International Journal of Organic Chemistry
Vol. 2 No. 2 (2012) , Article ID: 20377 , 9 pages DOI:10.4236/ijoc.2012.22020
The Reaction of 2-Amino-4,5,6,7-tetrahydrobenzo[b]thiophenes with Benzoyl-Isothiocyanate: Synthesis of Annulated Thiophene Derivatives and Their Antitumor Evaluations
1Department of Organic Chemistry, Faculty of Pharmacy, October University for Modern Sciences and Arts, 6th of October City, Egypt
2Pharmaceutical Chemistry Department, Faculty of Pharmacy (Girls), Al-Azhar University, Nasr City, Cairo, Egypt
3Higher Institute of Engineering and Technology, El-Tagammoe El-Khames, New Cairo, Egypt
Email: *karamsyn@yahoo.com
Received April 10, 2012; revised May 12, 2012; accepted May 17, 2012
Keywords: Tetrahydrobenzo[b]thiophene; Pyrimidine; Thiourea; Antitumor Activity
ABSTRACT
The reaction of the tetrahydrobenzo[b]thiophene derivatives 1a,b with benzoyliso thiocyanate (2) afforded the thiourea derivatives 3a,b. Cyclization of the latter products gave the annulated products 4a,b. Compounds 3a,b reacted with either hydrazine hydrate (5a) or phenyl hydrazine (5b) afforded compounds 7a-d which underwent cyclization for compounds 7a,c afforded compound 9. On the other hand compounds 4a,b reacted with either hydrazine hydrate (5a) or phenyl hydrazine (5b) afforded compounds 6a-d a second pathway was applied to synthesize compound 6c which underwent cyclization afforded compound 9. Also compounds 4a,b reacted with either phenacyl bromide (10) afforded compounds 11a,b or ethylchloroacetae (12) compounds 13a,b were produced. The latter products reacted with each hydrazine hydrate (5a) and phenyl hydrazine (5b) afforded compounds 14a-d. Their antitumor activities were tested using three different cell lines.
1. Introduction
