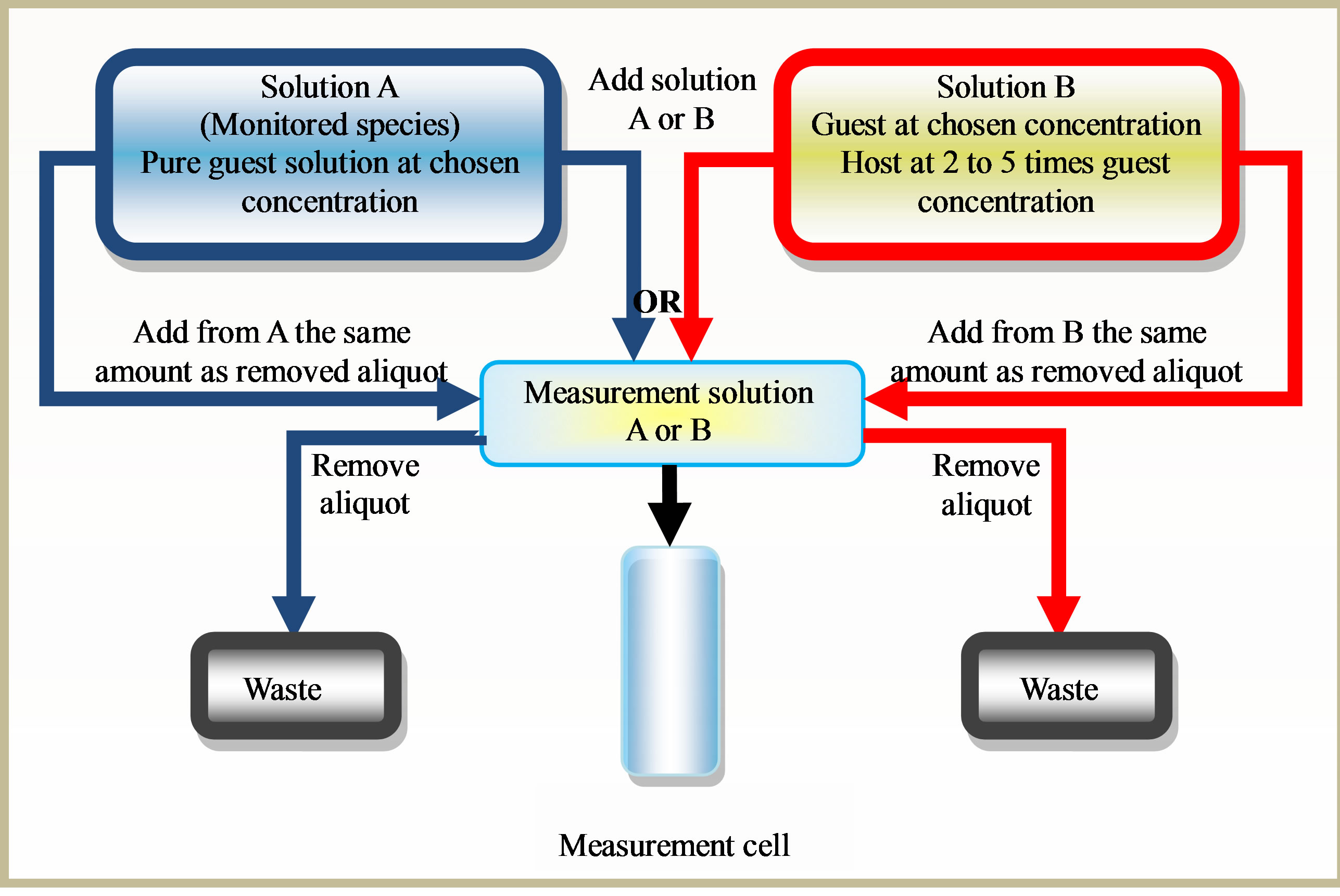
Figure 1. Titration method, schematic diagram showing the most effective ways to perform binding titration.
The binding constants for the interaction being measured here are K ~ 103 M–1, this implies a guest concentration of the order 10–3 M. The extinction coefficient of the 1Lb band is e ~1000 which gives A = 1 for a 10–3 M solution in a 1 cm cell. This appears to be reasonable conditions spectroscopically. To ensure a good molar excess the host concentration needs to be in the order of 5 × 10–3 M.
The solubility of β-CyD is 15.8 × 10–3 M (~18 mg/ml). In practice, the titrations were undertaken with solution B containing 15 × 10–3 M (17.03 mg/ml) β-CyD. Nevertheless, it is crucial to ensure that solution B is clear and not turbid due to the presence of undissolved β-CyD. This may involve extensive stirring and ultrasonication. Likewise, the aryl carboxylic acids as a class have limited solubility and it is essential to ensure that clear solutions are used. It cannot be over emphasised how important it is to have optically clear solutions and extremely clean cells. Therefore, the cells were soaked in nitric acid over night, and then washed thoroughly with distilled water and ethanol, and vacuum dried.
2.3. Mathematical Analysis
The mathematical analysis of the CD guest (ligand) binding data is based upon the Law of Mass Action, Beer’s Law, and the non-linear fitting of the binding equation expressed as the observed CD as a function of the guest and host concentrations. In order to monitor the CD of the guest, the guest must have an accessible ordinary electronic absorption. It is also important to ensure that the total absorbance of the measured solutions is in agreement with Beer’s Law. It should remain as close to 0.8 as possible. Fixing the concentration of the monitored chromophore makes this readily achieved. The free guest may or may not be chiral. That is the free guest may or may not have an associated natural CD in free solution. At the same time the host may or may not have a CD at the chosen wavelength. In this case, we are dealing with a chiral guest (2-PPA)/non-chiral host with a single binding site. Therefore, in the completely general case, the binding of a single Guest (ligand) to a single Host site is adequately described by the standard formalism:
Host (H) + Guest (G) [Host: Guest complex]
[Host: Guest complex]
Setting the initial (total) Host and Guest concentrations as HT and GT respectively, at equilibrium the complex concentration (HG) and free Host and Guest concentrations (Hf and Gf respectively) are related by:


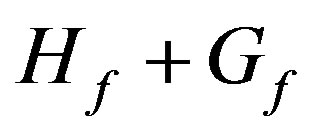 ,
,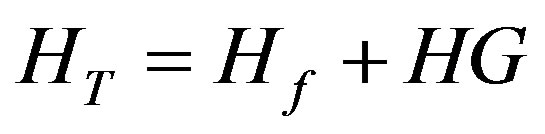
and 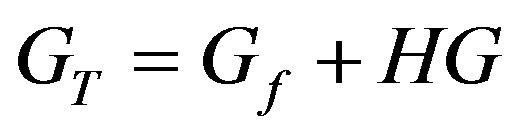
The association constant (Kass) (often referred to as the binding or affinity constant) is expressed as:
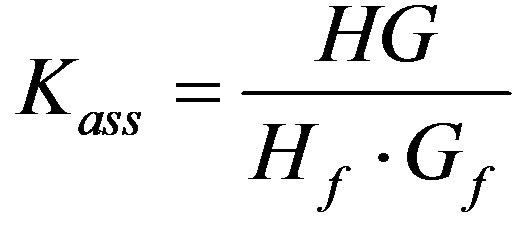 Giving:
Giving: 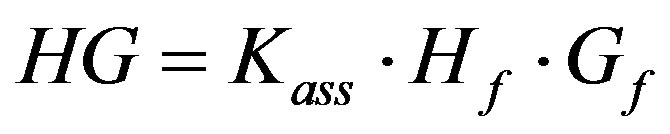
For the complete analysis and the deduced equation that was used in the origin program to obtain the binding constant see the published work by Aboel Dahab and ElHag, 2012 [36].
