International Journal of Organic Chemistry
Vol.3 No.3A(2013), Article ID:39076,8 pages DOI:10.4236/ijoc.2013.33A005
Improvement on the Synthesis of Primary Amino Sugar Derivatives via N-Benzyl Intermediates
1Dipartimento di Chimica, Università di Firenze, Florence, Italy
2Dipartimento di Farmacia, Università di Pisa, Pisa, Italy
Email: *massimo.corsi@unifi.it, *roberto.bianchini@unifi.it
Copyright © 2013 Massimo Corsi et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received September 2, 2013; revised October 4, 2013; accepted October 21, 2013
Keywords: Amino Sugars; Nucleophilic Substitution; Benzylamine; Primary Tosylates
ABSTRACT
Primary tosylates 1a-d were converted to the corresponding amino species 3a-d. Benzylamine was proved effective for the substitution of tosylates, using acetonitrile (MeCN) as the solvent of choice and citric acid to remove excess of the reagent from crude products 2a-d. Debenzylation was carried out at circa (ca.) atmospheric pressure of hydrogen gas in the presence of acetic acid (AcOH). The method was also demonstrated in a demo batch experiment for the synthesis of compound 3a on a 50 g scale of 1a.
1. Introduction
The importance of amino sugars in medical and biological applications have been long recognized [1], proving their essential value in studies about the mechanisms of cell-protein recognition [2] and interactions with nucleic acids [3]. In addition, antibiotic research has found fertile ground in this class of compounds [4] and the developments of potent inhibitors of glycosidases [5] have also gained credit along with the preparation of semisynthetic glycoconjugate vaccines for anticancer therapies [6].
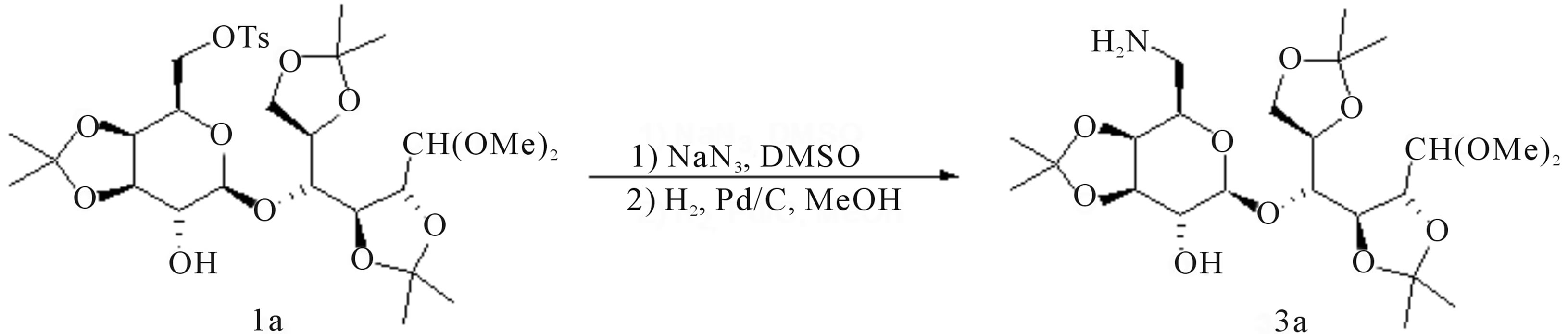
In the course of our program on the synthesis of sugar derived dyes [7,8], we needed to prepare a batch quantity of 6’-amino-6’-deoxy-2,3:5,6:3’,4’-tri-O-isopropylidenelactose dimethyl acetal 3a [9], in order to expand the library of naturalized dyes for textile dyeing studies [10]. Literature procedures indicated sodium azide as the most popular reagent to elaborate the primary tosylate 1a to the corresponding azido intermediate [11], which would be hydrogenated to 3a (Scheme 1).
The replacement of a tosylate group on a sugar type scaffold with sodium azide has been documented in polar aprotic solvents such as N,N-dimethylformamide (DMF) or dimethylsulfoxide (DMSO) at high temperature and on multigram scale without any safety concern [12,13]. However, the potential hazards associated to the use of sodium azide in substitution processes [14] prompted us to seek out a safer route to scale up the synthesis of 3a accordingly. Benzylamine is a routine chemical for the installation of a masked primary amino group on organic structures, generally unmasked under transition metal catalysed hydrogenation [15]: a two-step sequence which may well work for 1a. Some reports have shown the use of benzylamine on cyclodextrin tosylates [16-22] and only few others on monosaccharide type structures [23, 24].
In most cases, benzylamine or derivative thereof has been used as the reaction solvent [16-18,20,23,24] to accomplish the substitution of the tosylates. Here, we would like to propose a practical method for the elaboration of sugar derived primary tosylates to the corresponding amino species via N‑benzylamine intermediates, to provide a convenient protocol and a safer alternative to the use of hazardous sodium azide.
2. Results and Discussion
Compounds 1a-1d were chosen as model structures, to investigate the potential of benzylamine in promoting the substitution of the tosylate group (Figure 1). Isopropylidene acetals 1a [25], 1b [26] and 1c [10,27] were synthesized according to literature procedures. Whereas 1d was achieved from the elaboration of known dimethyl
Scheme 1. Synthesis of 3a.
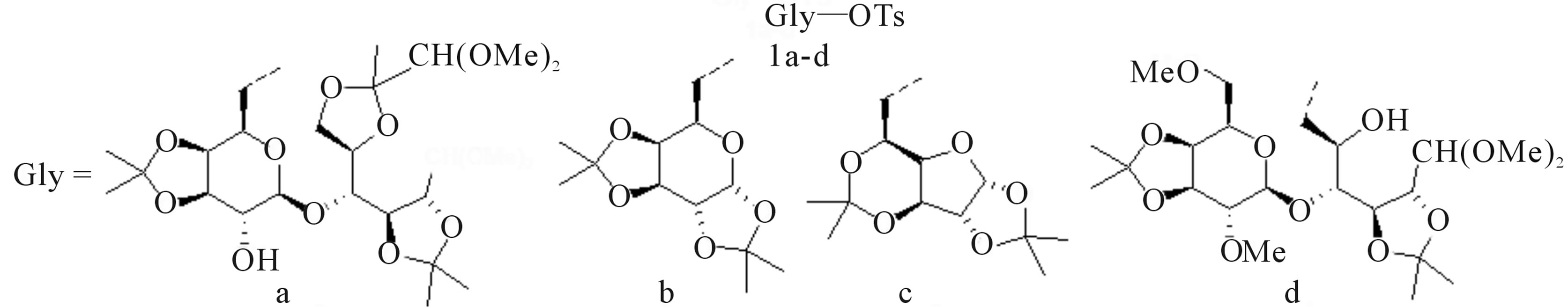
Figure 1. Model tosylates 1a-d.