Over the past few years some research groups and our group were interested to introduce a comprehensive study program towards the synthesis of thiophenes and their fused derivatives [1-4]. The importance of such compounds based on their uses as anti-inflammatory [5-7] thiophene derivatives was evaluated as antiprotozoal [8,9], on the other hand thiophene derivatives was applied as a new antitumor agents [10,11], in addition to fused thiophene derivatives was tested as templates for serine protease inhibition [12] and alternate substrate inhibitors of cholesterol esterase [13]. In this article we are using the 2-amino-4,5,6,7-tetrahydrobenzo[b] thiophene derivatives [14] 1a,b in the synthesis of fused derivatives of pharmaceutical interest.
2. Results and Discussion
Thus, the reaction of 1a,b with benzoylisothiocyanate (2) in 1,4-dioxane at room temperature gave the N-benzoylthiourea derivatives 3a and 3b, respectively. The structures of compounds 3a,b were based on analytical and spectral data. Thus, 1H NMR spectrum of 3a (as an example) showed beside the expected regular data for the cyclohexenyl moiety, two multiplet at δ 2.14 - 2.16 (4H, 2CH2), δ 2.23 - 2.26 (4H, 2CH2), a multiplet at δ 7.30 - 7.41 (5H), corresponding to aromatic protons. Two singlet at δ 8.26, 8.30 (2H) corresponding to 2NH. The reactions of isothiocyanates with NH2 compounds was reported earlier [15]. Compounds 3a,b underwent ready cyclization when heated in sodium ethoxide/ethanol solution in a boiling water bath to give the tetrahydro bezo[b]thieno [2,3-d]pyrimidine derivatives 4a and 4b, respectively. The 1HNMR spectrum of each compound which revealed, in case of 4a showed three multiplet at δ 1.64 - 1.72 (4H, 2CH2), δ 2.21 - 2.23 (4H, 2CH2) and multiplet at δ 7.32 - 7.44 (5H) corresponding to aromatic protons. Two singlet at δ 8.26, 8.29 (2H) corresponding to 2NH. Further confirmations for the structures of 4a and 4b were obtained through studying their reactivity towards some chemical reagents. Thus, the reaction of either 4a or 4b with either hydrazine