3. Results and Discussion
• 3.1. (R)-(−)-2-Phenylpropionic Acid
The pH titration of R-2PPA in the absence of cyclodextrin
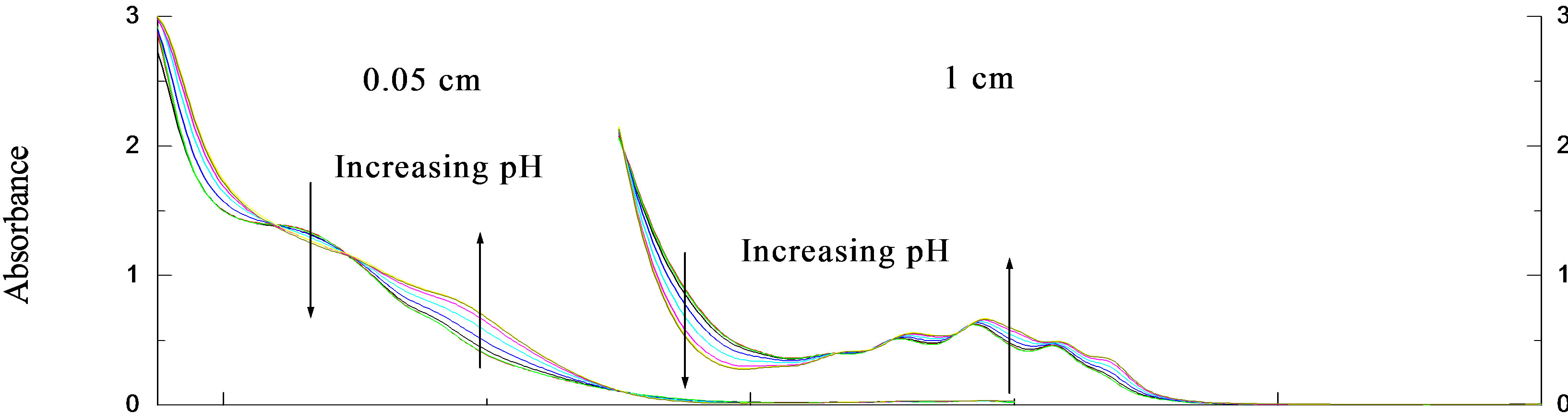
The UV and CD spectra for the pH titrations of R- 2PPA in the absence of β-CyD are presented in Figures 2(a) and (b) respectively. Isosbestic points are observed as a result of a two state system (ionised ↔ unionised). The absorption data show evidence of the 1Lb band around 258nm and the 1La/CT transitions between 230 and 200 nm. Below 205nm, the 1Ba,b transitions dominate. The CD 1Lb band is observed between 270 and 240 nm, with vibronic components at both acidic and basic pHs with negative and more intense positive CD for the protonated and ionised form respectively. The vibronic analysis of the 1Lb band is presented in Table 1. In the UV spectrum, the 0-0 vibrational peak is positioned at 267.7 and 267 nm for the ionised and the protonated form respectively with progressions of ~914 cm–1 vibrational quanta in the excited state. In the CD spectrum the 0-0 vibrational peak is positioned at the same wavelengths, however, the progression is only based upon the 0-0 origin and a ~965 cm–1 vibration. The spectra between 230 and 200 nm again show evidence of two transitions: the 1La and the assigned charge transfer (CT). At pH ~2.0, the UV absorption spectrum shows for both enantiomers a CT with ε215=3800 and a 1La transition with ε207=7400.
The corresponding CD spectrum shows a prominent negative CD peaking at 220 nm with De220 = –0.58 and a small residual positive CD associated with the 1La transition between 200 and 208 nm. At pH 7.0, the UV absorption spectrum shows a red-shifted CT with e218 = 5100 and a 1La transition with e207 = 7850 for both enantiomers. The corresponding CD spectra are very weak and relatively complex. The CT CD has apparently collapsed to give an extremely weak negative signal having an obvious zero CD at ~220 nm near the CT absorption wavelength maximum. The positive 1La CD is apparently slightly stronger but remains positive. Plots of DAl267 and Al267 against pH (Figure 2(c)) give mean pKa = 4.35 ± 0.018.
 (a)
(a)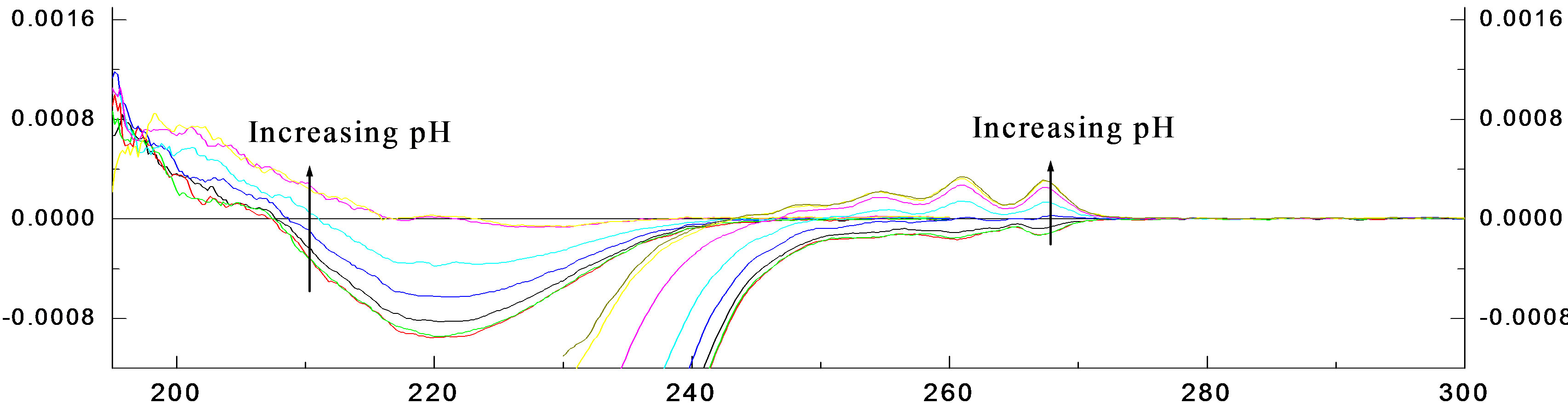 (b)
(b)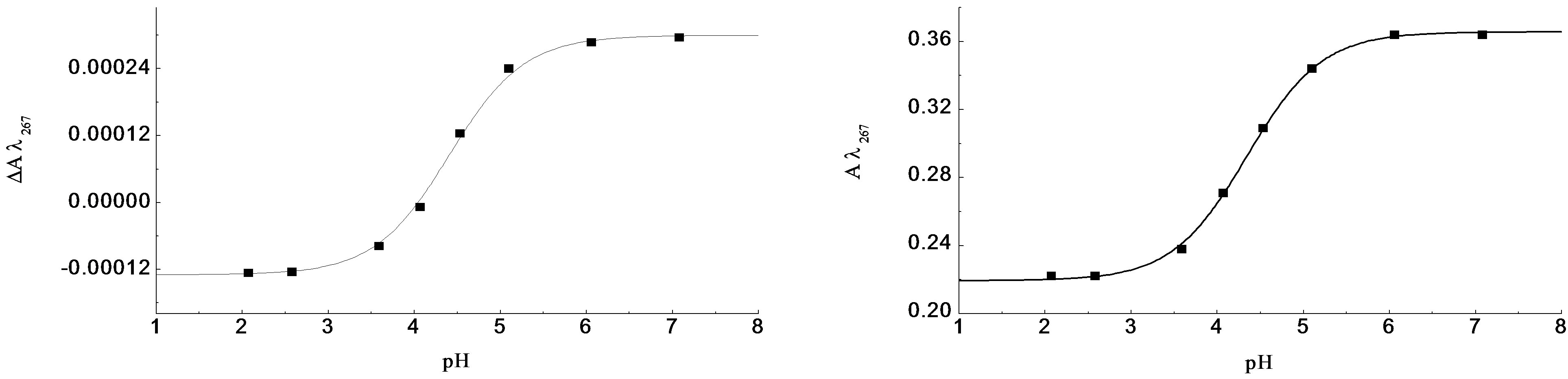 (c)
(c)
Figure 2. The pH titration of R-2PPA, pH titration of R-2PPA in H2O, with 1.0 cm cell (300 - 230 nm), 0.05 cm cell (260 - 190 nm).