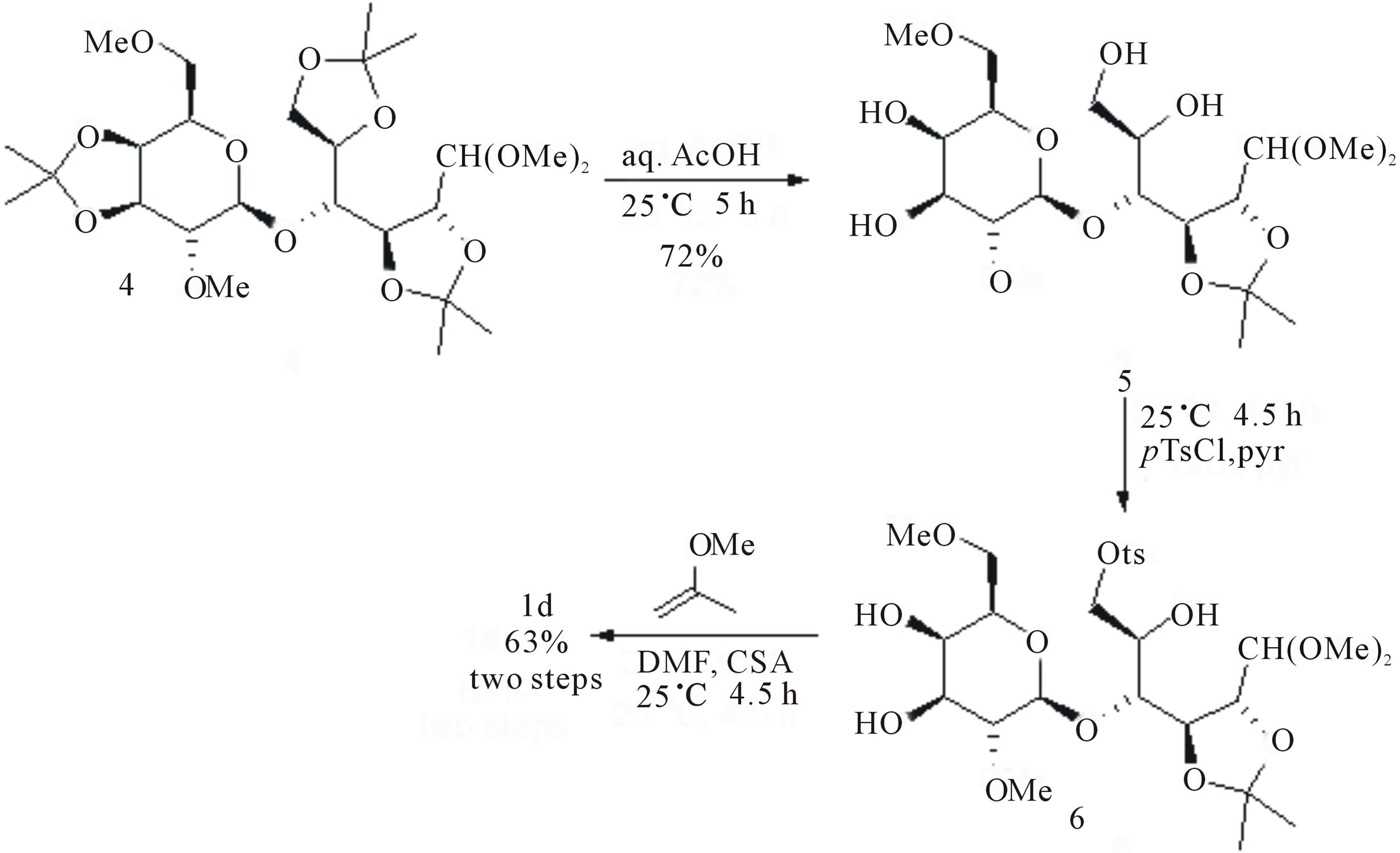
ether 4 [28] which was treated initially with aqueous (aq.) 60% acetic acid (AcOH) to obtain 5 in 72% yield [29]. Next, tosylation of the primary alcohol of 5 [30] led to the formation of 6, which upon protection of the cis‑diol moiety with 2-methoxypropene and camphor-sulphonic acid (CSA) [31] delivered the desired 1d in 63% yield for two steps from 5 (Scheme 2).
A first investigation into the conditions needed to replace the tosylate moiety with an N-benzyl radical, was started on 1a using a slight excess of benzylamine (1.2 eq.) in refluxing tetrahydrofuran: but regrettably, no reaction occurred. Increasing the polarity of the medium using acetonitrile (MeCN) did not aid the formation of 2a at the same temperature, circa (ca.) 67˚C. However, the process did proceed at the reflux temperature of MeCN (ca. 80˚C).
Interestingly, a trial carried out in DMF demonstrated that the polarity of the medium had relative influence on this substitution process, since 2a could be obtained at 90˚C. Thus, when less polar solvents such as toluene and chlorobenzene were used, formation of 2a was achieved as well at 110˚C and 130˚C, respectively. We also found that MeCN was the best medium for a clean process, as indicated by the monitoring of the reaction mixtures through thin layer chromatography (TLC) analysis. However, full conversion of 1a to 2a required a large excess (10 eq.) of benzylamine, posing an issue for the isolation of the desired compound. Therefore, MeCN was evaporated and the residue was partitioned between dichloromethane (DCM) and a solution of citric acid stoichiometric to the theoretical leftover of benzylamine, to retain this reagent in the aq. phase as the corresponding citrate salt. This work-up operation was followed by TLC analysis, which did not show any evidence of benzylamine in the organic solution containing 2a. Next, hydrogenation of 2a was carried out at ca. atmospheric pressure in methanol (MeOH) with Pd/C as catalyst (see experimental) and in the presence of AcOH, to recover amine 3a in excellent yield after an aq. NaOH work‑up. The two-step protocol was later transferred to species 1b-d, which produced amines 3b [32], 3c [33] and 3d respectively without changing the experimental conditions (Scheme 3).
The efficacy of the method was also proved in a scaleup demo batch experiment for the synthesis of compound 3a. Initially, 50 g of 1a were converted to the corresponding 2a derivative, following the procedure tested on small scale: but modifying slightly the part concerning the removal of the excess of benzylamine. In the relevant case, crude 2a was partitioned between the citric acid aq. solution and DCM at 0˚C, to allow the dissipation of the exotherm which may originate (see experimental). Then, hydrogenation of compound 2a was carried out without amending the procedure described on small scale: but, it required a longer reaction time (ca. 30 h) to complete. Compound 2a (35.4 g) was obtained in 94% yield.
3. Conclusions
We showed that benzylamine could be used on model tosylates 1a-d to obtain the corresponding N‑benzyl intermediates 2a-d. The reactions were carried out in MeCN as the solvent of choice and citric acid proved efficient to remove the excess of benzylamine. 2a-d were thus recovered with good purity for the next step. Standard hydrogenation of 2a-d in the presence of AcOH set the stage for the definition of a general two-step procedure to final 3a-d. A scale-up example concerning the synthesis of 3a was also provided, showing the potential of the method for process development on bulk scale.
It is envisaged that this synthetic method would be convenient for the synthesis of valuable primary amino
Scheme 2. Synthesis of 1d.
