International Journal of Organic Chemistry
Vol.04 No.04(2014), Article ID:52525,8 pages
10.4236/ijoc.2014.44029
11C-Labeling of the C(1)-C(10) Dihydroxy Acid Moiety for the Study on the Synthesis of Kulokekahilide-2 PET Tracer
Chunguang Han1, Hisashi Doi2, Junji Kimura3, Yoichi Nakao4, Masaaki Suzuki5*
1Research Center for Materials Science, Nagoya University, Nagoya, Japan
2RIKEN Center for Life Science Technologies, Kobe, Japan
3Department of Chemistry and Biological Science, College of Science and Engineering, Aoyama Gakuin University, Sagamihara, Japan
4Department of Chemistry and Biochemistry, School of Advanced Science and Engineering, Waseda University, Tokyo, Japan
5National Center for Geriatrics and Gerontology, Obu City, Japan
Email: *suzukims@ncgg.go.jp
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 7 October 2014; revised 20 November 2014; accepted 6 December 2014
ABSTRACT
11C-labeled C1-C10 partial structure of kulokekahilide-2 (1) was successfully synthesized based on Pd0-mediated rapid C-[11C]methylation using [11C]methyl iodide and pinacol alkenylboronate. The preparation of organoboron intermediate via olefin cross-metathesis is also a crucial procedure for the synthesis of 11C-labeling C1-C10 dihydroxy acid moiety of 1.
Keywords:
Kulokekahilide-2, Carbon-11, Boron, C-C Coupling, Isotopic Labeling

1. Introduction
Kulokekahilide-2 (1) was isolated from the Hawaiian marine mollusk Philinopsisspeciosa, as an aurilide-type metabolite to show potent cytotoxicity against several cell lines in nanomolar concentrations [1] . The 26-mem- bered form of 1 consisting of five amino acids (43-D-Ala, 37-L-Ile, 34-MeGly, 24-D-MePhe, 21-L-Ala) and two hydroxy acids {15-D-2-hydroxyisocaproic acid (15-D-Hica) and (5S,6S,7S,2E,8E)-2,6,8-trimethyl-5,7-dihy- droxy-2,8-decadienoic acid (Dtda), Figure 1}, possesses almost the same components as the aurilides [2] [3] and lagunamides [4] isolated from the Japanese sea hare Dolabellaauricularia and marine cyanobacteria respectively [5] . The aurilide also shows high-level cytotoxicity against renal and prostate cancer cell lines and its molecular target for inducing apoptosis in human cell lines was reported by Sato et al. [6] [7] . Recently, Kimura et al. revealed that kulokekahilide-2 shows potent cytotoxicity against 39 human cancer cell lines, suggesting a mechanism of action could be different from that of standard anticancer drugs, aurilides, palau’amide [8] [9] , and lagunamides [5] .
Positron emission tomography (PET), which uses specific probes radiolabeled with short-lived positron- emitting radionuclides (11C, 13N, 15O, 18F etc.), is a powerful non-invasive molecular imaging technique usable for highly accurate diagnoses and investigation of the in vivo biochemistry of bioactive compounds. In addition, it is strongly hoped that PET would be applied as a human microdosing study to an early-stage of drug development [10] . Carbon-11 (half-life = 20.4 min) is one of the most meritorious isotopes for PET research because carbon is included in all organic molecules. In recent years, efficient labeling methods of the 11C radioisotope into organic frameworks have continuously been developed by Suzuki et al. using palladium(0)-mediated rapid C-[11C]methylation (5 min reactions) consisting of the cross-coupling reactions of [11C]methyl iodide and the stannyl or boron substrates [11] [12] . Actually, the rapid cross-coupling reactions have successfully been applied for the syntheses of various disease-oriented PET tracers [13] -[16] . Our interest has been intrigued by extending the 11C labeling reactions to complex natural product. Described herein in the synthesis of 11C-incorporated C(1)-C(10) partial structure in [11C]kulokekahilide-2 focused on the 11C labeling at C10 carbon of 1 using a combination of olefin cross metathesis (CM) and rapid C-[11C]methylation.
2. Results and Discussion
Kulokekahilide-2 has several possible positions for 11C radiolabeling. Prior to actual synthesis of 11C-incorpo- rated 1, we investigated a model study using a partial structure. Here, we are particularly interested in introducing 11C onto the methylene group of 2 as a 11CH3 by our rapid cross-coupling reaction [12] , [17] between sp2vinyl- and sp3-carbon atoms using an organostannyl or boron precursor 3 (Scheme 1) where key step of synthetic strategy involves the preparation of precursor 3 derived from methyl ester 2.
