International Journal of Organic Chemistry
Vol.4 No.1(2014), Article ID:43422,11 pages DOI:10.4236/ijoc.2014.41005
Diastereoselective Catalytic Hydrogenation of Schiff Bases of N-Pyruvoyl-(S)-Proline Esters
Toratane Munegumi1*, Shokichi Ohuchi2, Kaoru Harada2
1Department Science Education, Naruto University of Education, Naruto, Japan
2Department of Chemistry, University of Tsukuba, Tsukuba, Japan
Email: *tmunegumi@naruto-u.ac.jp
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 14 December 2013; revised 20 January 2014; accepted 29 January 2014
ABSTRACT
Diastereoselective catalytic hydrogenation of pyruvic acid esters, amides, and their Schiff bases has been well studied over a long period to show that proline is one of the most effective chiral auxiliaries. Proline derivatives have been used as auxiliaries in the diastereoselective catalytic hydrogenation of pyruvamide Schiff bases. The diastereoselective hydrogenation resulted in up to a 78% enantiomeric excess of the amino acid derived from the hydrolysis of the dipeptide products. The chelation hypothesis explains the stereochemistry of the catalytic hydrogenation using (S)-proline esters in the amide moiety and the two chiral centers in the amide and Schiff base moieties.
Keywords
Diastereoselective; Catalytic Hydrogenation; Proline; Pyruvamide; Schiff Base

1. Introduction
Pyruvic acid is an important precursor for the biosynthesis of amino acids [1] [2] . In the pathway of amino acid biosynthesis, pyruvic acid possessing no chiral centers can be derived via a pyridoxal at the active center of the enzymes involved [1] [2] . This chiral induction may be explained by the role of the chiral active center of enzymes composed of a homochiral protein. The pyruvic acid intermediates and transition states always form a chiral complex at the active center of the enzyme proteins. The newly formed chiral amino acid and the enzyme active center including pyridoxal form a type of diastereomeric intermediate, specifically an L-amino acid plus L-protein and a D-amino acid plus L-protein. The former intermediate is often more stable than the latter and yields L-amino acids. The diastereomeric intermediate formation from pyruvic acid and L-protein is recognized as a diastereoselective reaction. The diastereoselectivity [3] is almost exclusive.
On the other hand, the diastereoselective hydrogenation of biosynthesis has been imitated by organic synthesis [4] . Model reactions using pyridoxal in vitro and catalytic hydrogenation exemplify this synthesis. Diastereoselective catalytic hydrogenation of pyruvic acid and related compounds has long been investigated and demonstrates that the chelation of a vicinal C=O plus C=N group or two vicinal C=O groups to palladium catalysts (Figure 1) affects higher diastereoselectivity [5] -[20] . The chiral center of pyruvic acid amide moiety [11] [15] -[20] can influence the configuration of the newly formed chiral center. In particular, proline and its derivatives as chiral auxiliaries have demonstrated their effectiveness [18] [20] in diastereoselective catalytic hydrogenation.
In this study, the diastereoselective hydrogenation process follows the formation of Schiff bases having a chiral center in the pyruvic acid amide moiety. We used (S)-proline derivatives as chiral auxiliaries in the pyruvic acid amide moiety and benzylamine in the Schiff base moiety. We also examined diastereoselective hydrogenation using chiral 1-phenylethylamines in the Schiff base moiety to examine the effect of a double chiral auxiliary. (S)-Proline is the same as L-proline, and we use R, S-nomenclature in preference to D, L-nomenclature to indicate the stereochemistry of chiral amines used in this study. Preparation and hydrogenation of substrates were conducted as shown in Figures 2 and 3.
2. Results and Discussion
2.1. Diastereoselective Catalytic Hydrogenation Using Achiral Benzylamine
Results of the catalytic hydrogenation of Schiff bases of N-pyruvoyl-(S)-proline esters using two methods: A (Pd(OH)2-C) and B (Pd-C and then Pd(OH)2-C) are shown in Table 1.
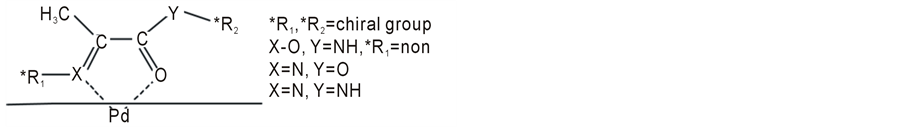
Figure 1. The chelation hypothesis of the diastereoselective catalytic hydrogenation.