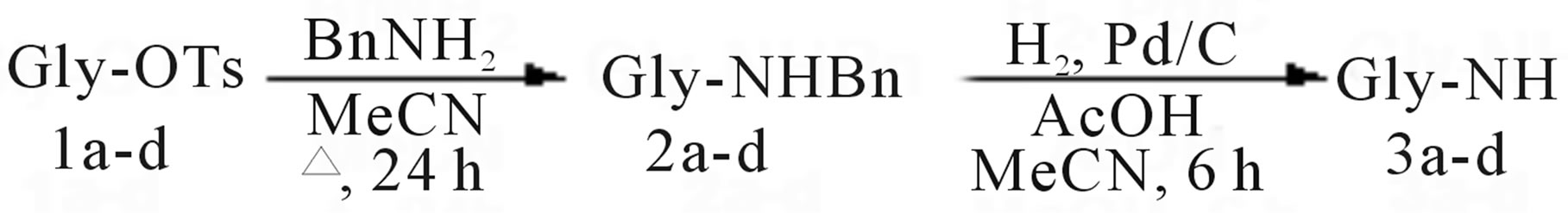
Scheme 3. Synthesis of 3a-d.
sugar species, where the N-benzyl group may be attractive within a suited synthetic plan.
4. Experimental
General Remarks. Commercially available reagents and solvents were purchased from SigmaAldrich and they were used directly. TLC analysis was performed using Fluka aluminium foils coated with 25 mm particle size silica gel matrix F254. TLC development involved either UV (254 nm) or visible light inspection, followed by either treatment with an acid solution of p-anisaldehyde or a basic solution of KMnO4 and heating. Flash chromatography was performed on Merck silica gel 60 (particle size 0.040 - 0.063 nm, 230 - 400 mesh ASTM) according to the procedure of Still [34]. Melting points were recorded on a Melting Point Apparatus SMP3- STUART SCIENTIFIC. Optical rotations were measured on a Jasco DIP-370 polarimeter using a 100 mm pathlength cell at 589 nm. Infra-red spectra were recorded in a KBr disk on a Perkin Elmer‑Spectrum BX FTIR system. Absorptions were quoted in wavenumbers (νmax, cm−1). 1H NMR and 13C NMR spectra were recorded with a Varian Gemini spectrometer operating at 200.13 (1H) and 50.3 MHz (13C) or on a Bruker Avance II 250 spec- trometer operating at 250.15 (1H) and 62.9 MHz (13C). Spin resonances were reported as chemical shifts (d) in parts per million (ppm) and referenced to the residual peak of the solvent employed as follows: CDCl3 7.27 ppm (1H NMR) and 77.0 ppm (13C NMR, central band), CD3OD 3.31 ppm (1H NMR, central band) and 49.0 ppm (13C NMR, central band). Spin multiplicity was indicated by s = singlet, d = doublet, t = triplet, dd = double doublet, m = multiplet, br = broad. Coupling constants J were reported in Hertz. Mass spectra were recorded on a ThermoScientific LCQ-Fleet mass spectrometer under electrospray ionisation (ESI, +c technique). Mass spectrometric analyses were quoted in the m/z form. Elemental analyses were recorded on a Perkin Elmer 240 C Elemental Analyzer.
General procedure A: synthesis of N‑benzyl intermediates 2a-d. A solution of benzylamine (10 mmol) in MeCN (20 mL) was brought to reflux and a solution of the appropriate tosylate 1a-d (1 mmol) in MeCN (5 mL) was added dropwise over 0.3 h. The resulting mixture was refluxed until the tosylate disappeared as indicated by TLC analysis and it was cooled to 20˚C. The solvent was evaporated in vacuo and the crude residue was dissolved in DCM (30 mL) washed with 0.3 M citric acid (10 mL), aq. saturated (sat.) NaHCO3 (30 mL) and brine (30 mL). The organic phase was separated, dried over Na2SO4 and concentrated to dryness under reduced pressure, to yield the desired product which was purified by flash chromatography.
General procedure B: synthesis of amino sugar derivatives 3a-d. The appropriate N‑benzyl intermediate 2a-d (1 mmol) was dissolved in MeOH (20 mL) and AcOH (3 mmol, 0.18 g) was added. The solution was degassed by three vacuum-N2 purge cycles, Pd/C catalyst [2% w/w, 50% wet Degussa type, (10% w/w loading, dry basis)] was added and the suspension was subjected to further three purge cycles. Then, a balloon of H2 gas was used to replace N2 by the same purging technique and the mixture was stirred at 20˚C under H2 at ca. 1 atm of pressure for 6 h. After replacing H2 with N2 as described above, the suspension was filtered and the filtrate was evaporated to dryness in vacuo. The residue was partitioned between DCM (30 mL) and aq. 1 M NaOH (10 mL) and the organic phase was separated, washed with brine and dried over Na2SO4 to yield compounds 3a-d, which were purified by flash chromatography.
4-O-(6-Benzylamino-6-deoxy-3,4-O-isopropylidene-β-d-galactopyranosyl)-2,3:5,6-di-O-isopropylidene- aldehydo-d-glucose dimethyl acetal 2a. The synthesis of 2a was carried out using benzylamine (1.1 g) tosylate 1a (0.66 g) and MeCN according to general procedure A. N-benzyl intermediate 2a (0.55 g, 92.1%) was isolated after the reported work-up and a flash column chromatography procedure [MeOH:ethyl acetate (EtOAc): DCM = 5:5:90] as off white foam, m. p. 48˚C - 50˚C; Rf (MeOH:EtOAc:DCM = 5:5:90) 0.26; [α]D27 +15.0 (c 0.95, CHCl3); IR (νmax): 3597, 3476 (br), 3064, 2990, 2936, 2907, 2836, 1454, 1382, 1373, 1245, 1217, 1158, 1123 and 1068 cm−1; 1H NMR (200.13 MHz, CDCl3): d 7.41-7.24 (5H, Ar), 4.53-4.40 (2H, m), 4.34-4.16 (3H, m), 4.08-3.75 (8H, m), 3.57-3.49 (1H, m), 3.39 (3H, s, OCH3), 3.29 (3H, s, OCH3), 3.12-3.02 (2H, m), 2.75 (1H, dd, J = 12.4 and J’ = 3.0 Hz), 1.98 [1H, s (br), OH], 1.51 [6H, s, (CH3 acetonide)2], 1.39 (3H, s, CH3 acetonide), 1.38 (3H, s, CH3 acetonide), 1.33 (3H, s, CH3 acetonide), and 1.31 ppm (3H, s, CH3 acetonide); 13C NMR (50.3 MHz, CDCl3): d 140.3, 128.2 (2C), 128.1 (2C), 126.8, 110.2, 110.1, 108.3, 106.8, 103.4, 79.3, 78.3, 77.8, 75.9, 75.7, 74.7, 74.2, 73.0, 64.5, 57.1, 54.3, 53.9, 49.8, 28.2, 27.3, 26.4, 26.3, 25.6 and 24.1 ppm; ESI (m/z, +c) Calcd for C30H48NO11 [M+H]+ 598.70, Found 598.71; Anal. Calcd for C30H47NO11 (597.69): C, 60.29; H, 7.93; N, 2.34. Found: C, 60.10; H, 7.86; N, 2.24.
