International Journal of Organic Chemistry
Vol.3 No.3(2013), Article ID:36604,4 pages DOI:10.4236/ijoc.2013.33025
Convenient Cleavage Reaction of 1-Acylhydantoin to Amide
Department of Applied Chemistry, Kanagawa Institute of Technology, Kanagawa, Japan
Email: *yamagu@chem.kanagawa-it.ac.jp
Copyright © 2013 Jun-ichi Yamaguchi et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received June 28, 2013; revised July 29, 2013; accepted August 14, 2013
Keywords: Amides; Acylhydantoin; Chiral Auxiliaries; Cleavage; Lewis Acid
ABSTRACT
A cleavage reaction of 1-acylhydantoin by an amine in tetrahydrofuran at elevated temperature provided the corresponding amide and hydantoin in high yield. It was found that the addition of diethylaluminum chloride accelerated the cleavage reaction rate. The amide yield depended on the steric hindrance of the amine.
1. Introduction
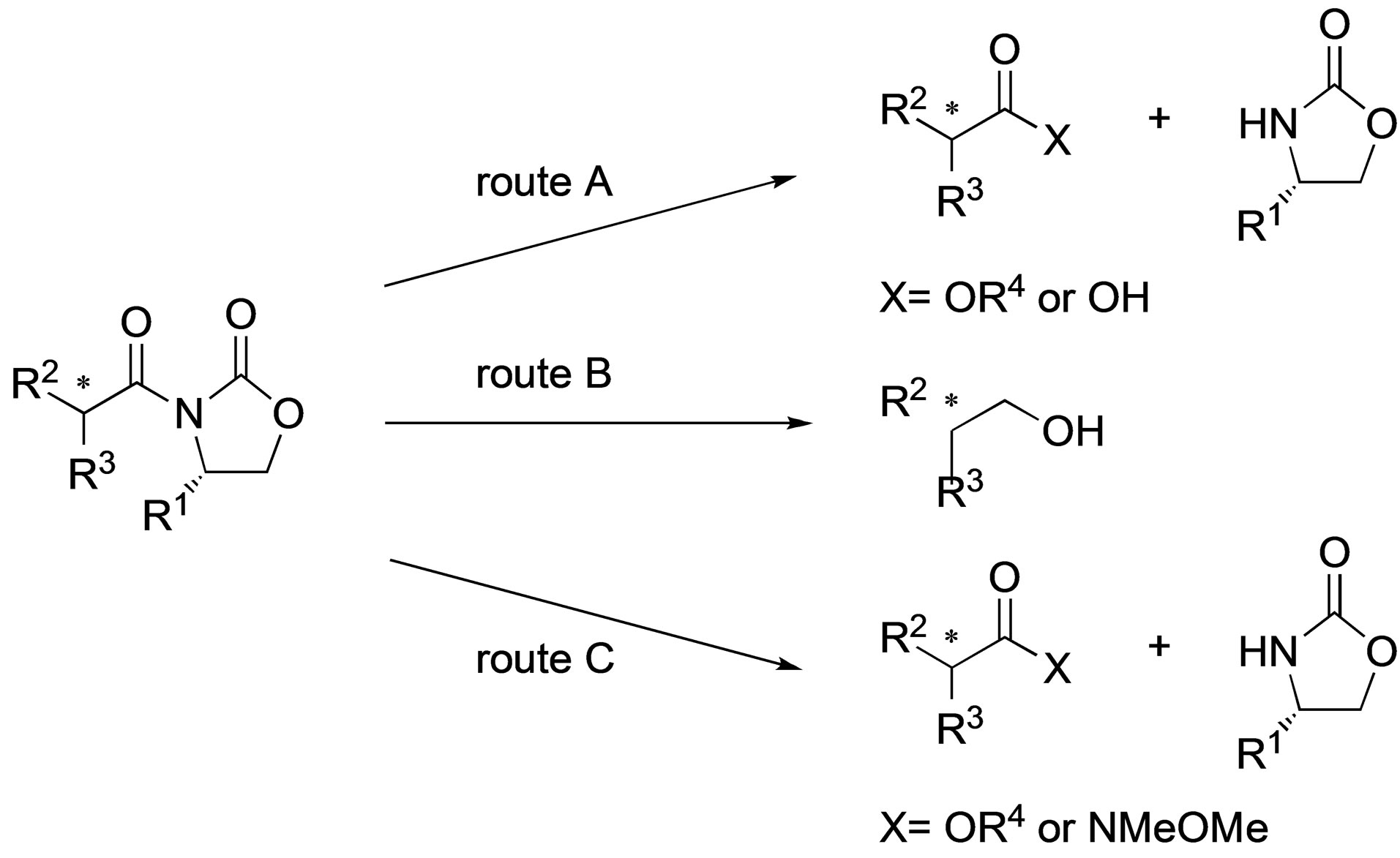
In the synthesis of an optically active compound utilizing a chiral auxiliary, cleaving the chiral auxiliary with concomitant reduction of the chiral center is an important step. Therefore, many methodologies for the cleavage of the chiral auxiliary such as the use of 2-oxazolidinone have been reported. The typical method for N-acyl-2- oxazolidinone cleavage is the treatment with alkoxide or lithium hydroperoxide to convert the substrate into a benzyl ester [1-3], a methyl ester [4], or a carboxylic acid [5-7], respectively (route A, Scheme 1). It is also wellknown that a reductive cleavage reaction using a reductant such as LiAlH4 [8-14] or LiBH4 [15] gives the corresponding alcohol (route B). On the other hand, only few reports of Lewis acid-promoted cleavage reactions of N-acyl-2-oxazolidinones to esters [16] or hydroxamic acid derivatives [17-19] have been published (route C).