Before synthesizing 11C-labeled 4a, we prepared the nonradioactive molecule 4b. Thus, the intermediary compound 5 was synthesized from (S)-3-propionyl-4-isopropyl-2-oxazolidinone according to the reported procedure [1] . Protection of the secondary alcohol 5 gave PMB ether 6 in 49% yield. Subsequent deprotection of the TBS group at C7 in 6 afforded the desired compound 4b in 78% yield after column chromatography on silica gel (Scheme 2).
The synthesis of the organostannyl or boron compound 3 started with an acylation of commercially available ox-azolidinone 7 with the treatment of LDA and EtCOCl to give aldol coupling precursor [18] . Subsequent an-

Figure 1. Chemical structure of kulokekahilide-2 (1).
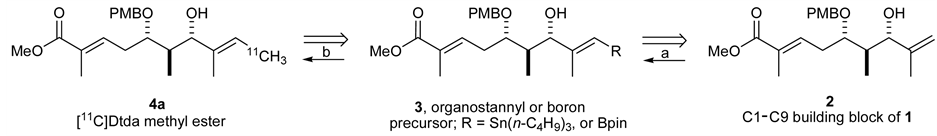
Scheme 1. Synthetic plan: Transformation from terminal alkene 2 to 11C-labeled 4a by (a) olefin cross metathesis and (b) rapid C-[11C]methylation.
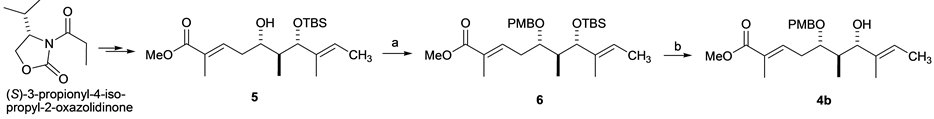
Scheme 2. Synthesis of 4b. Reagents and Conditions: (a) PMBTCA, Et2O, −78˚C added to CF3SO3H, then RT, 2 h, 49%; (b) HF・Py, pyridine, THF, RT, 40˚C, overnight, 78%.
ti-selective aldol reaction with methacrylaldehyde to provide the aldol product 8, which was converted into aldehyde 9 in three steps. Then the BF3・OEt2 mediated Mukaiyamaaldol reaction of 9 with silyl ketene acetal 10 afforded methyl ester (5R)-11 as a single diastereomer. Successful Moffatt oxidation of 11 to give ketone 12, and subsequent reduction of the resulting keto group in 12 with NaBH4 stereoselectively (S/R = 22/1) proceeded to give the desired alcohol (5S)-13 in 69% yield. PMB protection of the secondary hydroxy substituent at C5 in 14, followed by cleavage of the TBS protecting group at C7 with HF・Py, accomplishing the synthesis of 1,1-dis- ubstituted alkene 2 (Scheme 3).
As the key step for synthesis of organostannane or organoboron precursor of 2, cross metathesis was chosen because it has become a powerful and convenient synthetic technique for the preparation of functionalized alkenes in organic chemistry [19] . With this concern, hydroxy 1,1-disubstituted alkene 15 as a model compound for screening the most effective Ru complexes and cross partners in metathesis. Cross metathesis using 15 prepared by Grignard addition reaction [20] was investigated under various reaction conditions: Grubbs second- generation (G-II) [21] [22] or the Hoveyda-Grubbs second-generation complex (HG-II) [23] in CH2Cl2, benzene, and toluene at reflux or microware irradiation, and the use of an excess amount of cross partners such as vinylstannane 16a, or vinyl boronates (16b and 16c). These results are summarized in Table 1. Cross metathesis of 15 with vinylstannane 16a using HG-II (1.0 equiv) in CH2Cl2 or toluene at reflux did not give the desired organostannane precursor 17a presumably due to highly sterically hindered Sn(n-C4H9)3 group in 16a (Table 1, entries 1-2). The lower sterically hinderedvinyl dioxaborolane 16b compared with tetramethyl vinyl boronate 16c also did not afford the corresponding organoboron compound 17b with the use of HG-II or G-II catalysts in thermal or microware heating conditions [24] -[31] (entries 3-5). Grubbs II-catalyzed cross metathesis of 15 with vinyl pinacol boronate 16c (4.0 equiv) in benzene at refluxdid not afford desired 17c (entry 6). By contrast, when 15 was treated with more robust and powerful HG-II (25 mol%) in CH2Cl2 at reflux for one day, we observed a small amount of organoboron precursor 17c (entry 7). The increase of the catalyst to a stoichiometric amount under the same reaction conditions to give 17c in 35% yield (entry 8) as a single E-isomer as judged by the NOE observation.