hydrate (5a) or phenylhydrazine (5b) gave the corresponding compounds 6a-d (Scheme 1). On the other hand the reaction of either 3a or 3b with either hydrazine hydrate or phenylhydrazine gave the hydrazone derivatives 7a-d. The latter products 7a,c were cyclized when heated in DMF/piperdine solution to give the annulated derivative 9. Formation of the latter product took place through the intermediate triazole derivatives 8a,b. The structure of compound 9 was based on analytical and spectral data. Formation of such compound 9 from either 7a or 7c is explained in terms of the first addition of NH group to CN group followed by hydrolysis of the C=NH group [16,17], however the addition of NH on COOEt group was followed by loss of ethanol took place. Confirmation structure of compound 9 was carried out by applying the second pathway to synthesize such compound 9 through reaction of compound 4b with hydrazine hydrate (5a) in 1,4 dioxan at room temperature where the hydrazide derivative 6c was separated which underwent ready cyclization when heated in dimethylformamide solution to give the same product 9 (m.p. and mixed m.p.) (Scheme 2). The reaction of either compound 4a or 4b with ω-bromoacetophenone (10) in ethanol solution gave the thioether derivatives 11a and 11b, respectively.
Structures of the latter products were based on analytical and spectral data. On the other hand, the reaction of either 4a or 4b with ethyl chloroacetate (12) gave the thioether derivatives 13a and 13b respectively. The spectral data of the latter are in consistent with the proposed structures.
The reaction of either compounds 13a or 13b with either hydrazine hydrate or phenylhydrazine afforded the corresponding 2-hydrazino-4,5,6,7-tetrahydrobenzo[b] thieno-[2,3-d]pyrimidine derivatives 14a-d (Scheme 3). The analytical and spectral data of the latter products are in agreement with the proposed structures.