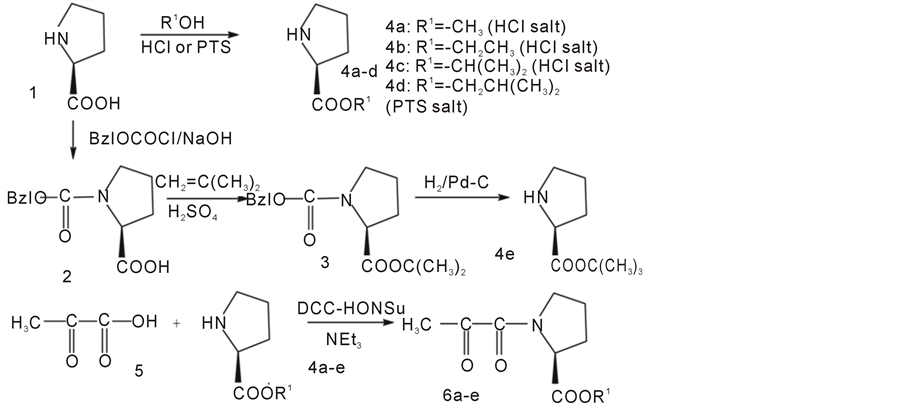
Figure 2. Preparation of N-pyruvoyl-(S)-proline esters. PTS: p-toluenesulfonic acid; Bzl: benzyl; DCC: N, N'-dicyclohexylcarbodiimide; HOSu: N-hydroxy-succinimide; NEt3: triethylamine.
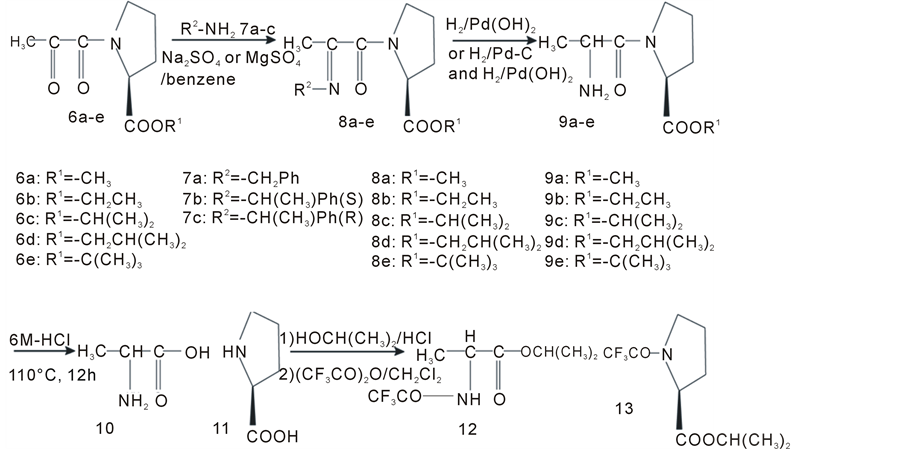
Figure 3. Preparation of Schiff bases of N-pyruvoyl-(S)-proline esters and their reaction process after hydrogenation.
The configuration of enantiomeric excess (e.e.) of alanine was (S)-configuration (20% to 69%). With the exception of the 2-methyl-2-propyl ester (No. 34 to 36), the e.e. of alanine did not cause a big difference (44% - 69%), because of the size of the ester moiety. The reactions gave a lower e.e. as follows: 22% in 2-propanol, 23% tetrahydrofuran, and 28% in ethyl acetate.
Although the e.e. obtained using method A was similar to that obtained using method B, the yield of the reaction using method A was slightly smaller than that using method B. Comparing entry No. 1 with 2, and entry No. 5 with 6, and so on provides typical examples. Yield of lactic acid is shown (10% to 40%) for several reactions. Lactic acid is formed by the hydrogenation of the remaining N-pyruvoyl-(S)-proline esters without the formation of Schiff bases.
2.2. Diastereoselective Catalytic Hydrogenation Using Achiral Benzylamine at Different Temperatures
Table 2 shows the results of asymmetric hydrogenation of Schiff bases of N-pyruvoyl-(S)-proline esters by method A (Pd(OH)2-C) at different temperatures.
The configuration of alanine in enantiomeric excess (e.e.) was (S)-configuration (23% to 78%). With the exception of 2-methyl-2-propyl ester (No. 10, 12), the e.e. of alanine did not cause a big difference (40% to 78%) because of the size of the ester moiety. The highest e.e.s from 8a and 8c were 75% in 2-propanol at –10˚C (No. 6) and 78% in ethanol at –10˚C (No. 13), respectively. The yield of the reactions was lower at lower temperatures and in 2-propanol, tetrahydrofuran, and ethyl acetate.
2.3. Plausible Stereochemistry of Diastereoselective Catalytic Hydrogenation of Schiff Bases of N-Pyruvoyl-(S)-Proline Esters
From the results shown in Tables 1 and 2, a plausible steric pathway is shown in Figure 4.