4-O-(6-Amino-6-deoxy-3,4-O-isopropylidene-β-d-ga-lactopyranosyl)-2,3:5,6-di-O-isopropylidene-aldehydo-d-glucose dimethyl acetal 3a. Compound 2a (0.60 g) was used in the synthesis of 3a along with Pd/C (12 mg), MeOH and H2 gas according to general procedure B. Amino derivative 3a (0.45 g, 89.0%) was isolated after the reported work-up and a flash column chromatography procedure [1% v/v conc. NH4OH/(MeOH:DCM = 10: 90)] as pale yellow foam, m. p. 54˚C - 56˚C; Rf [1% v/v conc. NH4OH/(MeOH:DCM = 10:90)] 0.45; [α]D27 + 25.9 (c 1.00, CHCl3), lit. [9] [α]D29 +29.8 (c 0.42, CHCl3); 1H NMR (200.13 MHz, CDCl3): d 4.56-4.42 (2H, m), 4.36-4.16 (3H, m), 4.07-3.89 (5H, m), 3.69-3.62 (1H, m), 3.58-3.49 (1H, m), 3.46 (3H, s, OCH3), 3.44 (3H, s, OCH3), 3.11 (1H, dd, J = 13.4 and J’ = 8.6 Hz, H2NCH2), 2.87 (1H, dd, J = 13.4 and J’ = 3.6 Hz, H2NCH2), 1.50 [6H, s, (CH3 acetonide)2], 1.39 (3H, s, CH3 acetonide), 1.38 (3H, s, CH3 acetonide) and 1.32 ppm [6H, s, (CH3 acetonide)2]; 13C NMR (50.3 MHz, CDCl3): d 110.1 (2C), 108.3, 106.5, 103.6, 79.4, 78.2, 77.8, 75.8, 75.7, 75.3, 74.5, 74.2, 64.5, 56.9, 54.2, 43.1, 28.1, 27.2, 26.3, 26.2, 25.6 and ppm 24.1; Anal. Calcd for C30H47NO11 (507.57): C, 54.43; H, 8.14; N, 2.76. Found: C, 54.36; H, 7.95; N, 2.69.
Scale up synthesis of 3a. In a three neck round bottomed flask equipped with a condenser, a thermometer and a dropping funnel, benzylamine (80.8 g, 0.75 mol) was dissolved in MeCN (500 mL) under nitrogen. The mixture was brought to reflux and a solution of compound 1a (50.0 g, 0.075 mol) in MeCN (50 mL) was added dropwise over 0.3 h. Reflux was maintained for 24 h, after which the solution was cooled to 20˚C and the solvent was evaporated under reduced pressure. The residue was dissolved in DCM (300 mL) and aq. 0.9 M citric acid (250 mL) was added at 0˚C under vigorous stirring, using a mechanical agitator. The two phase mixture was kept in ice for further 0.3 h: then it was warmed to 20˚C. The organic phase was separated, washed with aq. sat. NaHCO3 (2 × 300 mL) followed by brine (2 × 250 mL), dried over Na2SO4 and filtered. The solvent was evaporated in vacuo and crude compound 1b was dissolved in MeOH (450 mL). AcOH (0.23 mol, 13.8 g) was added at 0˚C and the resulting mixture was warmed to 20˚C, to carry out the hydrogenation process in the presence of Pd/C (0.9 g) following the protocol described in general procedure B. After 30 h, H2 gas was replaced by N2 and the suspension was filtered. The filtrate was evaporated to dryness and the residue was partitioned between DCM (300 mL) and aq. 1 M NaOH (230 mL). The organic phase was separated, washed with brine (2 × 250 mL) and dried over Na2SO4, to yield compound 3a (35.4 g, 94%) as off white foam.
6-Benzylamino-6-deoxy-1,2:3,4-di-O-isopropylidene-α-d-galactopyranose 2b. The synthesis of 2b was carried out using benzylamine (10 mmol, 1.1 g) tosylate 1b (1 mmol, 0.41 g) and MeCN according to general procedure A. N-Benzyl intermediate 2b (0.29 g, 83.4%) was isolated after the reported work-up and a flash column chromatography procedure [0.5% v/v conc. NH4OH/ (MeOH:DCM = 5:95)] as colorless syrup; Rf [0.5% v/v conc. NH4OH/(MeOH:DCM = 5:95)] 0.31; [α]D27 −61.9 (c 0.88, CHCl3); IR (νmax): 3665, 3065, 3029, 2991, 2935, 1495, 1454, 1384, 1373, 1256, 1212, 1166 and 1069 cm−1; 1H NMR (200.13 MHz, CDCl3): d 7.37-7.23 (5H, m, Ar), 5.56 (1H, d, J = 5.2 Hz, C1H), 4.61 (1H, dd, J = 7.8 and J’ = 2.2 Hz, C3H), 4.33 (1H, dd, J = 5.2 and J’ = 2.2 Hz, C2H), 4.21 (1H, dd, J = 7.8 and J’ = 1.8 Hz, C4H), 4.01-3.91 (1H, m, C5H), 3.91-3.76 [2H, m (AB system), PhCH2N-], 2.97 (1H, dd, J = 12.6 and J’ = 8.4 Hz, BnNHCH2), 2.79 (1H, dd, J = 12.6 and J’ = 4.4 Hz, BnNHCH2), 1.56 (3H, s, CH3 acetonide), 1.45 (3H, s, CH3 acetonide) and 1.34 ppm [6H, s, (CH3 acetonide)2]; 13C NMR (50.3 MHz, CDCl3): d 140.2, 128.3 (2C), 128.1 (2C), 126.8, 109.2, 108.4, 96.4, 72.0, 70.9, 70.6, 66.8, 53.7, 49.1, 26.2, 26.0, 25.0 and 24.4 ppm; ESI (m/z, +c) Calcd for C19H28NO5 [M+H]+ 350.43, Found 350.30; Anal. Calcd for C19H27NO5 (349.21): C, 65.31; H, 7.79; N, 4.01. Found: C, 65.26; H, 7.70; N, 4.32.