We have reported that hydantoin is conveniently derived from an amino acid amide without racemization [20] and can be used as a chiral auxiliary to create a chiral center at the β-position of an acyl group utilizing a conjugate addition system [21]. In order to develop hydantoin as a more useful tool as a chiral auxiliary, a new cleavage reaction of 1-acylhydantoin is required (Scheme 2). We report here that treatment of 1-acylhydantoin with an amine at an elevated temperature provides the corresponding amide and hydantoin in high yields. The use of a catalytic amount of diethylaluminum chloride accelerated the reaction rate.
2. Results and Discussion
The initial examinations began with treatment of N-(3- phenylpropionyl) hydantoin 1 with propylamine a in several solvents. As shown in Table 1, the cleavage reaction of 1 with 1.2 - 3.0 equimolar amounts of a in several solvents from 0˚C to room temperature proceeded to give the corresponding amide 2a and hydantoin 3 in moderate yields (Entries 1 - 3). The cleavage reaction in THF performed at elevated temperature resulted in improvement of the yields of 2a and 3 (Entry 4). Since propylamine has a low boiling point (<50˚C), the cleavage reaction using an equimolar amount of propylamine gave 2a in lower yield (Entry 5). In the case of benzylamine instead of n-propylamine, the yields of 2b and 3 were improved (Entry 6). Lewis acids are known to be effective additives in the cleavage reaction of N-acyl-2-oxazolidinone [7]. Therefore, we expected that the addition of a Lewis acid would also be effective for enhancing the present cleavage reaction. The cleavage reaction in the presence of 1.2 equimolar amounts of Et2AlCl was complete in only 1 h (Entry 7). Furthermore, a catalytic amount of Et2AlCl was enough to complete the cleavage reaction, and 2b and 3 were obtained in excellent yields (Entry 8).
In the cleavage reaction of N-acyl-2-oxazolidinone, Narasaka and co-workers reported its tolerance to the attack of primary amines and that the addition of a Cp2ZrHCl (Cp = cyclopentadienyl) catalyst was necessary for the amide conversion [22].
The cleavage reaction using primary amines or aniline also gave 2 and 3 in excellent yields in the presence of
Scheme 1. Conventional cleavage reactions of N-acyl-2-oxazolidinone.

Scheme 2.Concept of the present work.


Table 1. Cleavage reaction of 1 with propylamine or benzylamine.
diethylaluminum chloride (Table 2, Entries 1 - 4). In contrast, the reactivity using secondary amines depended on the steric hindrance of the amine (Entries 5 - 8). Thus, the cleavage reaction with the more sterically demanding diisopropylamine failed, whereas that with diethylamine succeeded (Entries 5 and 6). Among secondary amines, cyclic amines such as pyrrolidine or 1,2,3,4-tetrahydroisoquinoline resulted in better yields than acyclic seconddary amines (Entries 7 and 8).