Substrates 8a-e have two main rotatable bonds (C–C bond and C–N bond) as shown in Figure 4. There are four important conformers, including s-cis [21] and s-trans forms. These conformers may form intermediates (a-1), (b), (c), or (d) on the palladium catalyst. There is the equilibrium between the stable chelation intermediate (a-1) and the unstable intermediate (b), and between the stable intermediates (c) and the unstable intermediate (d). Intermediates (a-1) and (c) will be in a higher proportion than intermediates (b) and (d). The lower proportion of intermediates (b) and (d) results from their larger steric repulsion. Important conformers (a-1) and (c) can be hydrogenated to give (S)-alanyl-(S)-proline esters and (R)-alanyl-(S)-proline esters, respectively. The products

Table 1. Diastereoselective catalytic hydrogenation of Schiff bases (R1=Bzl) of N-pyruvoyl-(S)-proline esters at 30˚C.
a)2-PrOH, 2-propanol; THF, tetrahydrofuran; AcOEt, ethyl acetate; b)method A, H2/Pd(OH)2; method B, H2/Pd-C and then H2/Pd(OH)2-C; c)Yield of lactic acid in the bracket.
formed after passing through intermediate (a-1) can explain the actual results in Tables 1 and 2 better than that after passing through intermediate (c). Five-membered ring formation on palladium catalyst had been observed to make the chelation intermediate reasonable [22] .
2.4. Diastereoselective Catalytic Hydrogenation Using Chiral 1-Phenylethylamines
Table 3 shows the results of diastereoselective catalytic hydrogenation of chiral Schiff bases of N-pyruvoyl-(S)- proline esters in methanol.
Table 2. Diastereoselective catalytic hydrogenation of Schiff bases (R1=Bzl) of N-pyruvoyl-(S)-proline esters at different temperatures by method A.
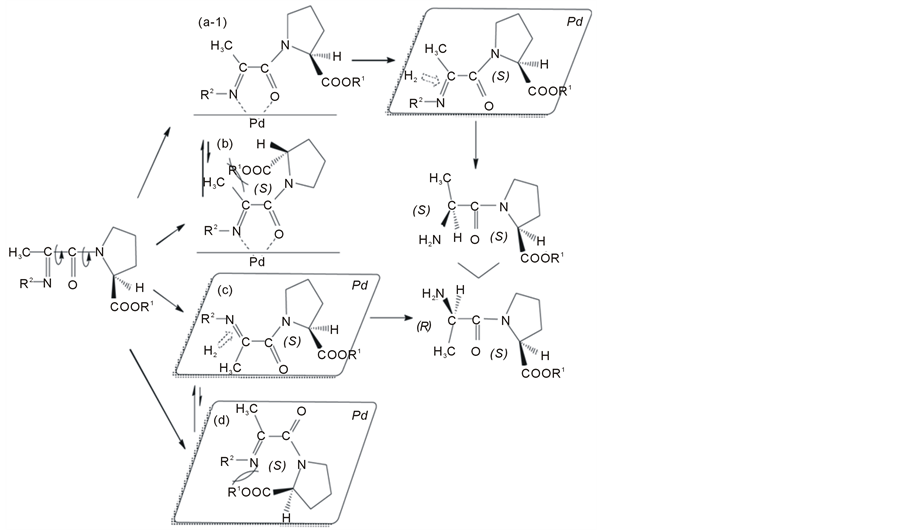
Figure 4. Plausible stereochemical course in the diastereoselective hydrogenation of Schiff bases of N-pyruvoyl-(S)-proline esters and benzylamine.
The configuration of alanine in enantiomeric excess (e.e.) was (S)-configuration. The e.e. was always larger when the configuration of 1-phenylethylamine was (R), rather than when the configuration of 1-phenylethylamine was (S). For example, No. 3 and No. 4 gave 67% and 54%, respectively. The results suggest that (R)- 1-phenylethylamine produces more stable intermediates, which yield (S)-alanyl-(S)-proline esters, than (S)- 1-phenylethylamine.
Figure 5 shows a plausible stereochemical pathway for the diastereoselective catalytic hydrogenation of Schiff bases between chiral amines and N-pyruvoyl-(S)-proline esters.
Table 3. Diastereoselective catalytic hydrogenation of chiral Schiff bases of N-pyruvoyl-(S)-proline esters in methanol.