6-Amino-6-deoxy-1,2:3,4-di-O-isopropylidene-α-dgalactopyranose 3b. Compound 2b (0.36 g) was used in the synthesis of 3a along with Pd/C (7.0 mg), MeOH and H2 gas according to general procedure B. Amino derivative 3b (0.27 g, 90.6%) was isolated after the reported work-up and a flash column chromatography procedure [1% v/v conc. NH4OH/(MeOH:DCM = 10:90)] as pale yellow syrup; Rf [1% v/v conc. NH4OH/(MeOH: DCM = 10:90)] 0.39; [α]D27 −50.2 (c 0.95, CHCl3), lit. [32] [α]D20 −53.1 (c 1.03, CHCl3); 1H NMR (200.13 MHz, CDCl3): d 5.55 (1H, d, J = 5.2 Hz, C1H), 4.60 (1H, dd, J = 8.0 and J’ = 2.4 Hz, C3H), 4.32 (1H, dd, J = 5.2 and J’ = 2.4 Hz, C2H), 4.21 (1H, dd, J = 8.0 and J’ = 1.8 Hz, C4H), 3.73-3.66 (1H, m, C5H), 2.99 (1H, dd, J = 13.2 and J’ = 7.8 Hz, H2NCH2), 2.82 (1H, dd, J = 13.2 and J’ = 5.0 Hz, H2NCH2), 1.44 [6H, s, (CH3 acetonide)2] and 1.33 ppm [6H, s, (CH3 acetonide)2]; 13C NMR (50.3 MHz, CDCl3): d 109.2, 108.4, 96.4, 71.8, 70.8, 70.6, 69.4, 42.3, 26.1, 26.0, 24.9 and 24.4 ppm; Anal. Calcd for C12H21NO5 (259.30): C, 55.58; H, 8.16; N, 5.40. Found: C, 55.45; H, 7.99; N, 5.52.
6-Benzylamino-6-deoxy-1,2:3,5-di-O-isopropylidene-α-d-glucofuranose 2c. The synthesis of 2c was carried out using benzylamine (10 mmol, 1.1 g) tosylate 1c (1 mmol, 0.41 g) and MeCN according to general procedure A. N-benzyl intermediate 2c (0.26 g, 74.4%) was isolated after the reported work-up and a flash column chromatography procedure (MeOH:DCM = 2:98) as colorless syrup; Rf (MeOH:DCM = 2:98) 0.36; [α]D27 +33.5 (c 0.97, CH3OH); IR (νmax): 3634, 3090, 3065, 2992, 2939, 2837, 1456, 1384, 1375, 1245, 1163, 1080 and 1019 cm−1; 1H NMR (200.13 MHz, CDCl3): d 7.36-7.19 (5H, m, Ar), 6.00 (1H, d, J = 3.6 Hz, C1H), 4.58 (1H, d, J = 3.6 Hz, C2H), 4.32 (1H, dd, J = 3.8 and J’ = 7.0 Hz, C4H), 4.19 (1H, d, J = 3.8 Hz, C3H), 3.82 (2H, s, PhCH2NH-), 3.79-3.69 (1H, m, C5H), 2.94 (1H, dd, J = 12.4 and J’ = 3.6 Hz, BnNHCH2), 2.76 (1H, dd, J = 12.4 and J’ = 7.8 Hz, BnNHCH2), 1.49 (3H, s, CH3 acetonide), 1.36 (3H, s, CH3 acetonide), 1.34 (3H, s, CH3 acetonide) and 1.32 ppm (3H, s, CH3 acetonide); 13C NMR (50.3 MHz, CDCl3): d 140.3, 128.4 (2C), 128.2 (2C), 126.9, 112.1, 106.4, 100.8, 84.1, 81.1, 75.0, 71.3, 53.8, 51.7, 27.1, 26.5, 24.2 and 24.1 ppm; ESI (m/z, +c) Calcd for C19H28NO5 [M+H]+ 350.43, Found 350.28; Anal. Calcd for C19H27NO5 (349.42): C, 65.31; H, 7.79; N, 4.01. Found: C, 65.07; H, 7.71; N, 4.27.
6-Amino-6-deoxy-1,2:3,5-di-O-isopropylidene-α-dglucofuranose 3c. Compound 2c (0.36 g) was used in the synthesis of 3c along with Pd/C (7.0 mg), MeOH and H2 gas according to general procedure B. Amino derivative 3c (0.23 g, 88.7%) was isolated after the reported work-up and a flash column chromatography procedure [1% v/v conc. NH4OH/(MeOH:DCM = 10:90)] as pale yellow syrup; Rf [1% v/v conc. NH4OH/(MeOH:DCM = 10:90)] 0.43; [α]D27 +41.3 (c 0.86, CHCl3), lit. [33] [α]D19 +39.2 (c 1.12, CHCl3); 1H NMR (200.13 MHz, CDCl3): d 6.00 (1H, d, J = 3.8 Hz, C1H), 4.58 (1H, d, J = 3.8 Hz, C2H), 4.27 (1H, dd, J = 7.0 and J’ = 3.6 Hz, C4H), 4.19 (1H, d, J = 3.6 Hz, C3H), 3.56-3.47 (1H, m, C5H), 3.00 (1H, dd, J = 13.2 and J’ = 3.6 Hz, H2NCH2), 2.80 (1H, dd, J = 13.2 and J’ = 7.6 Hz, H2NCH2), 1.50 (3H, s, CH3 acetonide), 1.37 [6H, s, (CH3 acetonide)2] and 1.33 ppm (3H, s, CH3 acetonide); 13C NMR (50.3 MHz, CDCl3): d 112.0, 106.3, 100.7, 84.0, 80.7, 75.0, 73.6, 44.7, 27.0, 26.4, 24.0 and 23.9 ppm; Anal. Calcd for C12H21NO5 (259.30): C, 55.58; H, 8.16; N, 5.40. Found: C, 55.37; H, 7.96; N, 5.53.
4-O-(2,6-Di-O-methyl-β-d-galactopyranosyl)-2,3-Oisopropylidene-aldehydo-d-glucose dimethyl acetal 5. A solution of dimethyl acetal 4 (1.0 g, 1.86 mmol) in 60% aq. AcOH (14.0 mL) was stirred at 20˚C for 20 h, after which TLC analysis (MeOH:EtOAc = 10:90) revealed the disappearance of 4 (Rf = 0.73) and the formation of a major product (Rf = 0.22). The reacting mixture was concentrated at reduced pressure and repeatedly coevaporated with toluene (5 × 20 mL). The crude residue was purified by flash chromatography (MeOH:EtOAc = 10:90) to obtain compound 5 (0.61 g, 72% yield) as off white solid, m. p. 136˚C - 139˚C; Rf (MeOH:EtOAc = 10: 90) 0.22; [α]D27 −19.6 (c 1.18, CHCl3); IR (νmax): 3445, 3378, 2934, 2877, 2835, 2834, 1455, 1384, 1370, 1240, 1201, 1138, 1112-1035 (br) and 1015 cm−1; 1H NMR (250.15 MHz, CD3OD): d 4.59 (1H, dd, J = 7.0 and J’ = 6.2 Hz), 4.49 (1H, d, J = 7.8 Hz), 4.39 (1H, d, J = 6.2 Hz), 4.33 (1H, d, J = 7.0 Hz), 3.88-3.75 (5H, m), 3.63-3.52 (3H, m), 3.55 (3H, s, OCH3), 3.48 (1H, dd, J 3.5 and J’ 9.9 Hz), 3.42 [6H, s, C1H(OCH3)2], 3.35 (3H, s, OCH3), 3.17 (1H, dd, J = 7.8 and J’ = 9.9 Hz), 1.42 (3H, s, CH3 acetonide) and 1.38 ppm (3H, s, CH3 acetonide); 13C NMR (62.8 MHz, CD3OD): d 111.1, 106.5, 104.1, 82.7, 78.8, 77.4, 76.7, 74.6, 74.4, 73.5, 72.6, 70.4, 63.5, 61.3, 59.4, 55.8, 53.8, 27.9 and 27.3 ppm; ESI (m/z, +c) Calcd for C19H36NaO12 [M + Na]+ 479.47, Found 479.29; Anal. Calcd for C19H36O12 (456.48): C, 49.99; H, 7.95. Found: C, 49.97; H, 7.93.