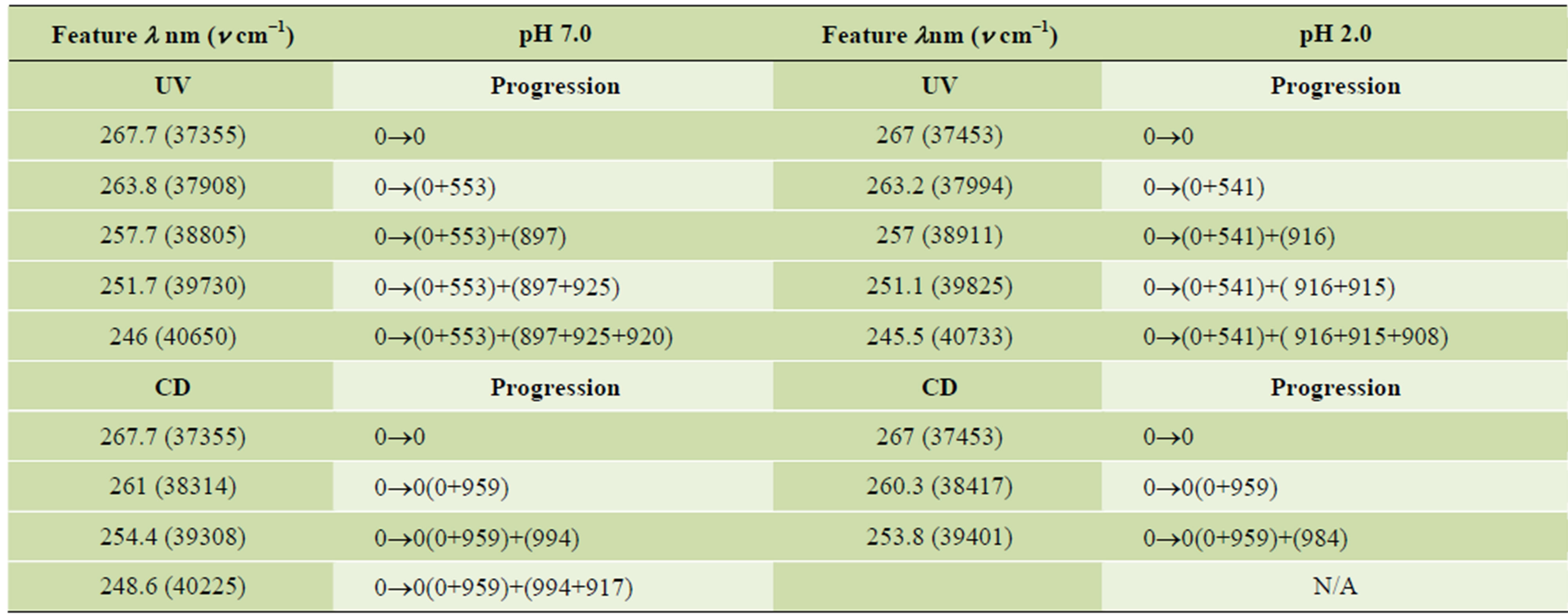
Table 1. Vibronic components of the 1Lb band of the ionised and non-ionised form of R-2PPA.
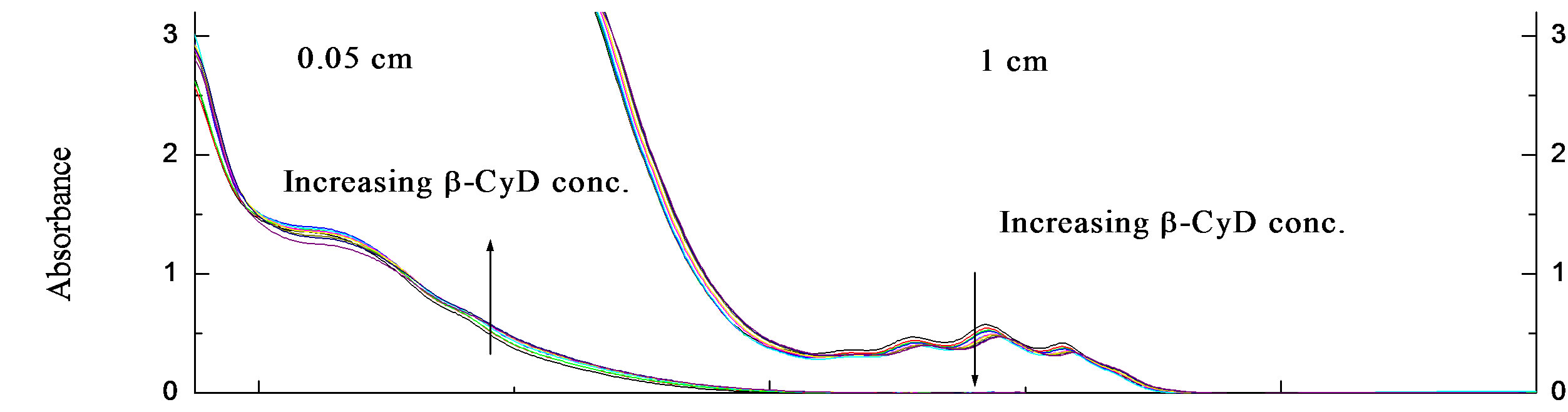
• R-2PPA/β-cyclodextrin binding at pH 2.0 The concentration of R-2PPA was fixed at (~3.3 × 10−3 M) and a manual titration was performed as explained before by effectively increasing the concentration of β- CyD gradually and recording the resulting spectra. Measurements were made using two different pathlengths, 0.05 and 1.0 cm. The absorbance and CD spectra of the binding of R-2PPA to β-CyD at pH 2.0 are shown in Figures 3(a) and (b) respectively. There are only small changes in the spectra and these are mostly associated with intensity. The A268 of the 1Lb band shows a slight decrease in intensity with increasing β-CyD concentration.
Also, the 0-0 vibrational peak at ~ 266.6 nm is less defined in the presence of β-CyD. Plots of DA267 and DA260, against β-CyD concentration (M) are shown in Figure 3(c), and they gave a mean K = 14.73 × 102 M–1 ± 48 with R2 = 0.99678 and 0.99787 for DA267 and DA260 respectively.
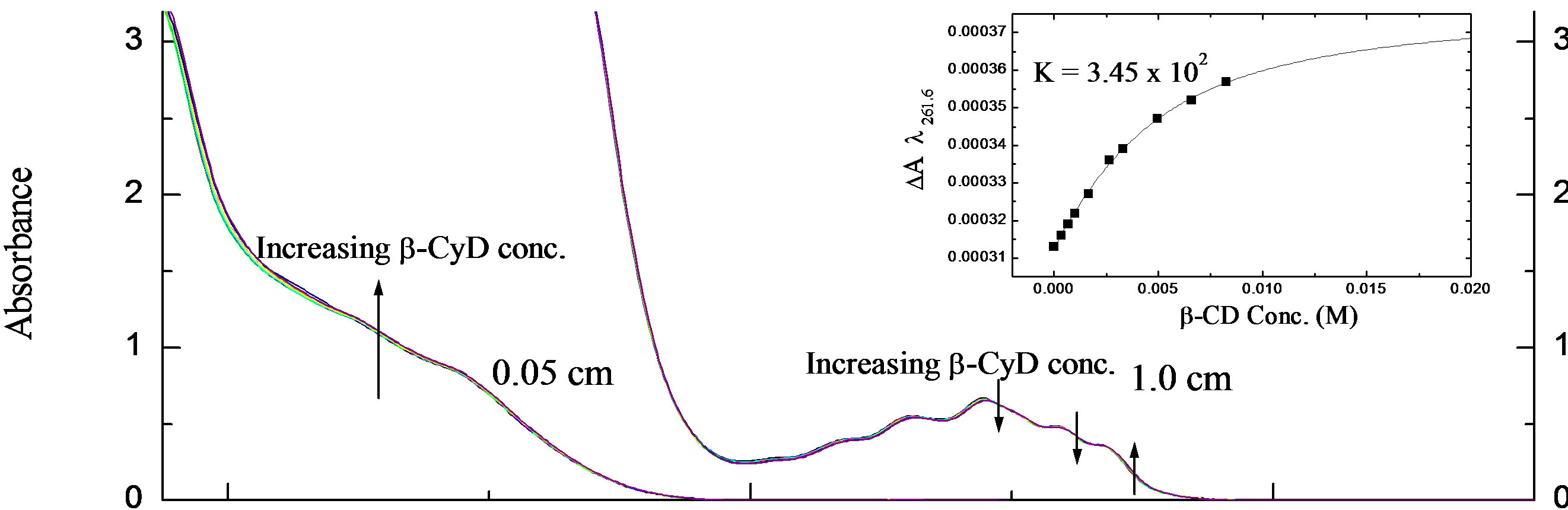
• R-2PPA/β-cyclodextrin binding at pH 7.0 R-2PPA was titrated with β-CyD at pH 7.0. The resulting UV and CD spectra are shown in Figures 4(a) and (b) respectively. Clearly, the changes in absorbance and CD are even smaller at pH 7.0 than at pH 2.0. The vibrational structure is less affected at pH 7.0 than at pH 2.0, and the 0-0 absorbance peak is positioned at ~267.7 nm as in the absence of β-CyD. At pH 7.0, the CD spectrum of R-2-PPA is relatively unaffected by the presence of β-CyD. The small change in CD around 261.6 nm gives a weak binding constant K = 3.45 × 102 M–1 ± 6.4 with R2 = 0.99889 as shown in the binding curve (Figure 4(a)). Although the changes in UV and CD spectra of R-2PPA as a result of interaction with β-CyD are small, the CD data indicate that the protonated form of R-2PPA binds more strongly (K = 14.73 × 102 M–1) to β-CyD than the ionised form.