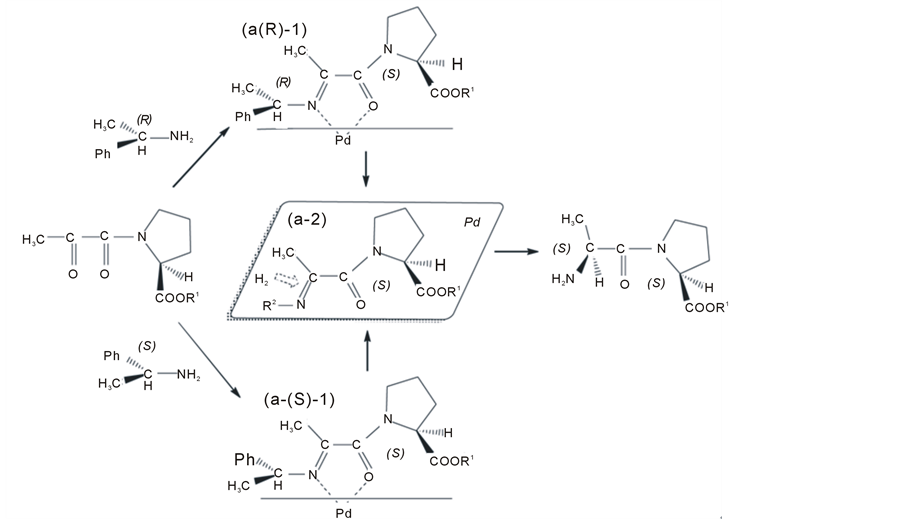
Figure 5. Plausible stereochemical course in the diastereoselective hydrogenation of Schiff bases of N-pyruvoyl-(S)-proline esters and benzylamine.
Figure 5 modifies Figure 4 to extend the chelation hypothesis to explain the double stereo-induction using two chiral centers.
N-Pyruvoyl-(S)-proline esters react with (R)-1-phenyl-ethylamine or (S)-1-phenylethylamine to afford their Schiff bases. These Schiff bases form chelation intermediates (a-(R)-1) and (a-(S)-1) during the hydrogenation. Schiff bases of N-pyruvoyl-(S)-proline esters with achiral amine and chiral amines preferentially yielded (S)- alanyl as shown in Tables 1-3. These results suggest that the stereochemistry of the catalytic hydrogenation is mainly controlled by the chirality of the proline ester moiety as distinct from the chiral amine moiety in Schiff bases. However, the e.e. of alanine moiety depends on the configuration of the amine moiety as shown in Table 3. (R)-1-Phenylethylamine yielded a higher e.e. of (S)-alanine than (S)-1-phenylethylamine did. The results may be caused by the difference in the orientation of the substituted group in the chelation intermediates. The chelation hypothesis suggests that the substituted groups of the amine moiety were in different positions in the intermediates (a-(R)-1) and (a-(S)-1).
Intermediate (a-(R)-1) directs the methyl group of the amine moiety to the opposite side, the phenyl group to the front side, and the hydrogen to the down side. The intermediate (a-(S)-1) directs the methyl group of the amine moiety to the front side, the phenyl group to the opposite side, and the hydrogen to the down side. Because phenyl is relatively more bulky than methyl, and carboxyl ester is relatively more bulky than hydrogen, we can compare the total bulkiness between the front and the opposite sides in the chelation intermediates.
Intermediate (a-(R)-1) directs methyl plus hydrogen groups to the opposite side and phenyl plus carboxyl ester groups to the front side. Therefore, the opposite side is the less bulky side and is more likely to be adsorbed as an intermediate (a-2) and hydrogenated on the catalyst. The intermediate (a-(S)-1) directs phenyl plus hydrogen groups to the opposite side and methyl and carboxyl ester groups to the front side. In that case, although the chirality of proline ester moiety controls the stereochemistry of the chiral induction, the adsorption from both sides competes to decrease the e.e. of alanine.
The conformation of the amine moiety in this chelation intermediate is different from the conformation proposed for Schiff bases of pyruvic esters [6] . It may be because the amine moieties in the chelation intermediates (a-(R)-1 and a-(S)-1) have narrower space from the catalyst than the amine moiety of Schiff bases of pyruvic acid esters have.
3. Experimental
3.1. Instrumentation
Gas chromatographic separation of enantiomeric alanine obtained by hydrolysis of hydrogenation products was achieved using a Hitachi 163 gas chromatograph equipped with a chiral capillary column (Chirasil-Val, 25 m ´ 0.3 mm I.D.). Nitrogen was used as the carrier gas at 30 mL/min. The column oven temperature was programmed to increase from 80˚C to 170˚C at 4˚C/min. A flame ionization detector was used.
Yield of the newly formed chiral amino acid was determined by using a high-performance liquid chromatography (HPLC) system composed of a Jasco UVdec-100-V UV spectrophotometer and Jasco TRI Rotor-V flow pump, equipped with a reversed phase C-18 column (TSK Inertsil ODS, TOSO, Tokyo, Japan) (4.6 mm ´ 250 mm). Water alone was used as the eluent. The flow rate was controlled at 0.5 ml/min. The spectrophotometer was set to detect absorption at 210 nm. The peaks on the chromatograms were integrated using a SIC Chromatocorder.
A Hitachi model 260-50 infrared spectrophotometer (Hitachi, Tokyo, Japan) was used to record the infrared spectra. A Jasco DIP-181 Digital Polarimeter was used to determine the optical rotation of the compounds synthesized from L-aspartic acid. An EX-270 NMR system (JEOL, Tokyo, Japan) was used to determine 1H NMR spectra.