We envisioned here that the E-selective olefin cross metathesis using HG-II catalyst in the reaction of 15 and vinyl pinacol boronate 16c could be applied for synthesis of the organoboron derivative of 1,1-disubstituted alkene 2, which is crucial precursor for the synthesis of the 11C-labeling dihydroxy acid moiety of 1. However, contrary to our expectation, cross metathesis using 2 did not proceed under above reaction conditions (Table 1, entry 8) with the notice of complete recovery of 2.The reaction was further conducted under more forcing the reaction conditions, giving the desired (E)-organoboron derivative 3 in 14% yield along with recovered 2 in 32% yield (Scheme 4). The geometry of the newly formed double bond was decided by NOE observation as shown below.
By using the precursor 3, we examined the Pd0-mediate rapid C-[11C]methylations protocol [12] for preparing the 11C-labeled partial structure of 1 under cold conditions (Scheme 4). The methylation of 3 was conducted
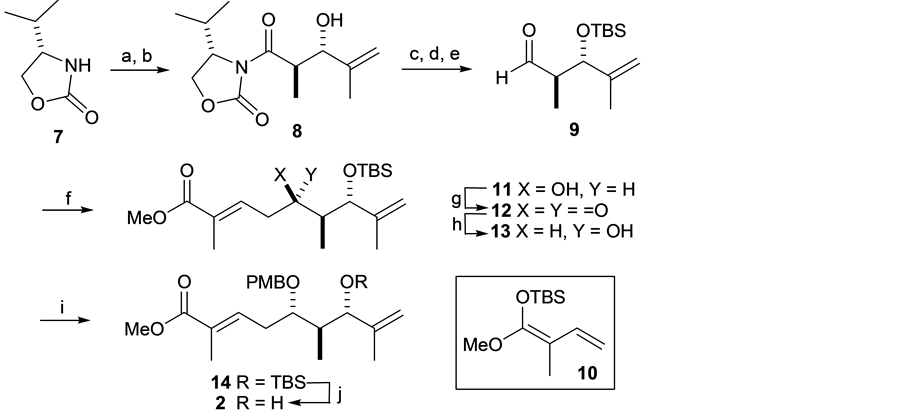
Scheme 3. Synthesis of 2. Reagents and Conditions: (a) EtCOCl, LDA, THF, −78˚C, 1 h, 95%; (b) methacylaldehyde, nBu2BOTf, DIPEA, CH2Cl2, −78˚C, 2 h, 47%; (c) MeNH(OMe)HCl, Me3Al, 0˚C-RT, 30 min, then 0˚C added to 8, warmed up to RT for overnight; (d) TBSCl, Imidazole, DMF, 94% for two steps; (e) DIBAL-H, THF, −78˚C, 1.5 h, 89%; (f) 10, BF3・OEt2, −78˚C, 1 h, 69%; (g) DCC, pyridine trifluoroacetate, DMSO/Et2O (1:1), RT, 1.5 h, 75%; (h) NaBH4, MeOH, −40˚C, 2 h, 69%; (i) PMBTCA, Et2O, −78˚C added to CF3SO3H, then RT, 2.5 h, 49%; (j) HF・Py, pyridine, THF, RT, 40˚C, overnight, 74%.
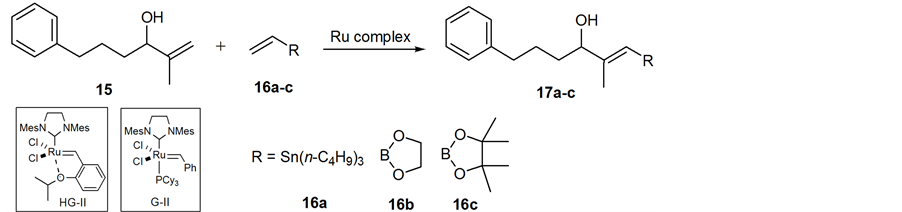

Table 1. Screening the reactions of Ru complexes and cross metathesis partnersa.
aReaction conditions: 15 (1 equiv), cross partner (4 equiv), in refluxing CH2Cl2 (0.02 M) for 24 h. bOnly the E isomer was observed in all cases; cIn toluene (0.02 M), reflux, 24 h; d In CH2Cl2. (0.1 M), microware heating, 60˚C, 30 min; eIn benzene (0.02 M), reflux, 24 h. fIsolated yield; HG-II: Hoveyda- Grubbs second-generation catalyst; G-II: Grubbs second-generation catalyst.
11C-labeled Dtda methyl ester 4a as C1-C10 building block of kulokekahilide-2 (1) has been successfully synthesized using a combination of olefin cross-metathesis/rapid C-[11C]methylation. Pinacol alkenyl boronate precursor 3 prepared via cross-metathesis is crucial for subsequent Pd0-mediated rapid C-[11C]methylation using [11C]CH3I. The 11C-labeling would be applied at later stage for the synthesis of 11C-incorporated 1.