4-O-(2,6-Di-O-methyl-3,4-O-isopropylidene-β-d-galactopyranosyl)-2,3-O-isopropylidene-6-O-tosyl-aldehy-do-d-glucose dimethyl acetal 1d. A solution of ptoluenesulfonyl chloride (0.89 g, 4.67 mmol) in dry pyridine (5 mL) was added dropwise to a solution of 5 (0.53 g, 1.17 mmol) in dry pyridine (5.6 mL) at 0˚C. The resulting mixture was stirred for 5 h, after which TLC analysis (MeOH:EtOAc = 10:90) indicated the quantitative conversion of 5 (Rf = 0.22) to a new product (Rf = 0.56). Therefore, the reaction mixture was warmed to 20˚C, MeOH (2.5 mL) was added and stirring continued for 0.5 h. The solvents were evaporated in vacuo and the crude residue was dissolved in dry DMF (3.4 mL) under argon and treated with 2-methoxypropene (0.3 mL, 3.0 mmol) and CSA (23 mg, 0.1 mmol) at 0˚C. The mixture was warmed to 20˚C and stirred for 6 h, after which triethylamine was added and the whole was concentrated under reduced pressure. The crude residue was purified by flash chromatography (EtOAc:hexane = 50:50) to obtain compound 1d (0.48 g, 63.2% overall) as a colorless syrup; Rf (EtOAc:hexane = 50:50) 0.45; [α]D27 +1.7 (c 1.12, CHCl3); IR (νmax): 3444, 2986, 2936, 2834, 1598, 1455, 1361, 1245, 1218, 1177 and 1099 cm−1; 1H NMR (200.13 MHz, CDCl3): d 7.82 [2H, d (AA’XX’), Ar], 7.34 [2H, d (AA’XX’), Ar], 4.51 (1H, dd, J = 10.8 and J’ = 3.0 Hz), 4.46-4.40 (1H, m), 4.37-4.34 (2H, m), 4.21 (1H, dd, J = 10.8 and J’ = 7.6 Hz), 4.11-3.96 (4H, m), 3.90-3.87 (1H, m), 3.82-3.75 (1H, m), 3.68-3.52 (2H, m), 3.46 (3H, s, OCH3), 3.41 (3H, s, OCH3), 3.40 (3H, s, OCH3), 3.35 (3H, s, OCH3), 3.14 (1H, dd, J = 8.2 and J’ = 6.6 Hz), 2.44 (3H, s, CH3Ph), 1.50 (3H, s, CH3 acetonide), 1.36 [6H, s, (CH3 acetonide)2] and 1.33 ppm (3H, s, CH3 acetonide); 13C NMR (50.3 MHz, CDCl3): d 144.7, 133.1, 129.7 (2C), 128.0 (2C), 110.3, 109.9, 104.9, 103.1, 82.7, 79.5, 79.2, 77.2, 75.4, 73.7, 72.2, 71.9, 71.6, 71.4, 59.4, 59.2, 55.6, 53.4, 28.0, 27.2, 26.5, 26.2 and 21.6 ppm; ESI (m/z, +c) Calcd for C29H46NaO14S [M + Na]+ 673.72, Found 673.24; Anal. Calcd for C29H46O14S (650.73): C, 53.53; H, 7.13. Found: C, 53.50; H, 7.12.
4-O-(2,6-Di-O-methyl-3,4-O-isopropylidene-β-d-galactopyranosyl)-6-benzylamino-6-deoxy-2,3-O-isopropylidene-aldehydo-d-glucose dimethyl acetal 2d. The synthesis of 2d was carried out using benzylamine (10 mmol, 1.1 g) tosylate 1d (1 mmol, 0.65 g) and MeCN according to general procedure A. N‑benzyl intermediate 1d (0.48 g, 81.9%) was isolated after the reported work-up and a flash column chromatography procedure [0.5% v/v conc. NH4OH/(MeOH:DCM = 10:90)] as colorless syrup; Rf [0.5% v/v conc. NH4OH/(MeOH:DCM = 10:90)] 0.38; [α]D27 +2.6 (c 0.90, CHCl3); IR (νmax): 3665, 3430 (br), 3028, 2990, 2935, 2835, 1454, 1383, 1372, 1245, 1219, 1202, 1154, 1101, 1076 and 1043 cm−1; 1H NMR (200.13 MHz, CDCl3): d 7.38-7.22 (5H, m, Ar) 4.51-4.44 (2H, m), 4.37 (1H, d, J = 6.0 Hz), 4.12-4.02 (3H, m), 3.93-3.74 (5H, m), 3.70-3.53 (2H, m), 3.53 (3H, s, OCH3), 3.48 (3H, s, OCH3), 3.41 (3H, s, OCH3), 3.36 (3H, s, OCH3), 3.13 (1H, dd, J = 8.0 and J’ = 6.3 Hz), 3.04-2.84 (2H, m), 1.51 (3H, s, CH3 acetonide), 1.42 (3H, s, CH3 acetonide), 1.41 (3H, s, CH3 acetonide) and 1.34 ppm (3H, s, CH3 acetonide); 13C NMR (50.3 MHz, CDCl3): d 140.2, 128.3 (2C), 128.1 (2C), 126.9, 110.2, 109.8, 105.0, 102.3, 82.8, 79.2, 78.5, 77.7, 75.6, 73.8, 72.0, 71.5 (2C), 59.8, 59.2, 55.4, 53.8, 53.5, 50.8, 28.1, 27.5, 26.7 and 26.3 ppm; ESI (m/z, +c) Calcd for C29H48NO11 [M+H]+ 586.69, Found 586.48; Anal. Calcd for C29H47NO11 (585.68): C, 59.47; H, 8.09; N, 2.39. Found: C, 59.23; H, 8.15; N, 2.69.