3.2. Chemicals
Solvents, (S)-proline (1), pyruvic acid (5), and benzylamine (7a) were purchased from Wako Pure Chemical Industry (Osaka, Japan). Optically active amines, (S)-1-phenylethylamine (7b, [a]D25 = +39.0 (neat), 96% e.e.) and (R)-1-phenylethylamine (7b, [a]D25 = –39.0 (neat), 96% e.e.) were purchased from Aldrich (Milwaukee, USA). N,N'-Dicyclohexylcarbodiimide and N-hydroxysuccinimide were purchased from Watanabe Chemical Industries (Hiroshima, Japan). 5% Palladium on charcoal was purchased from Nippon Engelhard. 10% Palladium hydroxide on charcoal was used.
3.2.1. Preparation of N-Benzyloxycarbonyl-(S)-Proline (2)
(S)-Proline (1) (11.5 g, 100 mmol) was dissolved in 0.5 M-sodium hydrogen carbonate solution (300 ml). To the cooled solution, benzyloxycarbonyl chloride (18.8 g, 110 mmol) was added dropwise. After the resulting suspension was stirred for 24 h at room temperature, the reaction mixture was extracted with ether. The remaining aqueous layer was acidified with concentrated hydrochloric acid to a pH of 2. The acidified aqueous solution was extracted with ethyl acetate. The obtained ethyl acetate layer was extracted with brine, and the remaining organic layer was dried with anhydrous magnesium sulfate. After removal of the magnesium sulfate by filtration, the filtrate was evaporated in vacuo, and the resulting precipitate was recrystallized with ethyl acetate to give 22.5 g (90%). M.p. 74˚C - 75˚C. [a]D20 –38.0 (c 1.09, methanol).
3.2.2. (S)-Proline 2-Methyl-2-Propyl Ester (3)
Sulfuric acid (2 ml) was added to a solution containing N-benzyloxycarbonyl-(S)-proline (2) (6.00 g, 24.0 mmol) and dichloromethane (50 ml) in a pressure bottle. To the resulting solution cooled in a dry ice-acetone mixture was added 2-butene liquid that had been cooled below boiling point until the weight of the reaction mixture increased by 5 g. The reaction mixture in the sealed pressure bottle was stirred for 3 days at room temperature. The reaction mixture was neutralized with sodium hydrogen carbonate and extracted with water. The resulting organic layer was dried with anhydrous magnesium sulfate and was evaporated in vacuo to give an oily N-benzyloxycarbonyl-(S)-proline 2-methyl-2-propyl ester (6.66 g, 91.0%). [a]D20 –45.3 (c 1.00, ethyl acetate).
3.2.3 Preparation of N-Pyruvoyl-(S)-Proline Methyl Ester (6a)
A suspension comprising (S)-Proline (1) (8.00 g, 69.4 mmol) and methanol (120 ml) was saturated with hydrogen chloride in an ice bath. The resulting suspension was stirred for 2 days, and then solvents were evaporated in vacuo to give (S)-proline methyl ester hydrogen chloride (4a) (11.80 g, 100%). [a]D20 –45.1 (c 1.00, methanol). (S)-Proline methyl ester hydrogen chloride (4a) (8.03 g, 48.4 mmol) was neutralized with triethylamine (4.98 g, 48.4 mmol) in ethyl acetate (40 ml) in an ice bath. The solution was added to a mixture of pyruvic acid (5) (4.26 g, 48.4 mmol), dicyclohexylcarbodiimide (11.0 g, 53.3 mmol) and N-hydroxysuccinimide (5.57 g, 48.4 mmol) in an ice bath. The reaction was continued for 3 h in the ice bath and for a further 18h at room temperature. After removal of N,N'-dicyclohexylurea, the reaction mixture was evaporated in vacuo to give an oily product. The oily product was loaded onto a silica gel column and chromatography conducted using a developing agent composed of benzene-ethyl acetate (12:1 (v/v)) to afford an oily pure product 3.17 g (33%). 1H-NMR (CDCl3, d): 1.50 - 2.50 (4H, m), 2.35 (3H, d), 3.64 (3H, s), 3.50 (2H, t), 4.75 (1H, m). IR (liquid, cm–1): 1740, 1710, 1640 (C=O). [a]D22 –75.1 (c 0.86, ethyl acetate). Elemental analysis: Calcd. for C9H13NO4: C, 54.65%; H, 6.83%; N, 6.76%. Found: C, 54.26%; H, 6.57%; N, 6.73%.