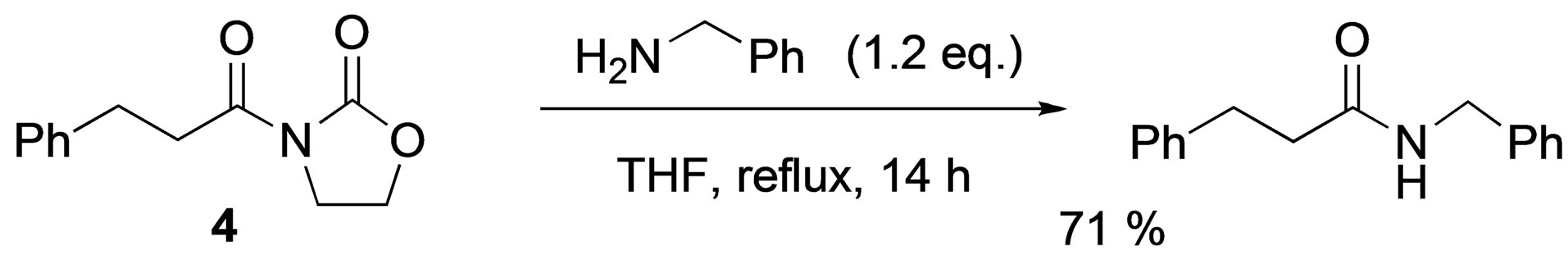
The cleavage reaction of N-(3-phenylpropionyl)-2- oxazolidinone 4 under similar reaction conditions was also examined (Scheme 3). Treatment of 4 with 1.2 equimolar amounts of benzylamine gave 2b in 71% yield. The reaction using hydantoin affords better results than that with 2-oxazolidinone.
3. Experimental
Typical Procedure for Cleavage Reaction of 1: Propylamine and a hexane solution of Et2AlCl were added to a THF solution of 1-acylhydantoin under Ar. After reflux, the reaction was quenched by addition of 1M HCl solution. The organic materials were extracted with CH2Cl2. The extract was washed with saturated NaCl solution and dried over Na2SO4. After removal of solvent under reduced pressure, the residue was separated by preparative TLC (hexane:EtOAc = 1:1) to give the amide and hydantoin.
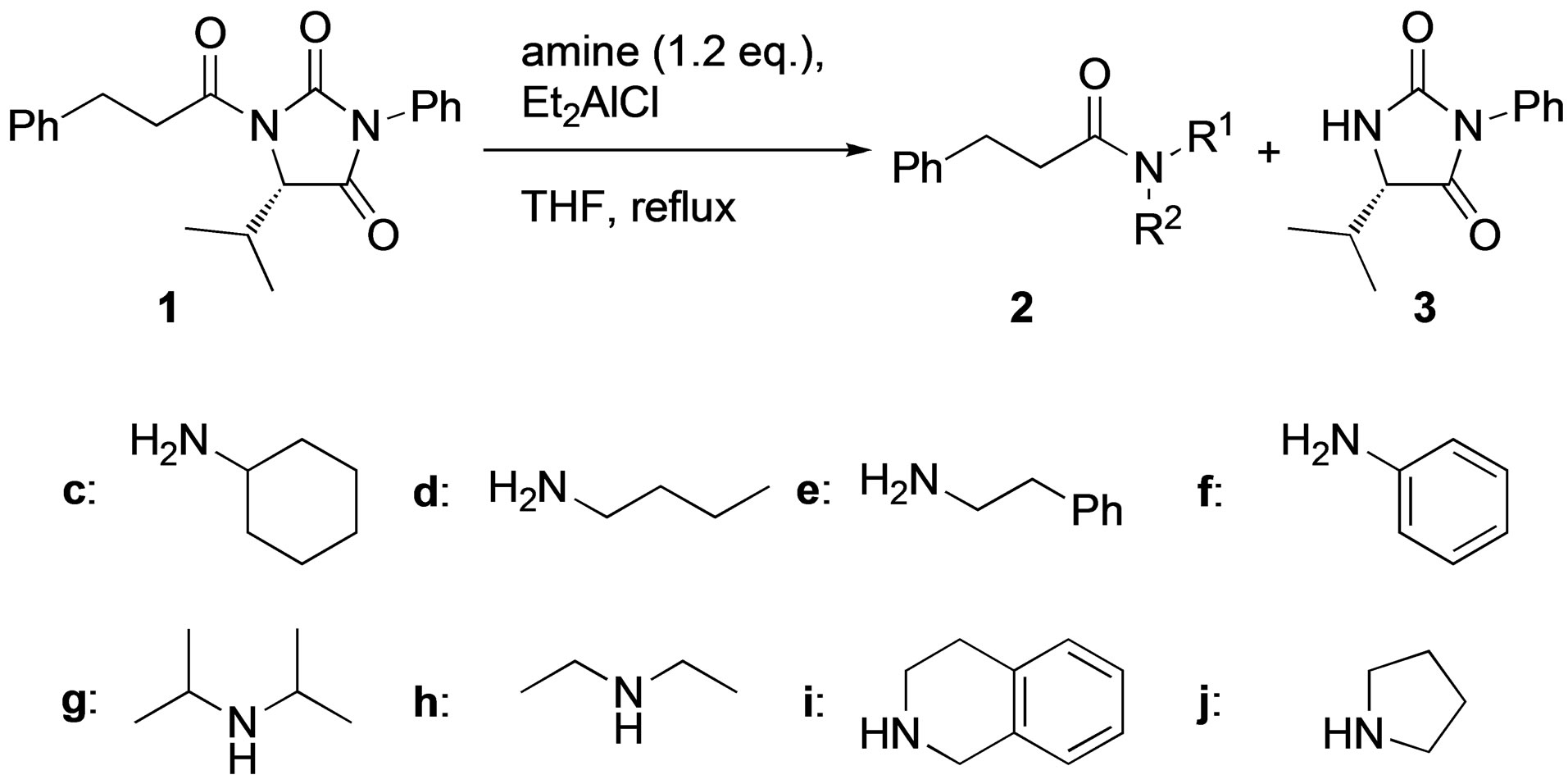

Table 2. Cleavage reactions of 1 with other amines.
Scheme 3.Conversion of N-dihydrocinnamoyl-2-oxazolidinone to amide.
N-Propyl-3-phenylpropionamide: 1H NMR (300 MHz, CDCl3) δ = 0.85 (3H, t, J = 7.3 Hz), 1.46 (2H, dq, J = 7.3 and 7.3 Hz), 2.46 (2H, t, J = 6.8 Hz), 2.96 (2H, t, J = 6.8 Hz), 3.16 (2H, q, J = 7.3 Hz), 5.48 (1H, br s), 7.18 - 7.38 (5H, m).