Based on the above-mentioned protocol, we preformed the synthetic of 11C-labeled Dtda methyl ester 4a, Thus, 3/Pd2(dba)3/P(o-tolyl)3/K2CO3 (2:1:4:10) dissolved in DMF under argon was mixed with [11C]CH3I, prepared as previously described [32] , the solution was heated at 70˚C for 5 min (Scheme 5). After the mixture was
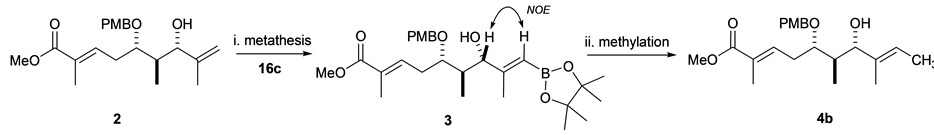
Scheme 4. Synthesis of 3 and 4b. Reagents and Conditions: (i) HG-II (1.3 equiv), toluene (0.01M), 80˚C, 24 h, 14%; (ii) MeI, Pd2(dba)3, P(o-tolyl)3, and K2CO3, DMF, 70˚C, 5 min, 78%.

Scheme 5. Synthesis of 11C-labeled Dtda methyl ester 4a.
poured into a separate vial containing a solution of ascorbic acid in acetonitrile, the resulting reaction mixture was submitted to HPLC, and then purified by reverse phase semi-preparative HPLC to give desired [11C]Dtda- methyl ester 4a in 72% as reverse phase HPLC analytical yield (Figure 2). Total synthesis time including HPLC purification was 33 min. The radioactivity of isolated 4a was 315 MBq and the radiochemical purity was >99%. The chemical identity of 4a was confirmed by co-injection with the authentic sample of 4b by analytical HPLC.
3. Conclusion
11C-labeled Dtda methyl ester 4a as C1-C10 building block of kulokekahilide-2 (1) has been successfully synthesized using a combination of olefin cross-metathesis/rapid C-[11C]methylation. Pinacol alkenyl boronate precursor 3 prepared via cross-metathesis is crucial for subsequent Pd0-mediated rapid C-[11C]methylation using [11C]CH3I. The 11C-labeling would be applied at later stage for the synthesis of 11C-incorporated 1.
4. Acknowledgements
This work was supported in part by a consignment expense for the Molecular Imaging Program on “Research Base for Exploring New Drugs” from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan. We thank Dr. Masakatsu Kanazawa (Central Research Lab. Hamamatsu Photonics K. K.), Ms. Mawatari Aya (RIKEN Center for Life Science Technologies) for experimental assistance and Mr. Masahiro Kurahashi (Sumitomo Heavy Industry Accelerator Service Ltd.) for operating the cyclotron.
5. Experimental
5.1. General
Nuclear magnetic resonance (NMR) spectra were recorded on a JEOL JNM-ECX400P spectrometer (400 MHz for 1H), and the chemical shifts in d (parts per million) were referenced to the solvent peaks of dH 7.26 for CHCl3. HR ESI-TOF-MS spectra were measured on an Applied Biosystems Mariner Biospectrometry Workstation using ABN as a calibration standard in the positive mode. Microwave irradiation was carried out in a Biotage Initiator™ (Tokyo, Japan) using a sealed vessel. [11C]Carbon dioxide was produced by a 14N(p, α)11C reaction by using a Sumitomo CYPRIS HM-12S cyclotron (Sumitomo Heavy Industries, Tokyo, Japan), and then converted to [11C]methyl iodide by treatment with lithium aluminum hydride followed by hydriodic acid using an automated synthesis system (Cupid, Sumitomo Heavy Industries). The obtained [11C]methyl iodide was used for palladium(0)-mediated rapid [11C]methylation shown in Scheme 5. The synthesis of 11C-labeled Dtda methyl ester 4a in Scheme 5 was conducted in a lead-shielded hot-cell with remote control of all operations in RIKEN CLST. Purification with HPLC was performed on a GL Science system (Tokyo, Japan). Radioactivity was quantified with an ATOMLAB™ 300 dose calibrator (Aloka, Tokyo, Japan). Analytical HPLC was performed on a Shimadzu system (Kyoto, Japan), and effluent radioactivity was measured with an RLC700 radio analyser

Figure 2. HPLC for purification of the reaction mixture. UV absorbance: 223 nm.
(Aloka). The column used for analytical and semi-preparative HPLC was Develosil ODS-HG-5 (Nomura Chemical, Japan).