3.2.4. Preparation of N-Pyruvoyl-(S)-Proline Ethyl Ester (6b)
(S)-Proline ethyl ester hydrochloride (4b) was prepared from (S)-proline (1) (8.00 g, 69.4 mmol) in ethanol (120 ml) in a manner similar to the preparation of methyl ester as an oily product (12.5 g, 100%). [a]D20 –46.1 (c 0.41, methanol). In a manner similar to that for the preparation of the methyl ester, N-pyruvoyl-(S)-proline ethyl ester was prepared (5.70 g, 55%) from corresponding (S)-proline ethyl ester (4b) (8.69 g, 48.4 mmol). 1H-NMR (CDCl3, d): 1.25 (3H, t), 1.50 - 2.50 (4H, m), 2.30 (3H, d), 3.60 (2H, m), 4.05 (2H, m), 4.70 (1H, m). IR (liquid, cm–1): 1740, 1710, 1640 (C=O). [a]D22 –75.9 (c 0.99, ethyl acetate). Elemental analysis: Calcd. for C10H15NO4: C, 56.32%; H, 7.09%; N, 6.56%. Found: C, 56.02%; H, 7.13%; N, 6.47%.
3.2.5. Preparation of N-Pyruvoyl-(S)-Proline 2-Propyl Ester (6c)
(S)-Proline-2-propyl ester hydrochloride (4c) was prepared from (S)-proline (1) (8.00 g, 69.4 mmol) in 2-propanol (120 ml) in a manner similar to that used for the preparation of the oily methyl ester (13.4 g, 100%). [a]D20 –47.8 (c 0.91, methanol). In a manner similar to that used for the preparation of the methyl ester, N-pyruvoyl-(S)-proline isopropyl ester (6c) was prepared (6.07 g, 55%) from corresponding (S)-proline 2-propyl ester (9.37 g, 48.4 mmol) in 2-propanol (120 mL). 1H-NMR (CDCl3, d): 1.15 - 1.30 (6H, dd), 1.80 - 2.25 (4H, br), 2.32 - 2.38 (3H, d), 3.40 - 3.90 (2H, m), 4.20 - 4.50 (1H, m). IR (liquid, cm–1): 1740, 1710, 1630 (C=O). [a]D22 –68.1 (c 1.09, ethyl acetate). Elemental analysis: Calcd. for C11H17NO4: C, 58.13%; H, 7.54%; N, 6.16%. Found: C, 57.88%; H, 7.60%; N, 6.22%.
3.2.6. Preparation of N-Pyruvoyl-(S)-Proline 2-Methyl-1-Propyl Ester (6d)
(S)-Proline (1) (5.76 g, 50.0 mmol), p-toluenesulfonic acid monohydrate (10.45 g, 55.0 mmol) and 2-methyl- 1-propanol (37.4 g, 500 mmol) were refluxed together in benzene (180 ml) for 12h with the azeotropic removal of water using a Dean-Stark apparatus. After refluxing, proline spot (Rf = 0.21) disappeared, but a product spot (Rf = 0.52) clearly appeared after thin layer chromatography (developing solvent: 1-butanol-acetic acid-water (volumetric ratio: 4-1-2) of the reaction mixture, which was evaporated in vacuo to give an oily product (4d).
A benzene solution (100 ml) of the product (4d) was washed three times with saturated sodium hydrogen carbonate. The resulting benzene layer was dried with anhydrous magnesium sulfate and evaporated to give an oily product. A part of the product (1.01 g, 5.9 mmol: free ester) was dissolved with pyruvic acid (0.52 g, 5.9 mmol) in 10 ml ethyl acetate. To the cooled solution was added N-hydroxysuccinimide (0.78 g, 6.8 mmol) and dicyclohexylcarbodiimide (1.34 g, 6.5 mmol) in ethyl acetate (10 ml). After stirring at 0˚C for 2 h and at room temperature for 11 h, the reaction mixture was filtered to give a pale yellow solution, which was washed with 0.5 M HCl (80 ml ´ 3), saturated sodium hydrogen carbonate (80 ml ´ 3), and brine. The resulted ethyl acetate layer was dried with anhydrous magnesium sulfate and evaporated in vacuo to give a brownish oily product, which was purified using flash chromatography (eluate: ethyl acetate-1-hexane (1 - 5)) to give an oil (0.61 g, 43%). 1H-NMR (CCl4, d): 0.87 - 0.99 (6H, d), 1.40 - 2.4 (4H, br), 2.29 - 2.35 (3H, d), 3.30 - 3.70 (1H, m), 3.73 - 3.84 (2H, d), 4.50 - 5.0 (1H, m). [a]D20 –51.8 (c 1.06, ethanol). Elemental analysis: Calcd. for C12H19NO4: C, 59.73%; H, 7.93%; N, 5.80%. Found: C, 59.33%; H, 7.93%; N, 6.18%.