N-Benzyl-3-phenylpropionamide: 1H NMR (300 MHz, CDCl3) δ = 2.51 (2H, t, J = 7.3 Hz), 2.99 (2H, t, J = 7.3 Hz), 4.39 (2H, d, J = 5.7 Hz), 5.64 (1H, br s), 7.10 - 7.30 (5H, m).
N-Butyl-3-phenylpropionamide: 1H NMR (300 MHz, CDCl3) δ = 0.89 (3H, t, J = 7.1 Hz), 1.25 (2H, sext, J = 7.1 Hz), 1.40 (2H, t, J = 7.1 and 5.9 Hz), 2.46 (2H, t, J = 7.8 Hz), 2.97 (2H, t, J = 7.8 Hz), 3.21 (2H, q, J = 5.9 Hz), 5.34 (1H, br s), 7.10 - 7.35 (5H, m).
N-Phenethyl-3-phenylpropionamide: 1H NMR (300 MHz, CDCl3) δ = 2.42 (2H, t, J = 7.3 Hz), 2.73 (2H, t, J = 7.1 Hz), 2.95 (82H, t, J = 7.3 Hz), 3.48 (2H, q, J = 7.1 Hz), 5.34 (1H, br s), 7.00 - 7.35 (10H, m).
N-Cyclohexyl-3-phenylpropionamide: 1H NMR (300 MHz, CDCl3) δ = 0.85 - 1.20 (3H, m), 1.25-1.45 (2H, m), 1.55-1.70 (3H, m), 1.80 - 1.85 (2H, m), 2.43 (2H, t, J = 7.3 Hz), 2.96 (2H, t, J = 7.3 Hz), 3.65 - 3.80 (1H, m), 5.16 (1H, br s), 7.15 - 7.35 (5H, m).
N-Phenyl-3-phenylpropionamide: 1H NMR (300 MHz, CDCl3) δ = 2.64 (2H, t, J = 7.6 Hz), 3.04 (2H, t, J = 7.6 Hz), 7.06 (1H, t, J = 7.8 Hz), 7.10-7.35 (8H, m), 7.43 (2H, d, J = 7.8 Hz).
N-(3-Phenylpropionyl)pyrrolidine: 1H NMR (300 MHz, CDCl3) δ = 1.65 - 1.90 (4H, m), 2.56 (2H, t, J = 7.6 Hz), 2.99 (2H, t, J = 7.6 Hz), 3.29 (2H, t, J = 6.6 Hz), 3.47 (2H, t, J = 6.6 Hz), 7.10 - 7.35 (5H, m).