5.2. Synthesis of Authentic Sample 4b
p-Methoxybenzyl ether (6): To a stirred solution of alcohol 5 (19 mg, 0.05 mmol) and p-methoxybenzyl- 2,2,2-trichloroacetimidate (72 mg, 0.26 mmol) in ether (1.6 mL) cooled at −78˚C was added trifluoromethanesulfonic acid (1.5 μL, 0.018 mmol). After the addition was completed, the ice bath was removed and the reaction mixture was warmed to room temperature. After being stirred at rt for 2 h, the reaction mixture was quenched by the addition of saturated aqueous NaHCO3. The separated aqueous layer was extracted with Et2O (8 mL × 3). The combined organic layer was concentrated under reduced pressure. The residue oil was purified by column chromatography on silica gel (9:1 hexane/ethyl acetate) to give 6 (12 mg, 49%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.31 (d, J = 6.8 Hz, 2H), 6.92 (d, J = 6.8 Hz, 2H), 6.88 (d, J = 8.4 Hz, 1H), 5.43 (q, J = 6.8 Hz, 1H), 4.57 (d, J = 11.2 Hz, 1H), 4.42 (d, J = 11.2 Hz, 1H), 3.90 (m, 1H), 3.87 (s, 3H), 3.81 (s, 3H), 3.76 (d, J = 9.2 Hz, 1H), 2.33 (m, 2H), 2.21 (m, 1H), 1.90 (s, 3H), 1.67 (d, J = 6.8 Hz, 3H), 1.61 (s, 3H), 0.92 (s, 9H), 0.80 (d, J = 6.8 Hz, 3H), 0.03 (s, 3H), 0.01 (s, 3H).
Authentic sample (4b): p-methoxybenzyl ether 6 (12 mg, 0.02 mmol) was dissolved in a 5:3:12 mixture of HF・Py, Pyridine and THF (0.6 mL). The solution was stirred at 40˚C for 12 h, diluted with EtOAc (2 mL), and poured into saturated aqueous NaHCO3 (6 mL) cooled at 0˚C. The mixture was extracted with EtOAc (4 mL × 3). The combined extracts were washed with brine dried (Na2SO4), and concentrated. The residue oil was purified by column chromatography on silica gel (3:1 hexane/ethyl acetate) to give desired 4b (7 mg, 78%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.26 (d, J = 6.4 Hz, 2H), 6.91 (t, J = 6.4 Hz, 1H), 6.87 (d, J = 6.4 Hz, 2H), 5.43 (q, J = 6.8 Hz, 1H), 4.57 (d, J = 11.2 Hz, 1H), 4.45 (d, J = 11.2 Hz, 1H), 3.83 (m, 1H), 3.80 (s, 3H), 3.72 (s, 3H), 3.69 (m, 1H), 2.59 (m, 1H), 2.43 (m, 1H), 1.96 (m, 1H), 1.87 (s, 3H), 1.61 (d, J = 6.8 Hz, 3H), 1.59 (s, 3H), 0.66 (d, J = 6.8 Hz, 3H). HRMS (ESI-TOF) m/z: [M + H]+Calcd for C22H33O5 377.2322; Found 377.2298.
5.3. Synthesis of Organoboron Precursor 3
Alcohol 2 was prepared from 14 by deprotection of the second TBS ether using HF・Py as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.26 (d, J = 8.8 Hz, 2H), 6.91 (t, J = 6.4 Hz, 1H), 6.87 (d, J = 8.8 Hz, 2H), 4.89 (s, 1H), 4.87 (s, 1H), 4.54 (d, J = 11.2 Hz, 1H), 4.45 (d, J = 11.2 Hz, 1H), 3.92 (d, J = 10 Hz, 1H), 3.80 (s, 3H), 3.75 (s, 3H), 3.72 (m, 1H), 2.57 (m, 1H), 2.43 (m, 1H), 1.92 (m, 1H), 1.87 (s, 3H), 1.71 (s, 3H), 0.73 (d, J = 6.8 Hz, 3H).
Organoboron precursor (3): To a solution of HG-II catalyst (63 mg, 0.1 mmol) and dry toluene (6.5 mL) in a round-bottomed flask equipped with a reflux condenser was added alcohol 2 (28 mg, 0.08 mmol) and vinyl boronate 16c (66 μL, 0.38 mmol). The solution was refluxed for roughly overnight. The mixture was then concentrated, and the products were purified by SiliaMetSÒ DMT. The filtrate was concentrated, and then the residue was separated by HPLC [Develosil ODS-HG-5 (Æ 10 × 250 mm), flow rate 4 mL/min, 60% - 80% aqMeCN, 40 min] to give 3 (5.3 mg, 14%; tR = 19.5 min,). 1H NMR (400 MHz, CDCl3): δ 7.25 (d, J = 8.4 Hz, 2H), 6.88 (t, J = 7.6 Hz, 1H), 6.86 (d, J = 8.4 Hz, 2H), 5.28 (s, 1H), 4.52 (d, J = 10.8 Hz, 1H), 4.44 (d, J = 10.8 Hz, 1H), 3.88 (d, J = 7.2 Hz, 1H), 3.80 (s, 3H), 3.75 (s, 3H), 3.71 (m, 1H), 2.57 (m, 1H), 2.43 (m, 1H), 1.97 (m, 1H), 1.95 (s, 3H), 1.86 (s, 3H), 1.26 (s, 12H), 0.74 (d, J = 7.2 Hz, 3H). HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C27H41BO7Na 511.2837; Found 511.2814.