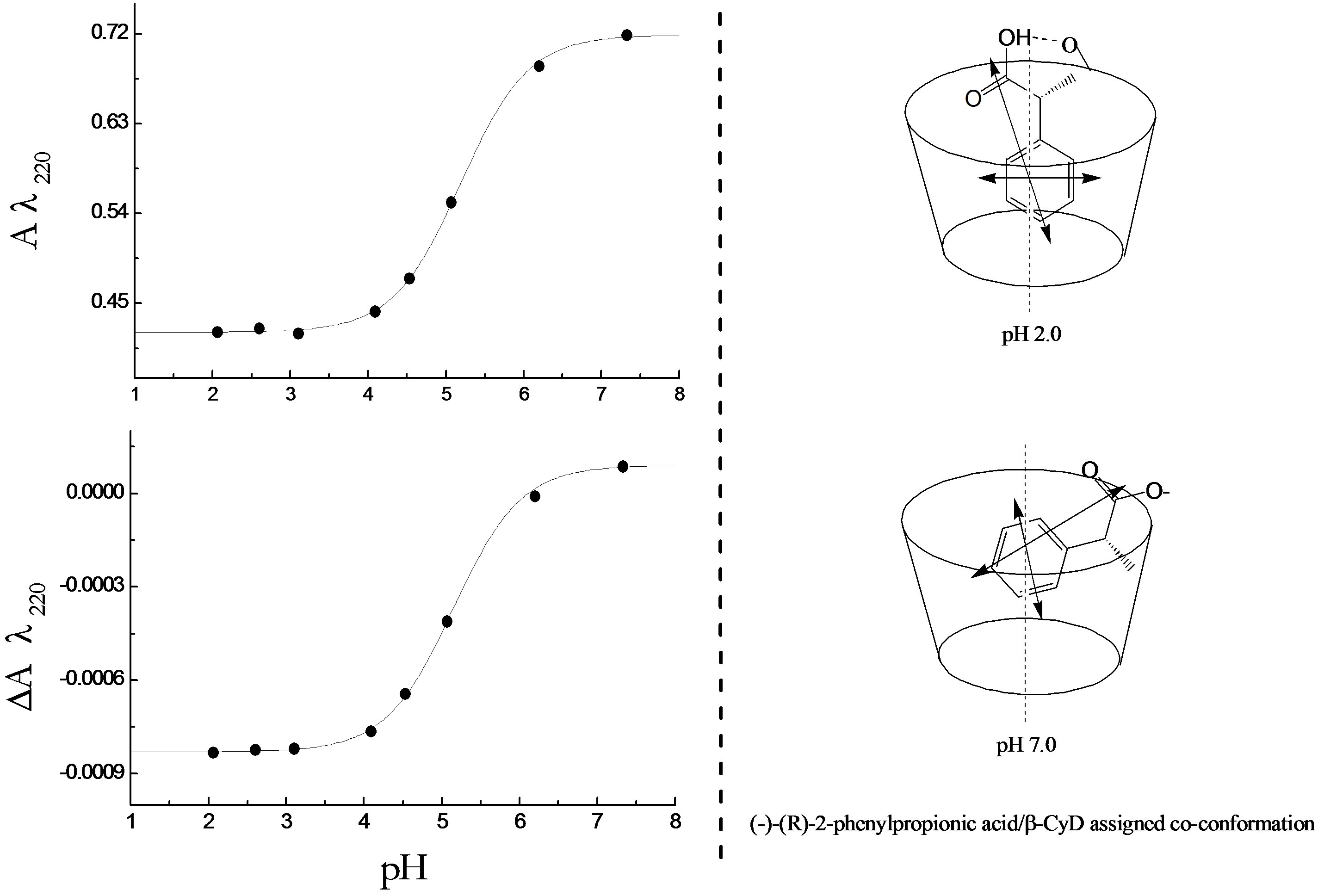
The result of R-2-PPA titration with β-CyD at pH 2.0 (Figure 3) shows a red shift of ~1 nm at 258 nm accompanied by a decrease in the intensity with increasing β-CyD. This observation suggests the occurrence of hydrogen bonding. R-2PPA is a chiral molecule and exhibits a negative CD at pH 2.0. The change in the CD signal upon complexation is an indication of the co-conformation of the guest-β-CyD complex. The small increase in intensity of the negative CD between ~270 - 240 nm indicates a negative co-conformation presumably resulting from a frontal inclusion with the phenyl ring inside the cavity restricted by the hydrogen bonding to the β- CyD rim. The decrease in CD intensity between 240 and 200 nm suggests a positive ICD upon complexation which could result from alignment of the transition moment along the β-CyD axis and immersing inside the cavity (according to Harata’s rule) [37], as shown in Figure 5(b). At pH 7.0, the hydrogen bonding is lost allowing more conformational freedom resulting in a positive increase in the CD intensity at all wavelengths (Figure 4) suggesting a tilted lateral co-conformation, less immersion in the cavity (Figure 5(b)) and a lower affinity (binding constant).
The pH titration of R-2-PPA in the presence of cyclodextrin
The UV and CD spectra (not shown) of the pH titration of R-2PPA/β-CyD complex at a molar ratio 1:3 were measured with two different pathlengths; 0.05 cm (260 - 190 nm) and 1.0 cm (300 - 240 nm). Examination of the spectra does not show any significant change in the intensity of either absorbance or CD bands compared to the titration in the absence of β-CyD. However, the pH titration curves (Figure 5(a)) were produced by plotting the values at A220 nm and DA220 against pH. The mean pKa value determined for the R-2PPA/β-CyD complex is
 (a)
(a)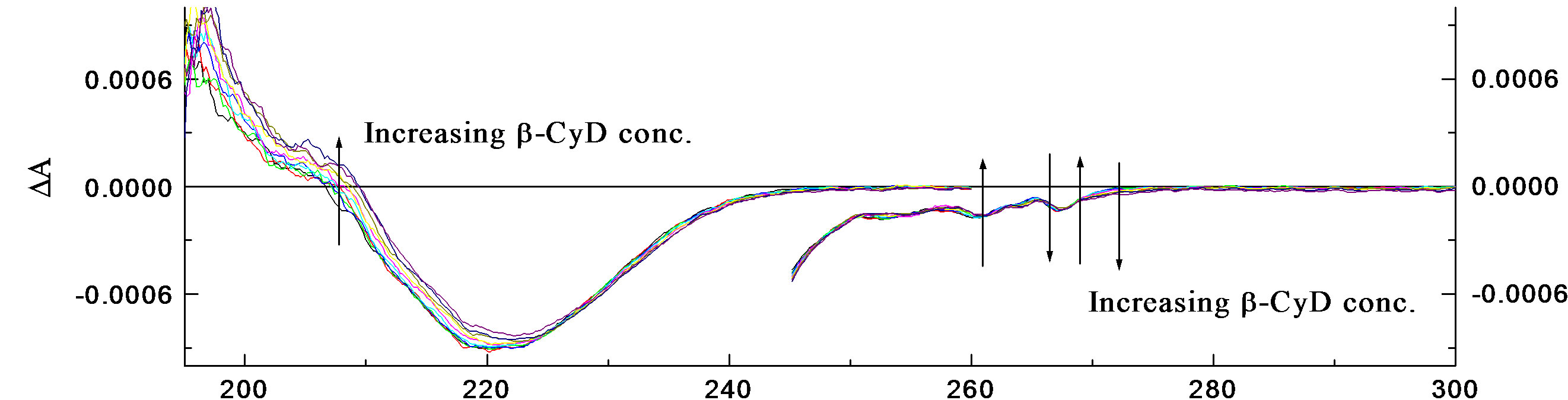 (b)
(b)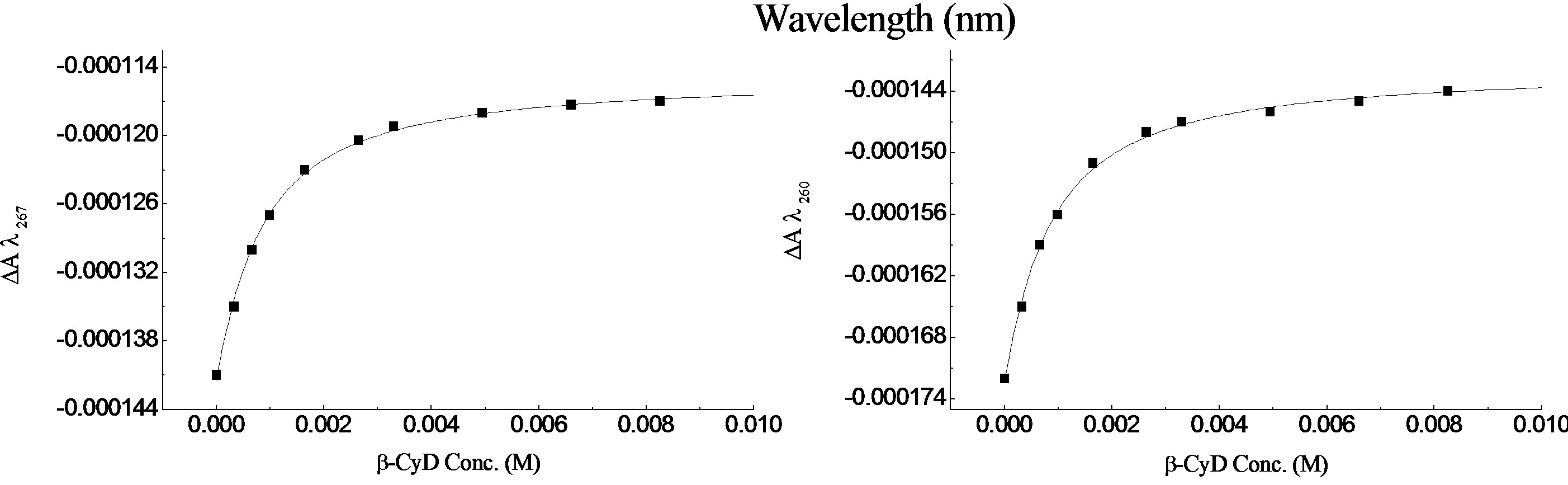 (c)
(c)
Figure 3. β-CyD titration at pH 2.0 in 1.0 cm cell (300 - 230 nm), 0.05 cm cell (260 - 190 nm). (a) UV spectra showing β-CyD titration of R-2PPA in H2O; (b) CD spectra; (c) Binding curves of β-CyD/R-2PPA complex.
 (a)
(a)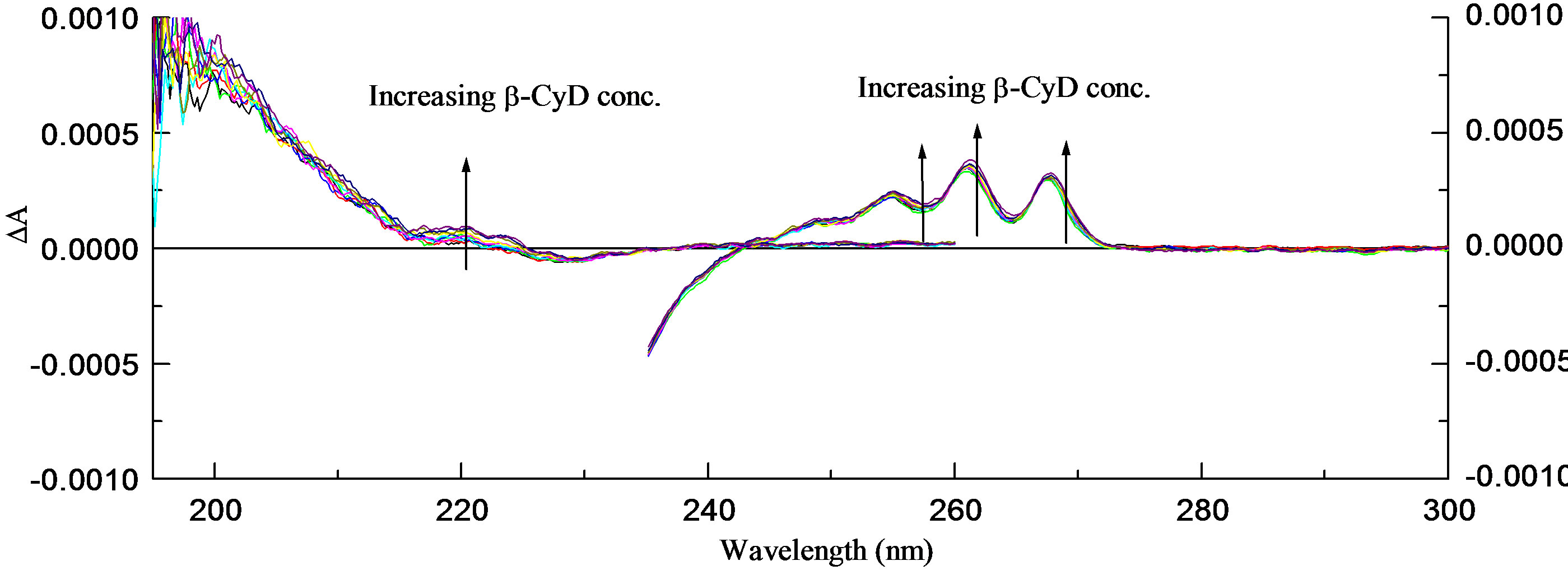 (b)
(b)
Figure 4. β-CyD titration at pH 7.0 in 1.0 cm cell (300 - 230 nm), 0.05 cm cell (260 - 190 nm). (a) UV spectra and a binding curve of β-CyD titration of R-2PPA; (b) CD spectra.
 (a) (b)
(a) (b)