N,N-Diethyl-3-phenylpropionamide: 1H NMR (300 MHz, CDCl3) δ = 1.10 (3H, t, J = 7.1 Hz), 1.11 (3H, t, J = 7.1 Hz), 2.60 (2H, t, J = 7.6 Hz), 2.99 (2H, t, J = 7.6 Hz), 3.23 (2H, q, J = 7.1 Hz), 3.38 (2H, q, J = 7.1 Hz), 7.15-7.35 (5H, m).
N-(3-Phenylpropionyl)-1,2,3,4-tetrahydroisoquinoline: A mixture of rotamers. 1H NMR (300 MHz, CDCl3) δ = 2.65 - 2.75 (2H, m), 2.80 - 2.90 (2H, m), 2.95 - 3.05 (2H, m), 3.60 and 3.84 (2H, t, J = 5.9 and 5.9 Hz), 5.54 and 4.75 (2H, s and s), 7.00 - 7.35 (8H, m).
4. Conclusion
We have developed the convenient cleavage reaction of 1-acylhydantoin to the corresponding amide. The conversion is successfully performed under mild conditions with a catalytic amount of Et2AlCl. The activation of the imide moiety accelerates the formation of the amide. Other methodologies for new and convenient conversions are now in progress.
REFERENCES
- D. A. Evans and D. J. Mathre, “Asymmetric Synthesis of the Enkephalinase Inhibitor Thiorpan,” The Journal of Organic Chemistry, Vol. 50, No. 11, 1985, pp. 1830-1835. doi:10.1021/jo00211a008
- W. H. Miles, P. M. Smiley and H. R. Brinkman, “The Reaction of Chiral Nucleophiles with Organomanganese Arene Complex,” Journal of the Chemical Society, Chemical Communications, No. 24, 1989, pp. 1897-1899. doi:10.1039/c39890001897
- D. A. Evans, R. L. Dow, T. L. Shih, J. M. Takacs, R. Zahler, “Total Synthesis of the Polyether Antibiotic Ionomycin,” Journal of the American Chemical Society, Vol. 112, No. 13, 1990, pp. 5290-5313. doi:10.1021/ja00169a042
- D. L. J. Clive, K. S. K. Murthy, A. G. H. Wee, J. S. Prasad, G. V. J. da Silva, M. Majewski, P. C. Anderson, C. F. Evans, R. D. Haugen, L. D. Heerze and J. R. Barrie, “Total Synthesis of Both (+)-Compactins. A General Strategy Based on the Use of a Special Titanium Reagent for Dicarbonyl Coupling,” Journal of the American Chemical Society, Vol. 112, No. 8, 1990, pp. 3018-3028. doi:10.1021/ja00164a024
- D. A. Evans, T. C. Britton and J. A. Ellman, “Contrasteric Carboximide Hydrolysis with Lithium Hydroperoxide,” Tetrahedron Letters, Vol. 28, No. 49, 1987, pp. 6141- 6144. doi:10.1016/S0040-4039(00)61830-0
- M. Nakano, S. Atsuumi, Y. Koike, S. Tanaka, H. Funabashi, J. Hashimoto, M. Ohkubo and H. Morishima, “Synthesis of a Homostatine-Containing Renin Inhibitor Which Incorporates a Sulfonemethylene Isostere at Its N-Terminus,” Bulletin of Chemical Society of Japan, Vol. 63, No. 8, 1990, pp. 2224-2232. doi:10.1246/bcsj.63.2224
- M. Trova, R. E. Babine, R. A. Byrn, W. T. Casscles, Jr., R. C. Hastings, G. C. Hsu, M. R. Jirousek, B. D. Johnson, S. S. Kerwar, S. R. Schow, A. Wissner, N. Zhang and M. M. Wick, “Synthesis and Biological Evaluation of a Series of HIV-1 Protease Inhibitors,” Bioorganic and Medicinal Chemistry Letters, Vol. 3, No. 8, 1993, pp. 1595- 1600. doi:10.1016/S0960-894X(00)80024-4
- P. E. Sonnet and R. R. Heath, “Aryl Substituted Diastereomeric Alkenes: Gas Chromatographic Behavior on a Non-polar Versus a Liquid Crystal Phase,” Journal of Chromatography, Vol. 321, 1985, pp. 127-136. doi:10.1016/S0021-9673(01)90429-7