5.4. Synthesis of 4b by the Rapid C-Methylation (Cold Conditions)
Iodomethane (0.25 μL, 4 mmol) dissolved in N,N-dimethylformamide (19.75 μL, 0.2 M) was added to a solution of Pd2(dba)3 (1.8 mg, 2.0 μmol), tri(o-tolyl)phosphine (2.4 mg, 7.9 μmol), potassium carbonate (2.8 mg, 20 μmol), and 3 (2.0 mg, 4.0 μmol) in anhydrous N,N-dimethylformamide (200 μL). The resulting mixture was heated at 70˚C for 5 min. The reaction solution was evaporated in vacuo and purified on ODS, eluting with MeCN-H2O (7:3) to afford 4b (1.2 mg) in78% yield.
5.5. Synthesis of 11C-Labeled Dtda Methyl Ester 4a
[11C]CH3I was transported into a vial where the organoboronprecuesor 3 (2.0 mg), Pd2(dba)3 (1.8 mg), P(o- tolyl)3 (2.4 mg), K2CO3 (2.8 mg) were dissolved in anhydrous DMF (200 μL) at room temperature. After the solution was heated at 70˚C for 5 min, the mixture was poured into a separated vial. Then the reaction vial was washed with MeCN/water (70:30, 800 μL) and the washing was added to the above mixture solution. The resulting mixture was purified to reverse-phase HPLC [Develosil ODS-HG-5 (Æ 10 × 250 mm), flow rate 5.0 mL/min, 70% aqMeCN, detection at 223 nm, 3a: tR = 11.4 min]. The desired fractions were collected in a flask containing 25% ascorbic acid (50 μL), and the organic solvent was removed under reduced pressure.
References
- Nakao, Y., Yoshida, W.Y., Takada, Y., Kimura, J., Yang, L., Susan, L.M. and Scheuer, P.J. (2004) Kulokekahilide-2, a Cytotoxic Depsipeptide from a Cephalaspidean Mollusk Philinopsisspeciosa. Journal of Natural Products, 67, 1332- 1340. http://dx.doi.org/10.1021/np049949f
- Suenaga, K., Mutou, T., Shibata, T., Itoh, T., Kigoshi, H. and Yamada, K. (1996) Isolation and Stereostructure of Aurilide, a Novel Cyclodepsipeptide from the Japanese Sea Hare Dolabellaauricularia. Tetrahedron Letters, 37, 6771-6774. http://dx.doi.org/10.1016/S0040-4039(96)01464-5
- Suenaga, K., Mutou, T., Shibata, T., Itoh, T., Fujita, T., Takada, N., Hayamizu, K., Takagi, M., Irifune, T., Kigoshi, H. and Yamada, K. (2004) Aurilide, a Cytotoxic Depsipeptide from the Sea Hare Dolabellaauricularia: Isolation, Structure Determination, Synthesis, and Biological Activity. Tetrahedron, 60, 8509-8527. http://dx.doi.org/10.1016/j.tet.2004.06.125
- Tripathi, A., Puddick, J., Prinsep, M.R., Rottmann, M. and Tan, L.T. (2010) Lagunamides A and B: Cytotoxic and Antimalarial Cyclodepsipeptides from the Marine Cyanobacterium Lyngbyamajuscule. Journal of Natural Products, 73, 1810-1814. http://dx.doi.org/10.1021/np100442x
- Umehara, M., Negishi, T., Maehara, Y., Nakao, Y. and Kimura, J. (2013) Stereochemical Analysis and Cytotoxicity of Kulokekahilide-2 and Its Analogues. Tetrahedron, 69, 3045-3053. http://dx.doi.org/10.1016/j.tet.2013.01.089
- Nagarajan, M., Maruthanayagam, V. and Sundararaman, M. (2012) A Review of Pharmacological and Toxicological Potentials of Marine Syanobacterial Metabolites. Journal of Applied Toxicology, 32, 153-185. http://dx.doi.org/10.1002/jat.1717
- Sato, S., Murata, M., Orihara, T., Shirakawa, T., Suenaga, K., Kigoshi, H. and Uesugi, M. (2011) Marine Natural Product Aurilide Activates the OPA1-Mediated Apoptosis by Binding to Prohibitin. Chemistry & Biology, 18, 131-139. http://dx.doi.org/10.1016/j.chembiol.2010.10.017