3.2.7. Preparation of N-Pyruvoyl-(S)-proline 2-Methyl-2-Propyl Ester (6e)
N-Benzyloxycarbonyl-(S)-proline 2-methyl-2-propyl ester (3) (6.10 g, 20.0 mmol) dissolved in ethyl acetate (80 ml) was hydrogenolyzed over 5% palladium on charcoal under hydrogen. After removal of the catalyst by filtration, the filtrate was evaporated in vacuo to give an oily product, (S)-proline t-butyl ester of 2.15 g (63%). This compound was all used for the synthesis of N-pyruvoyl-(S)-proline t-butyl ester (6e) in the same manner as for the synthesis of others (6a-c). 1H-NMR (CDCl3, d): 1.44 (9H, s), 2.07 (4H, m), 2.40 (3H, d), 3.50 - 3.83 (2H, m), 4.60 - 4.83 (1H, m). IR (liquid, cm–1): 1740, 1720, 1640 (C = O). [a]D22 –70.8 (c 0.84, methanol). Elemental analysis: Calcd. for C12H19NO4: C, 59.73%; H, 7.93%; N, 5.80%. Found: C, 59.81%; H, 8.11%; N, 6.08%.
3.3. Hydrogenolysis of Schiff Bases
N-Pyruvoyl-(S)-proline methyl ester (6a) (43.8 mg, 0.22 mmol) was dissolved in benzene (7a) (1 ml), (S)-1- phenylethylamine (7b), or (R)-1-phenylethylamine (7c). To the solution was added anhydrous sodium sulfate (500 mg) for method A or magnesium sulfate (300 mg) for method B under an atmosphere of nitrogen. A dehydration reaction was conducted at room temperature for 24 h. After removal of sodium sulfate or magnesium sulfate by filtration, the filtrate was evaporated in vacuo, and the resulting yellow oil was redissolved in methanol. The hydrogenation and hydrogenolysis were conducted using alternative methods, A and B.
Method A is as follows: the hydrogenation and subsequent hydrogenolysis were conducted over 15 mg of 10% hydroxyl palladium on charcoal under a hydrogen atmosphere at 30˚C for 4 days. Method B is as follows: the hydrogenation was conducted over 30 mg of 5% palladium on charcoal at 30˚C for 24 h, and then the filtrate was subsequently hydrogenolyzed over 30 mg of 10% palladium hydroxide on charcoal at 30˚C for 3 h. The work-up for the reaction mixtures resulting from methods A and B was same. The filtrate obtained by the removal of catalyst was evaporated in vacuo to give a precipitate, which was hydrolyzed in 5 ml 6 M HCl at 110˚C for 12 h.
3.4. Sample Preparation for Analysis of Hydrolysates of Hydrogenolysis Mixture
The resulting hydrolysate of hydrogenolysis mixture was diluted with pure water to 10 ml. An amount of 2 ml of the solution was analyzed by HPLC to determine yield. The remaining aqueous solution was evaporated in vacuo to dryness leaving a residue, to which was added 5 ml of 1.5 M HCl/2-propanol, and the solution was refluxed for 3 h. The 2-propanol solution was evaporated in vacuo, and the obtained residue was redissolved in a mixture of dichloromethane (2 ml) and trifluoroacetic anhydride (1 ml). The solution was refluxed for 1 h and was evaporated in vacuo to give an oily product. The oily product was dissolved in ethyl acetate. After washing with 1 M HCl, 4% sodium hydrogen sulfate and brine, the ethyl acetate layer was dried with anhydrous magnesium sulfate, later removed, and evaporated to an oily product for enantiomeric separation by gas chromatography.
4. Conclusions
1) Diastereoselective catalytic hydrogenation of Schiff bases of N-pyruvoyl-(S)-proline esters was conducted to afford the highest e.e. of 78% of (S)-alanine from 8c in ethanol at –10˚C.
2) Preference of (S)-alanine-(S)-proline ester was explained by the chelation hypothesis based on the two-step adsorption process.
3) Double stereo-induction by the chiral centers in the (S)-proline ester and the chiral amine in the Schiff moiety was conducted, and the stereochemistry can be explained by the chelation hypothesis [5] .
References
- Voet, D. and Voet, J.G. (2010) Biochemistry. 4th Edition, Wiley, Hoboken.
- McMurry, J.E. and Begley, T.P. (2005) The Organic Chemistry of Biological Pathways. Roberts and Company Publishers, Englewood.
- Zimmerman, S.C. and Breslow, R. (1984) Asymmetric Synthesis of Amino Acids by Pyridoxamine Enzyme Analogs Utilizing General Base-Acid Catalysis. Journal of the American Chemical Society, 106, 1490-1491. http://dx.doi.org/10.1021/ja00317a054
- Michael, B.S. and March, J. (2007) March’s Advanced Organic Chemistry. 6th Edition, John Wiley & Son, Inc., Hoboken.