- D. F. Taber, E. H. Petty and K. Raman, “Enantioselective Ring Construction: Synthesis of (+)-a-Cuparenone,” Journal of the American Chemical Society, Vol. 107, No. 1, 1985, pp. 196-199. doi:10.1021/ja00287a035
- R. E. Zelle, M. P. DeNinno, H. G. Selnick and S. J. Danishefsky, “A Systematic Degradation of Zincophorin: A Stereoselective Synthesis of the C17-C-25 Fragment,” The Journal of Organic Chemistry, Vol. 51, No. 25, 1986, pp. 5032-5036. doi:10.1021/jo00375a060
- D. J. Porubek, H. Barnes, L. J. Theodore and T. A. Baillie, “Enantioselective Synthesis and Primary Metabolic Studies of the Optical Isomers of 2-n-Propyl-4-pentanoic Acid, a Hepatotoxic Metabolite of Valproic Acid,” Chemical Research in Toxicology, Vol. 1, No. 6, 1988, pp. 343-348. doi:10.1021/tx00006a004
- M. T. Crimmins, D. M. Bankaitis-Davis and W. G. Hollis, Jr., “Total Synthesis of Milbemycin b3,” The Journal of Organic Chemistry, Vol. 53, No. 3, 1988, pp. 652-657. doi:10.1021/jo00238a031
- M. T. Crimmins and R. O’Mahony, “A Synthesis of (−)- Talaromycin A,” The Journal of Organic Chemistry, Vol. 54, No. 5, 1989, pp. 1157-1161. doi:10.1021/jo00266a030
- C. Santini, R. G. Bal and G. D. Berger, “Absolute Stereochemistry of the Squalene Synthase Inhibitor Zaragozic Acid C,” The Journal of Organic Chemistry, Vol. 59, No. 8, 1994, pp. 2261-2266. doi:10.1021/jo00087a054
- T. D. Penning, S. W. Djuric, R. A. Haack, V. J. Kalish, J. M. Miyashiro, B. W. Rowell and S. S. Yu, “Improved Procedure for the Reduction of N-Acyloxazolidinones,” Synthetic Communications, Vol. 20, No. 2, 1990, pp. 307- 312. doi:10.1080/00397919008052299
- A. Orita, Y. Nagano, J. Hirano and J. Otera, “Lewis AcidPromoted Transesterification of N-Acyl Oxazolidinones under Mild Conditions,” Synlett, No. 5, 2001, pp. 637- 639.
- D. A. Evans, H. P. Ng and D. L. Rieger, “Total Synthesis of the Macrolide Antibiotic Rutamycin B,” Journal of the American Chemical Society, Vol. 115, No. 24, 1993, pp. 11446-11459. doi:10.1021/ja00077a049
- V. Rodeschini, P. Van de Weghe, C. Tarnus and J. Eustache, “A Simple Spiroepoxide as Methionine Aminopeptidase-2 Inhibitor: Synthetic Problems and Solutions,” Tetrahedron Letters, Vol. 46, No. 39, 2005, pp. 6691- 6695. doi:10.1016/j.tetlet.2005.07.136
- T. K. Jones, R. A. Reamer, R. Desmond and S. G. Mills, “Chemistry of Tricarbonyl Hemiketals and Application of Evan’s Technology to the Total Synthesis of the Immunosuppressant (−)-FK-506,” Journal of the American Chemical Society, Vol. 112, No. 8, 1990, pp. 2998-3017. doi:10.1021/ja00164a023
- J. Yamaguchi, M. Harada, T. Kondo, T. Noda and T. Suyama, “A Facile Method for Preparation of Optically Active Hydantoin,” Chemistry Letters, Vol. 32, No. 4, 2003, pp. 372-373. doi:10.1246/cl.2003.372
- J. Yamaguchi, M. Harada, T. Narushima, A. Saitoh, K. Nozaki and T. Suyama, “Diastereoselective Conjugate Addition of 1-(a,b-Unsaturated acyl)hydantoin with Nucleophiles,” Tetrahedron Letters, Vol. 46, No. 38, 2005, pp. 6411-6415. doi:10.1016/j.tetlet.2005.07.116
- K. Narasaka, N. Iwasawa, M. Inoue, T. Yamada, M. Nakashima, J. Sugimori, “Asymmetric Diels-Alder Reaction by a Chiral Titanium Reagent,” Journal of the American Chemical Society, Vol. 111, No. 14, 1989, pp. 5340-5345. doi:10.1021/ja00196a045
NOTES
*Corresponding author.