Figure 5. (a) The pH titration curves of R-PPA/β-CyD complex; (b) The possible structural assignments of R-PPA/β-CyD complexes at pH 2.0 and pH 7.0.
5.15 ± 0.016 which is markedly increased in comparison to the free uncomplexed form of R-2PPA (pKa = 4.35).
• 3.2. (S)-(+)-2-Phenylpropionic Acid
The pH titration of S-2-PPA in the absence of cyclodextrin
Similarly, the UV and CD spectra (not shown) as a function of pH were measured in 0.05 and 1.0 cm cells.
Comparison with Figure 2 shows that, as expected, there is no change in either the position or the intensity of the three absorption bands 1Lb, CT, and 1La in comparison to the (R)-enantiomer.
The absorbance spectra are practically identical, however, as expected; the CD bands sign is reversed. The pH titration curves (A218 and DA262 versus pH) gave pKa = 4.35 ± 0.02, the same value as the R-enantiomer.
S-2PPA/β-cyclodextrin titration
At pH 2.0, β-CyD titration of S-2PPA was carried out covering a wavelength range between 300 nm to 190 nm (spectra not shown). The results showed no significant change in the CD signal, however, the addition of β-CyD results in a decrease in the absorbance intensity of the 1Lb band and a “loss” of one of the vibronic components at ~l267. There is also a slight increase in the CD and UV intensities of the CT and 1La bands with increasing β-CyD concentration. The change in DA was too small to warrant further analysis. However, the value at A263 was plotted against β-CyD concentration (Figure 6(a)) giving K = 3.59 × 102 M−1 ± 22 for the binding constant. This indicate that the change in conformation have a significant effect on the binding causing the S-enantiomer to act in an opposite way to the R-enantiomer and binds less strongly to β-CyD at pH 2.0.
At pH 7.0, like the previous measurements, the CD and UV spectra (not shown) of the (S)-enantiomer exhibited a small change in the presence of β-CyD. Nevertheless, plotting DA262.2 against β-CyD concentration gave K = 9.73 × 102 M−1 ± 33 (Figure 6(b)). Apparently, the S-enantiomer binds to β-CyD more strongly at pH 7.0 than at pH 2.0, contrary to the R-enantiomer which interacts more strongly at pH 2.0.
The UV and CD spectra (not shown) of the pH titration of S-2PPA/β-CyD complex showed that spectra of both enantiomers are effectively identical, again, bearing in mind that the CD have opposite signs.
Plotting A220 and DA262 nm against pH gave pKa = 5.15 ± 0.02, which is significantly higher than the pKa of the free compound.