- Williams, P.G., Yoshida, W.Y., Quon, M.K., Moore, R.E. and Paul, V.J. (2003) The Structure of Palau’amide, a Potent Cytotoxin from a Species of the Marine Cyanobacterium Lyngbya. Journal of Natural Products, 66, 1545-1549. http://dx.doi.org/10.1021/np034001r
- Sugiyama, H., Watanabe, A., Teruya, T. and Suenaga, K. (2009) Synthesis of Palau’amide and Its Diastereomers: Con- firmation of Its Stereostructure. Tetrahedron Letters, 50, 7343-7345. http://dx.doi.org/10.1016/j.tetlet.2009.10.059
- Rowland, M. (2012) Microdosing: A Critical Assessment of Human Data. Journal of Pharmaceutical Sciences, 101, 4067-4074. http://dx.doi.org/10.1002/jps.23290
- Suzuki, M., Doi, H., Bjӧrkman, M., Andersson, Y., Långstrӧm, B., Watanabe, Y. and Noyori, R. (1997) Rapid Coupling of Methyl Iodide with Aryltributylstannanes Mediated by Palladium(0) Complexes: A General Protocol for the Synthesis of 11CH3-Labeled PET Tracers. Chemistry―A European Journal, 3, 2039-2042. http://dx.doi.org/10.1002/chem.19970031219
- Doi, H., Ban, I., Nonoyama, A., Sumi, K., Kuang, C., Hosoya, T., Tsukada, H. and Suzuki, M. (2009) Palladium(0)- Mediated Rapid Methylation and Fluoromethylation on Carbon Frameworks by Reacting Methyl and Fluoromethyl Iodide with Aryl and AlkenylBoronic Acid Esters: Useful for the Synthesis of [11C]CH3-C- and [18F]FCH2-C-Con- taining PET Tracers (PET=Positron Emission Tomography). Chemistry―A European Journal, 15, 4165-4171. http://dx.doi.org/10.1002/chem.200801974
- Kanazawa, M., Furuta, K., Doi, H., Mori, T., Minami, T., Ito, S. and Suzuki, M. (2011) Synthesis of an Acromelic Acid A Analog-Based 11C-Labeled PET Tracer for Exploration of the Site of Action of Acromelic Acid A in Allodynia Induction. Bioorganic & Medicinal Chemistry Letters, 21, 2017-2020. http://dx.doi.org/10.1016/j.bmcl.2011.02.018
- Takashima-Hirano, M., Takashima, T., Katayama, Y., Wada, Y., Sugiyama, Y., Watanabe, Y., Doi, H. and Suzuki, M. (2011) Efficient Sequential Synthesis of PET Probes of the COX-2 Inhibitor [11C]Celecoxib and Its Major Metabolite [11C]SC-62807 and in Vivo PET Evaluation. Bioorganic & Medicinal Chemistry, 19, 2997-3004. http://dx.doi.org/10.1016/j.bmc.2011.03.020
- Suzuki, M., Takashima-Hirano, M., Koyama, H., Yamaoka, T., Sumi, K., Nagata, H., Hidaka, H. and Doi, H. (2012) Efficient Synthesis of [11C]H-1152, a PET Probe Specific for Rho-Kinases, Highly Potential Targets in Diagnostic Medicine and Drug Development. Tetrahedron, 68, 2336-2341. http://dx.doi.org/10.1016/j.tet.2012.01.033
- Suzuki, M., Takashima-Hirano, M., Ishii, H., Watanabe, C., Sumi, K., Koyama, H. and Doi, H. (2014) Synthesis of 11C-Labeled Retinoic Acid, [11C]ATRA, via an Alkenylboron Precursor by Pd(0)-Mediated Rapid C-[11C]Methylation. Bioorganic & Medicinal Chemistry Letters, 24, 3622-3625. http://dx.doi.org/10.1016/j.bmcl.2014.05.041
- Suzuki, M., Doi, H., Koyama, H., Zhang, Z., Hosoya, T., Onoe, H. and Watanabe, Y. (2014) Pd0-Mediated Rapid Cross- Coupling Reactions, the Rapid C-[11C]Methylations, Revolutionarily Advancing the Syntheses of Short-Lived PET Molecular Probes. The Chemical Record, 14, 516-541. http://dx.doi.org/10.1002/tcr.201400002
- Evans, D.A., Dow, R.L., Shih, T.L., Takacs, J.M. and Zahler, R. (1990) Total Synthesis of the Polyether Antibiotic Ionomycin. Journal of the American Chemical Society, 112, 5290-5313. http://dx.doi.org/10.1021/ja00169a042