4-O-(2,6-Di-O-methyl-3,4-O-isopropylidene-β-dgala-ctopyranosyl)-6-amino-6-deoxy-2,3-Oisopropylidene-aldehydo-d-glucose dimethyl acetal 3d. Compound 2d (0.60 g) was used in the synthesis of 3d along with Pd/C (12 mg), MeOH and H2 gas according to general procedure B. Amino derivative 3d (0.45 g, 91.8%) was isolated after the reported work-up and a flash column chromatography procedure [1% v/v conc. NH4OH/(MeOH:DCM = 10:90)] as pale yellow syrup; Rf [1% v/v conc. NH4OH/(MeOH:DCM = 10:90)] 0.52; [α]D27 +1.3 (c 0.80, CHCl3); IR (νmax): 3689, 3606, 3414 (br), 2990, 2935, 2835, 1455, 1382, 1372, 1244, 1219, 1202, 1155, 1098, 1075 and 1045 cm−1; 1H NMR (200.13 MHz, CDCl3): d 4.50-4.36 (3H, m), 4.13-4.02 (3H, m), 3.87-3.62 (8H, m), 3.58 (3H, s, OCH3), 3.43 (3H, s, OCH3), 3.42 (3H, s, OCH3), 3.36 (3H, s, OCH3), 3.20- 3.13 (1H, m), 3.06-2.82 (2H, m), 2.37-2.14 (3H, br), 1.52 (3H, s, CH3 acetonide), 1.43 (3H, s, CH3 acetonide), 1.41 (3H, s, CH3 acetonide) and 1.34 ppm (3H, s, CH3 acetonide); 13C NMR (50.3 MHz, CDCl3): d 110.2, 109.9, 105.1, 102.5, 82.9, 79.3, 79.1, 77.6, 75.7, 73.8 (2C), 72.2, 71.6, 59.8, 59.2, 55.5, 53.5, 43.7, 28.0, 27.4, 26.6 and 26.2 ppm; ESI (m/z, +c) Calcd for C22H42NO11 [M + H]+ 496.57, Found 496.41; Anal. Calcd for C22H41NO11 (495.56): C, 53.32; H, 8.34; N, 2.83. Found: C, 53.49; H, 8.00; N, 2.71.
5. Acknowledgements
The authors are indebted to the Regional Council of Tuscany for fellowship funding to M.C. and M.B and to Ente Cassa di Risparmio di Firenze, who granted the present research and funded the purchase of the mass spectrometer ThermoFisher LCQ fleet ion trap instrument, try “R. B. G. thanks”.
REFERENCES
- S. F. Kuan, J. C. Byrd, C. Basbaum and Y. S. Kim, “Inhibition of Mucin Glycosylation by Aryl-N-Acetyl-α-Galactosaminides in Human Colon Cancer Cell,” Journal of Biological Chemistry, Vol. 264, No. 32, 1989, pp. 19271- 19277.
- D. Kahne, C. Leimkuhler, W. Lu and C. Walsh, “Glycopeptide and Lipoglycopeptide Antibiotics,” Chemical Reviews, Vol. 105, No. 2, 2005, pp. 425-448. http://dx.doi.org/10.1021/cr030103a
- B. Lindberg, “Components of Bacterial Polysaccharides,” Advances in Carbohydrate Chemistry and Biochemistry, Vol. 48, 1990, pp. 279-318. http://dx.doi.org/10.1016/S0065-2318(08)60033-5
- B. R. Griffith, C. Krepel, X. Fu, S. Blanchard, A. Ahmed, C. E. Edmiston and J. S. Thorson, “Model for Antibiotic Optimization via Neoglycosylation: Synthesis of Liponeoglycopeptides Active against VRE,” Journal of the American Chemical Society, Vol. 129, No. 26, 2007, pp. 8150-8155. http://dx.doi.org/10.1021/ja068602r
- H. Liu, X. Liang, H. Søhoel, A. Bülow and M. Bols, “Noeuromycin, A Glycosyl Cation Mimic that Strongly Inhibits Glycosidases,” Ibid, Vol. 123, No. 21, 2001, pp. 8150-8155. http://dx.doi.org/10.1021/ja010240u
- Q. Wang, Z. Zhou, S. Tang and Z. Guo, “CarbohydrateMonophosphoryl Lipid A Conjugates Are Fully Synthetic Self-Adjuvanting Cancer Vaccines Eliciting Robust Immune Responses in the Mouse,” ACS Chemical Biology, Vol. 7, No. 1, 2012, pp. 235-240. http://dx.doi.org/10.1021/cb200358r
- R. Bianchini, M. Rolla, J. Isaad, G. Catelani, L. Guazzelli, M. Corsi and M. Bonanni, “Efficient Double Glycoconjugation to Naturalize High Molecular Weight Disperse Dyes,” Carbohydrate Research, Vol. 356, 2012, pp. 104- 109. http://dx.doi.org/10.1016/j.carres.2011.10.036
- R. Bianchini, M. Bonanni, M. Corsi and A. S. Infantino, “Viable and Straightforward Approach to the Preparation of Water Soluble Pyrazol-5-One Derivatives through Glycoconjugation,” Tetrahedron, Vol. 68, No. 41, 2012, pp. 8636-8644. http://dx.doi.org/10.1016/j.tet.2012.07.074
- G. Fontana, M. Abbate, G. Casella, C. Pellerito, A. Longo and F. Ferrante, “Synthesis, Chemical Characterization and Preliminary in Vitro Antitumor Activity Evaluation of New Rruthenium(II) Complexes with Sugar Derivatives,” Polyhedron, Vol. 30, No. 10, 2011, pp. 1671-1679. http://dx.doi.org/10.1016/j.poly.2011.03.046
- G. Bartalucci, R. Bianchini, G. Catelani, F. D’Andrea and L. Guazzelli, “Naturalised Dyes: A Simple Straightforward Synthetic Route to a New Class of Dyes-Glycoazodyes (GADs),” European Journal of Organic Chemistry, Vol. 2007, No. 4, 2007, pp. 588-595. http://dx.doi.org/10.1002/ejoc.200600686
- J. Isaad, M. Rolla and R. Bianchini, “Synthesis of WaterSoluble Large Naturalised Dyes Through Double Glycoconjugation,” European Journal of Organic Chemistry, Vol. 2009, No. 17, 2009, pp. 2748-2764. http://dx.doi.org/10.1002/ejoc.200801302
- J. Yang, X. Fu, Q. Jia, J. Shen, J. B. Biggins, J. Jiang, J. Zhao, J. J. Schmidt, P. G. Wang and J. S. Thorson, “Studies on the Substrate Specificity of Escherichia coli Galactokinase,” Organic Letters, Vol. 5, No. 13, 2003, pp. 2223-2226. http://dx.doi.org/10.1021/ol034642d
- J. A. F. Joosten, B. Evers, R. P. Summeren, J. P. Kamerling and J. F. G. Vliegenthart, “Synthesis of β-d-Galp-(1⇄4)-β-d-GlcpNAc-(1⇄2)-α-d-Manp-(1⇄O)(CH2)7CH3 Mimics to Explore the Substrate Specificity of Sialyltransferases and trans-Sialidases,” European Journal of Organic Chemistry, Vol. 2003, No. 18, 2003, pp. 3569‑3586. http://dx.doi.org/10.1002/ejoc.200300293
- J. P. Scott, M. Alam, N. Bremeyer, A. Goodyear, T. Lam, R. D. Wilson and G. Zhou, “Mitsunobu Inversion of a Secondary Alcohol with Diphenylphosphoryl azide. Application to the Enantioselective Multikilogram Synthesis of a HCV Polymerase Inhibitor,” Organic Process Research & Development, Vol. 15, No. 5, 2011, pp. 1116- 1123. http://dx.doi.org/10.1021/op200002u
- P. N. Rylander, “Catalytic Hydrogenation in Organic Synthesis,” Academic Press, New York, 1979.