The pKa change is the same for both enantiomers. Analysis of the binding curves for all forms 2-PPA indicates a 1:1 stoichiometry for the inclusion complex. Figure 7 shows an example of the stoichiometry determination for S-2PPA/β-CyD complex at pH 7.0.
S-2PPA shows similar interaction with β-CyD in respect to spectral changes. The difference in binding of the ionised and unionised form of both enantiomers indicates that the only obvious factor that can contribute to this difference is the different conformational assignments of the two enantiomers.
This suggests that the R-enantiomer can form intermolecular hydrogen bonds most effectively than the S-enantiomer, explaining the week binding of the S-enantiomer at pH 2.0, which in turn can affect the co-conformation of the guest-host complex as can be assumed in Figure 6 (c). There is a significant change in the pKa value for both enantiomers which increased by ~0.8 pKa. This decrease in acidity can be attributed to disturbance of the resonance on the CO2H group by interaction with the OH on the β-CyD molecule [38]. Reduction of electron density on the carboxyl group makes the loss of the proton easier in the complex form than it is in the free form, resulting in an increased pKa value. The co-conformation of the molecule in β-CyD can also have a significant effect on the acidity, because of the change in the entropy.
4. Conclusions
Thermodynamic, kinetic, and structural aspects of complexation by cyclodextrins play an essential role in the understanding of their functions and for the development of prospective applications. A fast kinetics, for example, is desirable for catalytic activity, and a regulated release of the guest is essential in drug delivery. On the other hand, a strong and selective binding may be of interest for analytical or environmental purposes. Finally, the solution structures are intimately related to molecular recognition, and the specific location of functional groups in the CyD cavity, in particular in solution, may be the prerequisite for enzymatic specificity.
The changes in intensity and the wavelength shift of the UV spectrum upon addition of β-CyD are evidence of interaction between these compounds and β-CyD. Also, change in the vibronic structure indicates a change in the environment of the ring system [39]. This can result from the ring being in a non-polar environment inside the β-CyD cavity.
In the case of the R-enantiomer, its conformation and spatial arrangement allows the carboxylic form to form hydrogen bonding and bind to β-CyD with higher affinity more than its carboxylate partner. In the case of carboxylate ion, when its orientation is pointing out intoI solution and thus hydrated, it is understandable that, if carboxylate/β-CyD binding is controlled by an induced dipole-dipole mechanism, the carboxylate should bind more tightly in the cavity than its carboxylic partner. However, the requirement for solvation of the charged carboxylate group by water may prohibit the aromatic
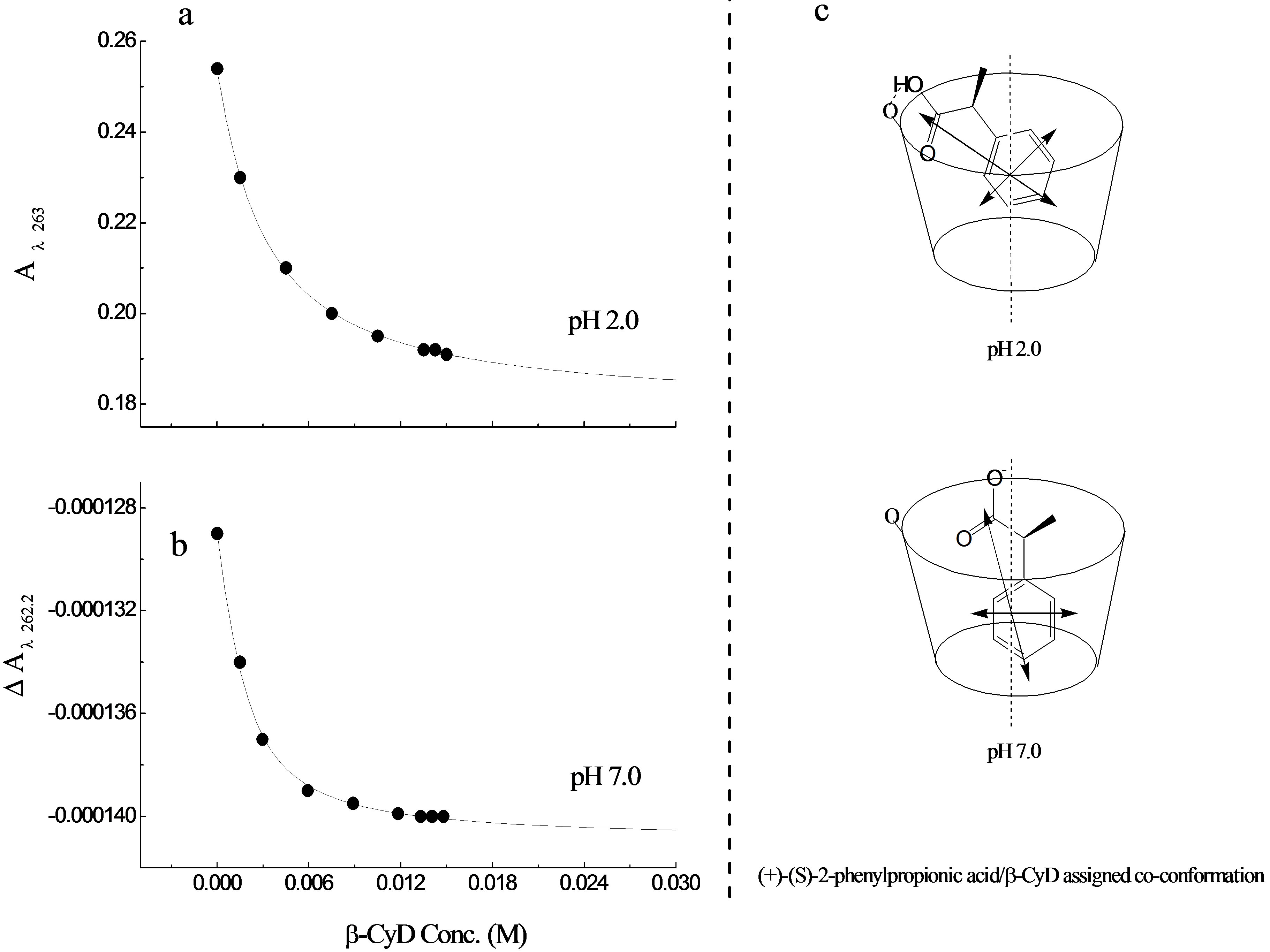
Figure 6. Binding curves for S-PPA/β-CyD complex (a) binding curve of S-PPA/β-CyD complex at pH 2.0 (b) at pH 7.0 (c) possible structural assignments of S-PPA/β-CyD complex at pH 2.0 and pH 7.0.