- Harada, K. and Munegumi, T. (1991) Reduction of C=X to CHXH by Catalytic Hydrogenation. In: B. M. Trost and I. Fleming, Eds., Comprehensive Organic Synthesis, Elsevier Science Ltd., Oxford, 139-158. http://dx.doi.org/10.1016/B978-0-08-052349-1.00222-5
- Matsumoto, K. and Harada, K. (1966) Stereoselective Syntheses of Optically Active Amino Acids from Menthyl Esters of α-Keto Acids. Journal of Organic Chemistry, 31, 1956-1958. http://dx.doi.org/10.1021/jo01344a064
- Harada, K. and Matsumoto, K. (1967) Sterically Controlled Syntheses of Optically Active Organic Compounds. V. Sterically Controlled Synthesis of Optically Active α-Amino Acids from α-Keto Acids by Reductive Amination. Journal of Organic Chemistry, 32, 1794-1800. http://dx.doi.org/10.1021/jo01281a020
- Harada, K. and Matsumoto, K. (1968) Sterically Controlled Syntheses of Optically Active Organic Compounds. VI. Solvent Effect in a Sterically Controlled Synthesis of Optically Active α-Amino Acids from α-Keto Acids by Hydrogenolytic Asymmetric Transamination. Journal of Organic Chemistry, 33, 4467-4470. http://dx.doi.org/10.1021/jo01276a036
- Harada, K. and Yoshida, T. (1970) A Temperature Effect in an Asymmetric Synthesis by Hydrogenolytic Asymmetric Transamination. Journal of the Chemical Society D: Chemical Communications, 1970, 1071-1072. http://dx.doi.org/10.1039/c29700001071
- Harada, K. and Yoshida, T. (1970) Syntheses of Optically Active α-Amino Acids from Esters of α-Keto Acids by Hydrogenolytic Asymmetric Transamination, a Solvent Effect. Bulletin of the Chemical Society of Japan, 43, 921-925. http://dx.doi.org/10.1246/bcsj.43.921
- Harada, K. and Matsumoto, K. (1971) Sterically Controlled Syntheses of Optically Active Organic Compounds. XIV. Syntheses of Dipeptides from N-(α-Ketoacyl)-α-Amino Acid Esters. Bulletin of the Chemical Society of Japan, 44, 1068-1071. http://dx.doi.org/10.1246/bcsj.44.1068
- Harada, K. and Kataoka, Y. (1978) Asymmetric Synthesis of Alanine by Hydrogenolytic Asymmetric Transamination. Tetrahedron Letters, 19, 2103-2106. http://dx.doi.org/10.1016/S0040-4039(01)94761-6
- Harada, K. and Kataoka, Y. (1978) The Temperature Dependence of Hydrogenolytic Asymmetric Transamination between Esters of Optically Active Phenylglycine and Pyruvic Acid. Chemistry Letters, 7, 791-794. http://dx.doi.org/10.1246/cl.1978.791
- Harada, K. and Tamura, M. (1979) Asymmetric Synthesis of Alanine by Hydrogenolytic Asymmetric Transamination between (R)-2-Amino-2-Phenylethanol and Ethyl Pyruvate. Bulletin of the Chemical Society of Japan, 52, 1227-1228. http://dx.doi.org/10.1246/bcsj.52.1227
- Harada, K., Munegumi, T. and Nomoto, S. (1981) Asymmetric Hydrogenation of Chiral Pyruvamides. Tetrahedron Letters, 22, 111-114. http://dx.doi.org/10.1016/0040-4039(81)80162-1
- Harada, K. and Munegumi, T. (1983) Asymmetric Hydrogenations of N-Pyruvoyl-(S)-Amino Acids Esters. Bulletin of the Chemical Society of Japan, 56, 2774-2777. http://dx.doi.org/10.1246/bcsj.56.2774
- Harada, K. and Munegumi, T. (1984) Asymmetric Catalytic Hydrogenations of Chiral α-Keto Amides. Bulletin of the Chemical Society of Japan, 57, 3203-3232. http://dx.doi.org/10.1246/bcsj.57.3203
- Munegumi, T., Fujita, M., Maruyama, T., Shiono, S., Takasaki, M. and Harada, K. (1987) Asymmetric Catalytic Hydrogenation of N-Pyruvoyl-(S)-Proline Esters. Bulletin of the Chemical Society of Japan, 60, 249-253. http://dx.doi.org/10.1246/bcsj.60.249
- Munegumi, T. and Harada, K. (1988) Asymmetric Catalytic Hydrogenations of Oximes and Benzylimino Derivatives of Chiral Pyruvamides. Bulletin of the Chemical Society of Japan, 61, 1425-1427. http://dx.doi.org/10.1246/bcsj.61.1425
- Munegumi, T., Maruyama, T., Takasaki, M. and Harada, K. (1990) Diastereoselective Catalytic Hydrogenations of Nα-Pyruvoyl-(S)-Prolinamide. Bulletin of the Chemical Society of Japan, 63, 1832-1834. http://dx.doi.org/10.1246/bcsj.63.1832
- Dorman, D.E. and Bovery, F.A. (1973) Carbon-13 Magnetic Resonance Spectroscopy. Spectrum of Proline in Oligopeptides. Journal of Organic Chemistry, 38, 2379-2383. http://dx.doi.org/10.1021/jo00953a021
NOTES

*Corresponding author.