- Chatterjee, A.K., Choi, L., Sanders, D.P. and Grubbs, R.H. (2003) A General Model for Selectivity in Olefin Cross Metathesis. Journal American Chemical Society, 125, 11360-11370. http://dx.doi.org/10.1021/ja0214882
- Dubowchik, G.M., Vrudhula, V.M., Dasgupta, B., Ditta, J., Chen, T., Sheriff, S., Sipman, K., Witmer, M., Tredup, J., Vyas, D.M., Verdoorn, T.A., Bollini, S. and Vinitsky, A. (2001) 2-Aryl-2,2-difluoroacetamide FKBP12 Ligands: Synthesis and X-Ray Structural Studies. Organic Letters, 3, 3987-3990. http://dx.doi.org/10.1021/ol0166909
- Trnka, T.M. and Grubbs, R.H. (2001) The Development of L2X2Ru=CHR Olefin Metathesis Catalysts: An Organo- metallic Success Story. Accounts of Chemical Research, 34, 18-29. http://dx.doi.org/10.1021/ar000114f
- Scholl, M., Ding, S., Lee, C.W. and Grubbs, R.H. (1999) Synthesis and Activity of a New Generation of Ruthenium- Based Olefin Metathesis Catalysts Coordinated with 1,3-Dimesityl-4,5-dihydroimidazol-2-ylidene Ligands. Organic Letters, 1, 953-956. http://dx.doi.org/10.1021/ol990909q
- Garber, S.B., Kingsbury, J.S., Gray, B.L. and Hoveyda, A.H. (2000) Efficient and Recyclable Monomeric and Dendritic Ru-Based Metathesis Catalysts. Journal American Chemical Society, 122, 8168-8179. http://dx.doi.org/10.1021/ja001179g
- Morrill, C. and Grubbs, R.H. (2003) Synthesis of Functionalized Vinyl Boronates via Ruthenium-Catalyzed Olefin Cross- Metathesis and Subsequent Conversion to Vinyl Halides. Journal of Organic Chemistry, 68, 6031-6034. http://dx.doi.org/10.1021/jo0345345
- Morrill, C., Funk, W. and Grubbs, R.H. (2004) Synthesis of Tri-Substituted Vinyl Boronates via Ruthenium-Catalyzed Olefin Cross-Metathesis. Tetrahedron Letters, 45, 7733-7736. http://dx.doi.org/10.1016/j.tetlet.2004.08.069
- Funk, T., Efskind, J. and Grubbs, R.H. (2005) Chemoselective Construction of Substituted Conjugated Dienes Using an Olefin Cross-Metathesis Protocol. Organic Letters, 7, 187-190. http://dx.doi.org/10.1021/ol047929z
- Fuwa, H., Saito, A. and Sasaki, M. (2010) A Concise Total Synthesis of (+)-Neopeltolide. Angewandte Chemie International Edition, 49, 3041-3044. http://dx.doi.org/10.1002/anie.201000624
- Fuwa, H., Suzuki, T., Kubo, H., Yamori, T. and Sasaki, M. (2011) Total Synthesis and Biological Assessment of (-)- Exiguolide and Analogues. Chemistry―A European Journal, 17, 2678-2688. http://dx.doi.org/10.1002/chem.201003135
- Coquerel, Y. and Rodriguez, J. (2008) Microwave-Assisted Olefin Metathesis. European Journal of Organic Chemistry, 7, 1125-1132. http://dx.doi.org/10.1002/ejoc.200700696
- Michaut, A., Boddaert, T., Coquerel, Y. and Rodriguez, J. (2007) Reluctant Cross-Metathesis Reactions: The Highly Beneficial Effect of Microwave Irradiation. Synthesis, 18, 2867-2871. http://dx.doi.org/10.1055/s-2007-983825
- Fuwa, H., Noto, K. and Sasaki, M. (2010) Stereoselective Synthesis of Substituted Tetrahydropyrans via Domino Olefin Cross-Metathesis/Intramolecular Oxa-Conjugate Cyclization. Organic Letters, 12, 1636-1639. http://dx.doi.org/10.1021/ol100431m
- Suzuki, M., Sumi, K., Koyama, H., Siqin, Hosoya, M., Takashima-Hirano, M. and Doi, H. (2009) Pd0-Mediated Rapid Coupling between Methyl Iodide and Heteroarylstannanes: An Efficient and General Method for the Incorporation of a Positron-Emitting 11C Radionuclide into Heteroaromatic Frameworks. Chemistry A European Journal, 15, 12489-12495. http://dx.doi.org/10.1002/chem.200901145
NOTES

*Corresponding author.