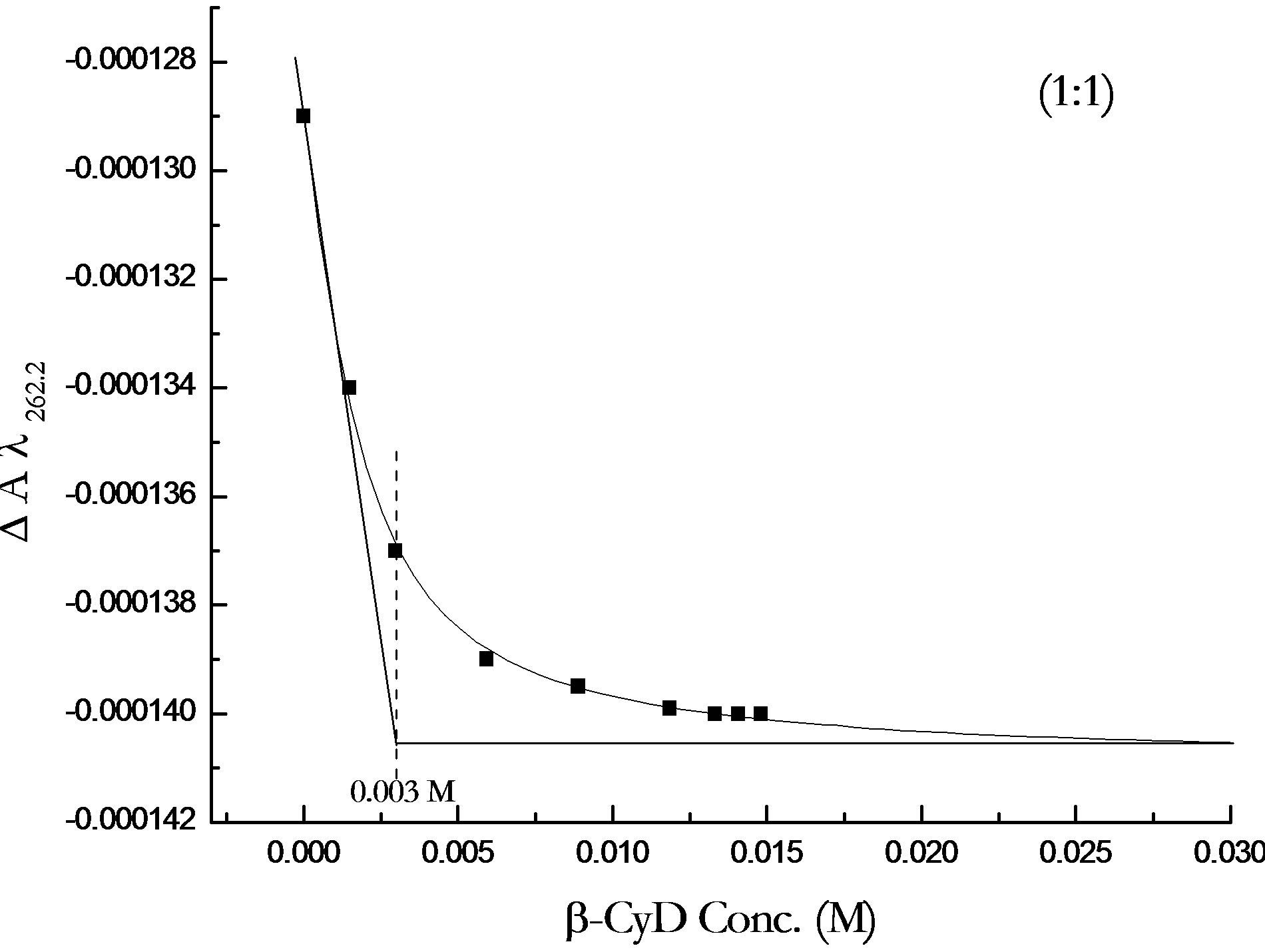
Figure 7. Binding curve of β-CyD titration of S-PPA at pH 7.0 showing 1:1 stoichiometry of 2-PPA/β-CyD complex.
ring from fully penetrating the β-CyD cavity. When the carboxylate is in the β-CyD cavity, out of solution and partially disolvated, other factors must be considered regarding the stability of the neutral vs. charged guest/β- CyD complexes. Therefore, the energy required in transferring a charged species such as the carboxylate ion from a medium of high dielectric to a medium of low dielectric seems to be a major factor controlling the stability of the complexes.
In the case of the S-enantiomer, it seems that the carboxylate ion is not restricted by hydrogen bonding and its conformation helps to adopt a more favourable co-conformation within the β-CyD cavity resulting in stronger affinity than it is in the carboxylic form. The change in acidity of these compounds upon complexation with β- CyD reflects the importance of the role of the carboxylic group on the binding of these compounds. In general, the major factors that control chiral drug/β-CyD complex are ( and not limited to) the molecular spatial arrangement, the ionisation state and the charge on the molecule.
The method for determining the concentration of free and bound compound, only allows precise interpretation in terms of binding constants when a single site is involved. When more than one site contributes to the ICD or the change in the CD signal, the method is less precise. The observed ICD is a function of the amounts bound at the individual sites and the associated molar ellipticities. If the binding constants of the primary and secondary sites are very different from one another and the ICD of the secondary sites are small, then reasonable estimates of the primary binding constant can be obtained, the values for the secondary sites being less reliable. It is the first site that is of major clinical importance and is the one of interest in this work. These problems are not peculiar to the CD technique, they are shared by other spectroscopic techniques where the intensive factor for the second and higher binding sites may be impossible to obtain.
The binding constants are determined by the direct titration as described earlier. From simple mathematical operations with the K values obtained in this work, one can estimate the initial quantity of free and available drug in the system, when a normal dose of a drug is administered encapsulated by β-CyD at biological pH. For example, and according to the result of this work, S-PPA, if administered encapsulated by β-CyD at biological pH, leaves in the medium an initial quantity of free and available S-PPA- about 25% - 30% of the total amount of the administered drug. What is more important, as long as the organism “makes use” of the drug, the equilibrium (β-CyD + Drug β-CyD/drug complex) is shifted by mass action toward the release of the active component, keeping and regulating its presence in the medium. These estimations rebound the advantages of the use of cyclodextrins as drug carriers, widely used in drug delivery.
β-CyD/drug complex) is shifted by mass action toward the release of the active component, keeping and regulating its presence in the medium. These estimations rebound the advantages of the use of cyclodextrins as drug carriers, widely used in drug delivery.
5. Acknowledgements
We would like to thank Molecular Biophysics group at King’s College London and Applied Photophysics Ltd. for their support.
REFERENCES
- C. Varas-Lorenzo, A. Maguire, J. Castellsague and S. PerezGutthann, “Quantitative Assessment of the Gastrointestinal and Cardiovascular Risk-Benefit of Celecoxib Compared to Individual NSAIDs at the Population Level,” Pharmacoepidemiology and Drug Safety, Vol. 16, No. 4, 2007, pp. 366-376. doi:10.1002/pds.1299
- M. M. Krause, M. D. Brand, S. Krauss, C. Meisel, H. Vergin, G. R. Burmester and F. Buttgereit, “Nonsteroidal Antiinflammatory Drugs and a Selective Cyclooxygenase 2 Inhibitor Uncouple Mitochondria in Intact Cells,” Arthritis & Rheumatism, Vol. 48, No. 5, 2003, pp. 1438- 1444. doi:10.1002/art.10969
- M. Zanocchi, V. Tibaldi, D. Amati, F. Francisetti, E. Martinelli, M. Gonella, F. Cerrato, E. Ponte, A. Luppino, B. Bardelli, A. Canade, F. Gariglio, C. Moiraghi and M. Molaschi, “Adverse Drug Reactions as Cause of Visit to the Emergency Department: Incidence, Features and Outcomes,” Recenti Progressi in Medicina, Vol. 97, 2006, pp. 381-388.
- G. Singh, “Triadafilopoulus G Epidemiology of NSAIDInduced GI Complications,” Journal of Rheumatology, Vol. 26, 1999, pp. 18-24.
- M. Filippa, M. I. Sancho and E. Gasull, “Encapsulation of Methyl and Ethyl Salicylates by Beta-Cyclodextrin HPLC, UV-Vis and Molecular Modeling Studies,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 48, No. 3, 2008, pp. 969-973. doi:10.1016/j.jpba.2008.06.005
- T. Loftsson and D. Duchene, “Cyclodextrins and Their Pharmaceutical Applications,” International Journal of Pharmaceutics, Vol. 329, No. 1-2, 2007, pp. 1-11. doi:10.1016/j.ijpharm.2006.10.044
- K. Miyake, H. Arima, F. Hirayama, M. Yamamoto, T. Horikawa, H. Sumiyoshi, S. Noda and K. Uekama, “Improvement of Solubility and Oral Bioavailability of Rutin by Complexation with 2-Hydroxypropyl-Beta-Cyclodextrin,” Pharmaceutical Development and Technology, Vol. 5, 2000, pp. 399-407.
- K. Uekama, “Design and Evaluation of Cyclodextrin-Based Drug Formulation,” Chemical and Pharmaceutical Bulletin, Vol. 52, No. 8, 2004, pp. 900-915. doi:10.1248/cpb.52.900
- R. Challa, A. Ahuja, J. Ali and R. K. Khar, “Cyclodextrins in Drug Delivery: An Updated Review,” AAPS PharmSciTech, Vol. 6, No. 2, 2005, pp. E329-E357. doi:10.1208/pt060243
- T. Loftsson, S. B. Vogensen, M. E. Brewster and F. Konradsdottir, “Effects of Cyclodextrins on Drug Delivery through Biological Membranes,” Journal of Pharmaceutical Sciences, Vol. 96, No. 10, 2007, pp. 2532-2546. doi:10.1002/jps.20992
- K. Uekama, F. Hirayama and H. J. Arima, “Recent Aspect of Cyclodextrin-Based Drug Delivery System,” Vol. 56, 2006, pp. 3-8.
- M. A. Mintzer and E. E. Simanek, “Nonviral Vectors for Gene Delivery,” Chemical Reviews, Vol. 109, No. 2, 2009, pp. 259-302. doi:10.1021/cr800409e
- M. Singh, R. Sharma and U. C. Banerjee, “Biotechnological Applications of Cyclodextrins,” Biotechnology Advances, Vol. 20, 2002, pp. 341-359.