- K. Harata, K. Y. Takenaka and N. Yoshida, “Crystal Structures of 6-Deoxy-6-Monosubstituted β-Cyclodextrins. Substituent-Regulated One-Dimensional Arrays of Macrocycles,” Journal of the Chemical Society, Perkin Transactions 2, Vol. 2001, No. 9, pp. 1667-1673. http://dx.doi.org/10.1039/b101521o
- F. Hacket, S. Simova and H.-J. Schneider, “The Complexation of Peptides by Aminocyclodextrins,” Journal of Physical Organic Chemistry, Vol. 14, No. 3, 2001, pp. 159‑170. http://dx.doi.org/10.1002/poc.348
- Y. Liu, C.-C. You, S.-Z. Kang, C. Wang, F. Chen and X.-W. He, “Synthesis of Novel β‑Cyclodextrin and Calixarene Derivatives and Their Use in Gas Sensing on the Basis of Molecular Recognition,” European Journal of Organic Chemistry, Vol. 2002, No. 4, pp. 607-613. http://dx.doi.org/10.1002/1099-0690(200202)2002:4<607::AID-EJOC607>3.0.CO;2-I
- R. Kaliappan and V. Ramamurthy, “Chiral Photochem.- istry within Natural and Functionalized Cyclodextrins: Chiral Induction in Photocyclization Products from Carbonyl Compounds,” Journal of Photochemistry and Photobiology A: Chemistry, Vol. 207, No. 1, 2009, pp. 144-152. http://dx.doi.org/10.1016/j.jphotochem.2009.03.004
- Y. Wang, T. S. Chung and H. Wang, “Polyamide-Imide Membranes with Surface Iimmobilized Cyclodextrin for Butanol Isomer Separation via Pervaporation,” AIChE Journal, Vol. 57, No. 6, 2011, pp. 1470-1484. http://dx.doi.org/10.1002/aic.12360
- M. Zengerle, F. Brandhuber, C. Schneider, F. Worek, G. Reiter and S. Kubik, “Highly Efficient Cyclosarin Degradation Mediated by a β-Cyclodextrin Derivative Containing an Oxime‑Derived Substituent,” Beilstein Journal of Organic Chemistry, Vol. 7, 2011, pp. 1543‑1554. http://dx.doi.org/10.3762/bjoc.7.182
- H. Yamamura, K. Suzuki, K. Uchibori, A. Miyagawa, M. Kawai, C. Ohmizob and T. Katsub, “Mimicking an Antimicrobial Peptide Polymyxin B by Use of Cyclodextrin,” Chemical Communications, Vol. 48, No. 6, 2012, pp. 892-894. http://dx.doi.org/10.1039/c1cc16369h
- T. Michaud, J. Chanet-Ray, S. Chou and J. Gelas, “Synthesis of New Enantiomerically Pure Polyhydroxylated Azetidines from Monosaccharides,” Carbohydrate Research, Vol. 299, No. 1, 1997, pp. 253-269. http://dx.doi.org/10.1016/S0008-6215(97)00141-9
- J. Cai, B. E. Davison, C. R. Ganellin, S. Thaisrivongs and K. S. Wibley, “Potential HIV Protease Inhibitors: Preparation of di-N-Alkylated 2-, 6-, and 2,6-Aminodeoxy-Derivatives of d-Glucose by Direct Displacement and by a Novel Reductive-Alkylation Procedure,” Ibid, Vol. 300, No. 2, 1997, pp. 109-117. http://dx.doi.org/10.1016/S0008-6215(97)00039-6
- G. Catelani, A. Corsaro, F. D’Andrea, M. Mariani, V. Pistara and E. Vittorino, “Convenient Preparation of l-Arabino-Hexos-5-ulose Derivatives from Lactose,” Ibid, Vol. 388, No. 22, 2003, pp. 2349-2358. http://dx.doi.org/10.1016/j.carres.2003.08.001
- S. B. Ferreira, A. C. R. Sodero, M. F. C. Cardoso, E. S. Lima, C. R. Kaiser, F. P. Jr. Silva and V. F. Ferreira, “Synthesis, Biological Activity, and Molecular Modeling Studies of 1H-1,2,3-Triazole Derivatives of Carbohydrates as α-Glucosidases Inhibitors,” Journal of Medicinal Chemistry, Vol. 53, No. 6, 2010, pp. 2364‑2375. http://dx.doi.org/10.1021/jm901265h
- J.-C. Lee, S.-W. Chang, C.-C. Liao, F.-C. Chi, C.-S. Chen, Y.-S. Wen, C.-C. Wang, S. S. Kulkarni, R. Puranik, Y.-H. Liu and S.-C. Hung, “From d-Glucose to Biologically Potent l‑Hexose Derivatives: Synthesis of α-l-Iduronidase Fluorogenic Detector and the Disaccharide Moieties of Bleomycin A2 and Heparan Sulfate,” Chemistry: A European Journal, Vol. 10, No. 2, 2004, pp. 399-415. http://dx.doi.org/10.1002/chem.200305096
- T. Yoshino, G. Reuter, S. Kelm and R. Schauer, “Facile Synthesis of 2’-Substituted Lactoses,” Glycoconjugate Journal, Vol. 3, No. 1, 1986, pp. 7-14. http://dx.doi.org/10.1007/BF01108607
- G. Attolino, G. Catelani and F. D’Andrea, “Regiospecific Synthesis of 4-Deoxy-d-threo-Hex-3-enopyranosides by Simultaneous Activation–Elimination of the Talopyranoside Axial 4-OH with the NaH/Im2SO2 System: Manifestation of the Stereoelectronic Effect,” European Journal of Organic Chemistry, Vol. 2006, No. 23, pp. 5279-5292. http://dx.doi.org/10.1002/ejoc.200600526
- G. W. Kabalka, M. Varma, R. S. Varma, P. C. Srivastava and F. F. Jr. Knapp, “The Tosylation of Alcohols,” Journal of Organic Chemistry, Vol. 51, No. 12, 1986, p. 2386. http://dx.doi.org/10.1021/jo00362a044
- P. J. Kocieński, “Protecting Groups,” 3rd Edition, Thieme, Stuttgart, 2005, pp. 120-132.
- B. Streicher and B. Wünsch, “Synthesis of Amino-Substituted Hexoand Heptopyranoses from-d-Galactose,” Carbohydrate Research, Vol. 338, No. 22, 2003, pp. 2375-2385. http://dx.doi.org/10.1016/S0008-6215(03)00382-3
- B. Coxon, “Studies of 15N-Labeled Amino Sugars1. The Synthesis and Mass Spectrometry of Derivatives of 6-Amino-6-deoxy-d-Glucose-6-15N,” Ibid, Vol. 19, No. 2, 1971, pp. 197-210. http://dx.doi.org/10.1016/S0008-6215(00)81620-1
- W. C. Still, M. Kahn and A. Mitra, “Rapid Chromatographic Technique for Preparative Separations with Moderate Resolution,” Journal of Organic Chemistry, Vol. 43, No. 14, 1978, pp. 2923-2925. http://dx.doi.org/10.1021/jo00408a041
NOTES
*Corresponding authors.