- V. J. Stella and R. A. Rajewski, “Cyclodextrins: Their Future in Drug Formulation and Delivery,” Pharmaceutical Research, Vol. 14, 1997, pp. 556-567. doi:10.1023/A:1012136608249
- T. Loftsson, F. Konradsdottir and M. Masson, “Influence of Aqueous Diffusion Layer on Passive Drug Diffusion from Aqueous Cyclodextrin Solutions through Biological Membranes,” Pharmazie, Vol. 61, 2006, pp. 83-89.
- T. J. Edkins, P. C. Meier, R. D. Shah, D. R. Bobbitt, H. Saranadasa and R. D. Lodevico, “Quantitative Analysis of Incomplete HPLC Resolution of Enantiomers. Fit of Polarimetric Detection for Dand L-Phenylalanine to a Gaussian Function,” Enantiomer, Vol. 7, No. 1, 2002, pp. 11- 22. doi:10.1080/10242430210703
- M. F. Landoni and A. Soraci, “Pharmacology of Chiral Compounds: 2-Arylpropionic Acid Derivatives,” Current Drug Metabolism, Vol. 2, No. 1, 2001, pp. 37-51. doi:10.2174/1389200013338810
- K. Lin, F. Zhang, S. Zhou, W. Liu, J. Gan and Z. Pan, “Stereoisomeric Separation and Toxicity of the Nematicide Fosthiazate,” Environmental Toxicology and Chemistry, Vol. 26, No. 11, 2007, pp. 2339-2344. doi:10.1897/07-255R.1
- F. Jamali, R. Mehvar and F. M. Pasutto, “Enantioselective Aspects of Drug Action and Disposition: Therapeutic pitfalls,” Journal of Pharmaceutical Sciences, Vol. 78, No. 9, 1989, pp. 695-715. doi:10.1002/jps.2600780902
- C.-J. Nunez-Agüero, C.-M. Escobar-Llanos, D. Dıaz, C. Jaime and R. Garduno-Juarez, “Chiral Discrimination of Ibuprofen Isomers in b-Cyclodextrin Inclusion Complexes: Experimental (NMR) and Theoretical (MD, MM/GBSA) Studies,” Tetrahedron, Vol. 62, No. 17, 2006, pp. 4162- 4172. doi:10.1016/j.tet.2006.02.010
- K. B. Lipkowitz, “Applications of Computational Chemistry to the Study of Cyclodextrins,” Chemical Reviews, Vol. 98, No. 5, 1998, pp. 1829-1874. doi:10.1021/cr9700179
- Z. Gao and X. Zhao, “Enhancing Electrorheological Behaviors with Formation of β-Cyclodextrin Supramolecular Complex,” Polymer, Vol. 44, No. 16, 2003, pp. 4519- 4526. doi:10.1016/S0032-3861(03)00416-6
- J. Geczy, J. Bruhwyler, J. Scuvee-Moreau, V. Seutin, H. Masset, J. C. Van Heugen, A. Dresse, C. Lejeune, E. Decamp, L. Szente, J. Szejtli and J. F. Liegeois, “The Inclusion of Fluoxetine into Gamma-Cyclodextrin Increases Its Bioavailability: Behavioral, Electrophysiological and Pharmacokinetic Studies,” Psychopharmacology, Vol. 151, No. 4, 2000, pp. 328-334. doi:10.1007/s002130000512
- F. Djedaini, S. Z. Lin, B. Perly and D. Wouessidjewe, “High-Field Nuclear Magnetic Resonance Techniques for the Investigation of a Beta-Cyclodextrin: Indomethacin Inclusion Complex,” Journal of Pharmaceutical Sciences, Vol. 79, No. 7, 1990, pp. 643-646. doi:10.1002/jps.2600790721
- C. Yujuan and L. Runhua, “1H NMR Titration and Quantum Calculation for the Inclusion Complexes of cis-Cyclooctene, cis, cis-1, 3-Cyclooctadiene and cis, cis-1, 5-Cyclooctadiene with Beta-Cyclodextrin,” Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, Vol. 73, No. 4, 2009, pp. 713-718. doi:10.1016/j.saa.2009.03.012
- Y. Nievergelt, “Exact Equations for the Equilibrium Constants of Single Intermolecular Complexes in Terms of Spectrophotometric Data,” Analyst, Vol. 119, No. 1, 1994, pp. 145-151. doi:10.1039/an9941900145
- H. Fuchs and R. Gessner, “The Result of EquilibriumConstant Calculations Strongly Depends on the Evaluation Method Used and on the Type of Experimental Errors,” Biochemical Journal, Vol. 359, 2001, pp. 411-418. doi:10.1042/0264-6021:3590411
- P. F. da Silva, J. C. Lima, A. A. Freitas, K. Shimizu, A. L. Macanita and F. H. Quina, “Charge-Transfer Complexation as a General Phenomenon in the Copigmentation of Anthocyanins,” Journal of Physical Chemistry A, Vol. 109, No. 32, 2005, pp. 7329-7338. doi:10.1021/jp052106s
- N. Sadlej-Sosnowska, L. Gasinski and W. P. Oziminski, “Fluorometric Determination of Stability Constants of Inclusion Complexes of Naproxen and Charged Cyclodextrins in Aqueous Solutions. Nonlinear vs Linear Data Processing,” Polish Journal of Chemistry, Vol. 77, 2003, pp. 1039-1048.
- K. A. Connors, “Optical Absorption Spectroscopy,” In: Binding Constants, The Measurement of Molecular Complex Stability, Wiley-Interscience, New York, 1987.
- C. D. Tran, M. S. Baptista and T. Tomooka, “Determination of Binding Constants by Flow Injection Gradient Technique,” Langmuir, Vol. 14, No. 24, 1998 pp. 6886- 6892. doi:10.1021/la9805894
- Y. Ikeda, F. Hirayama, H. Arima, K. Uekama, Y. Yoshitake and K. Harano, “NMR Spectroscopic Characterization of Metoprolol/Cyclodextrin Complexes in Aqueous Solution: Cavity Size Dependency,” Journal of Pharmaceutical Sciences, Vol. 93, No. 7, 2004, pp. 1659-1671. doi:10.1002/jps.20077
- E. Junquera, M. Martin-Pastor and E. Aicart, “Molecular Encapsulation of Flurbiprophen and/or Ibuprophen by Hydroxypropyl-b-Cyclodextrin in Aqueous Solution Potentiometric and Molecular Modelling Studies,” The Journal of Organic Chemistry, Vol. 63, 1998, pp. 4349-4358. doi:10.1021/jo980104g
- P. Mura, S. Furlanetto, M. Cirri, F. Maestrelli, G. Corti and S. Pinzauti, “Interaction of Naproxen with Ionic Cyclodextrins in Aqueous Solution and in the Solid State,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 37, No. 5, 2005, pp. 987-994. doi:10.1016/j.jpba.2004.06.016
- A. A. Dahab, D. El-Hag and A. F. Drake, “Simultaneous Determination of Photometric Accuracy during Circular Dichroism Measurements,” Analytical Methods, Vol. 2, No. 7, 2010, pp. 929-935. doi:10.1039/b9ay00216b
- A. A. Dahab and D. El-Hag, “Rapid Analysis of NSAIDs Binding to b-Cyclodextrin Using the Simultaneous Measurement of Absorption and Circular Dichroism with a Novel Multi-Cell Low Volume Device,” Analytical and Bioanalytical Chemistry, 2012, in press.
- K. Harata, “Induced Circular Dichroism of cycloamylose Complexes with Metaand Para-Disubstituted Benzenes,” Bioorganic Chemistry, Vol. 10, No. 3, 1981, pp. 255-265. doi:10.1016/0045-2068(81)90004-3
- T. D. Thomas, “Acidity of Carboxylic Acids: Resonance Delocalization or Induction?” Journal of the Chemical Society, Perkin Transactions, Vol. 2, 1994, pp. 1945-1948.
- J. B. Lambert, H. F. Shurvell, D. A. Lightner and R. G. Cooks, “Electronic Absorption and Chiroptical Spectroscopy,” In: J. Challice, Ed., Organic Structural Spectroscopy, Prentice Hall, New Jersey, 2001, pp. 274-303
NOTES
*Corresponding author.
*Complexation of R and S-2-phenylpropionic acid with -cyclodextrin