2.1. Antitumor Activity Tests
Reagents: Fetal bovine serum (FBS) and L-glutamine, were from Gibco Invitrogen Co. (Scotland, UK). RPMI- 1640 medium was from Cambrex (New Jersey, USA). Dimethyl sulfoxide (DMSO), doxorubicin, penicillin, streptomycin and sulforhodamine B (SRB) were from Sigma Chemical Co. (Saint Louis, USA).
Cell cultures: Three human tumor cell lines, MCF-7 (breast adenocarcinoma), NCI-H460 (non-small cell lung cancer), and SF-268 (CNS cancer) were used. MCF-7 was obtained from the European Collection of Cell Cultures
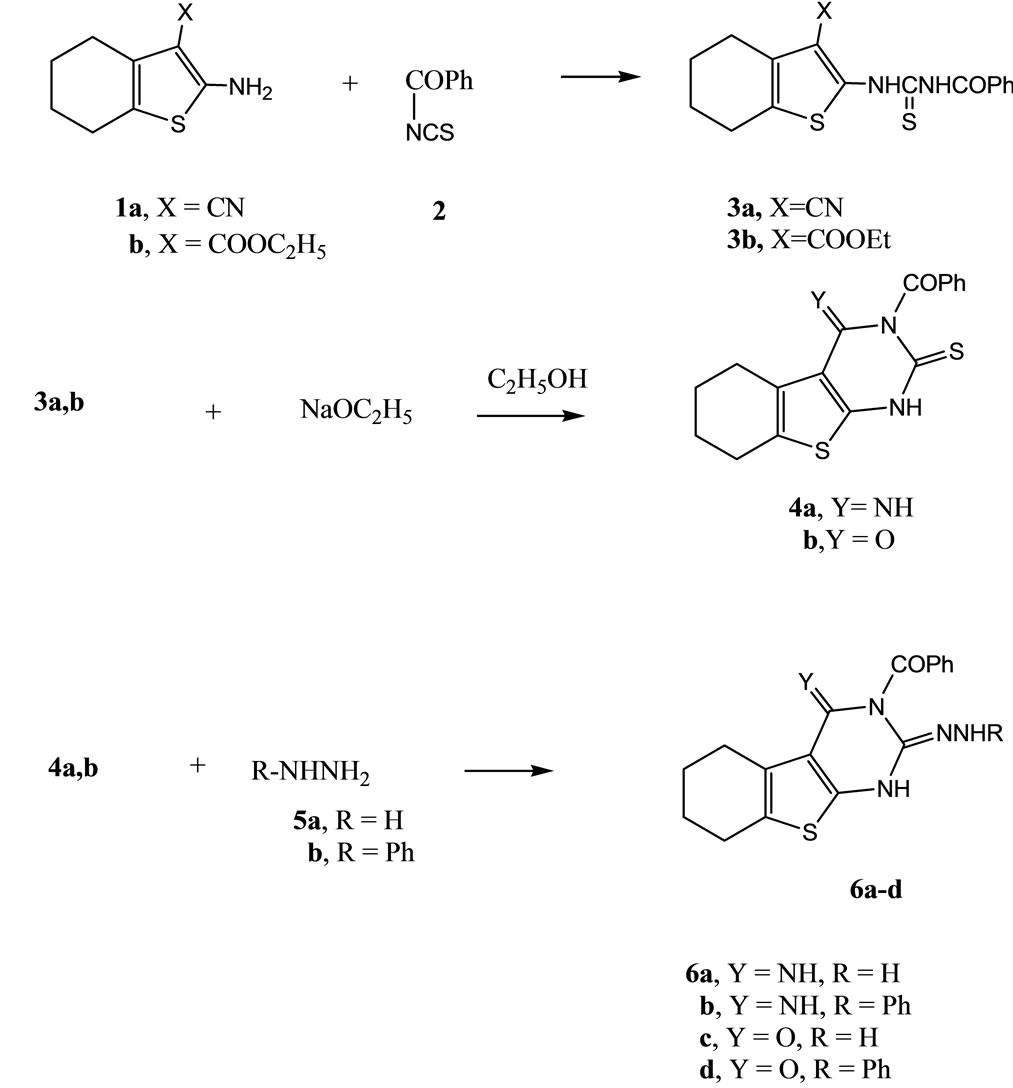
Scheme 1. Synthesis of compounds 3a,b-6a-d.
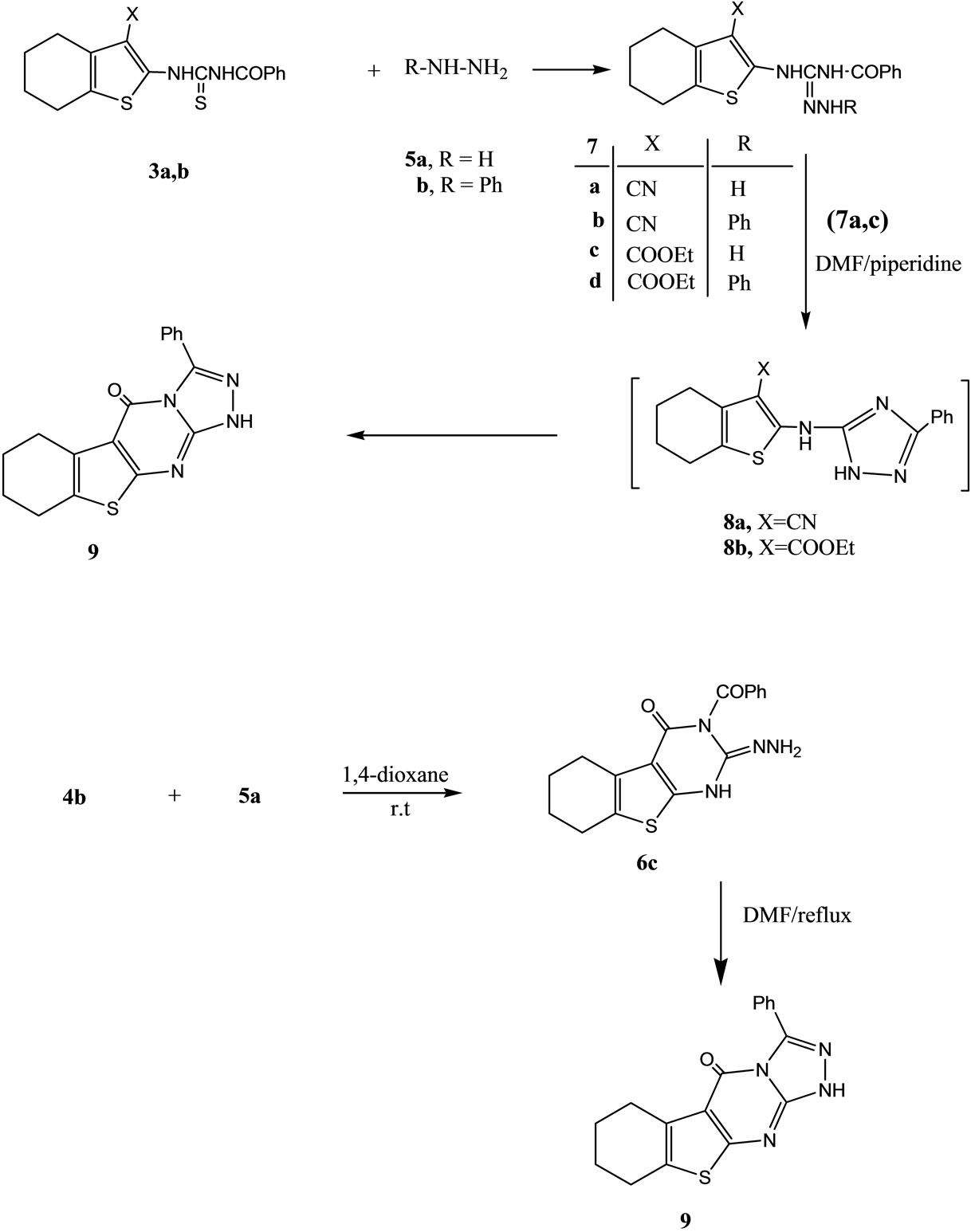
Scheme 2. Synthesis of compounds 7a-d-9.
(ECACC, Salisbury, UK) and NCI-H460 and SF268 were kindly provided by the National Cancer Institute (NCI, Cairo, Egypt). They grow as monolayer and routinely maintained in RPMI-1640 medium supplemented with 5% heat inactivated FBS, 2 mM glutamine and antibiotics (penicillin 100 U/mL, streptomycin 100 µg/mL), at 37˚C in a humidified atmosphere containing 5% CO2. Exponentially growing cells were obtained by plating 1.5 × 105 cells/mL for MCF-7 and SF-268 and 0.75 × 104 cells/mL for NCI-H460, followed by 24 h of incubation. The effect of the vehicle solvent (DMSO) on the growth of these cell lines was evaluated in all the experiments by exposing untreated control cells to the maximum concentration (0.5%) of DMSO used in each assay.
Tumor cell growth assay: The effects of 3a-14d on the in vitro growth of human tumor cell lines were evaluated according to the procedure adopted by the National Cancer Institute (NCI, USA) in the “In vitro Anticancer Drug Discovery Screen” that uses the protein-binding dye sulforhodamine B to assess cell growth [18]. Briefly, exponentially, cells growing in 96-well plates were then exposed for 48 h to five serial concentrations of each compound, starting from a maximum concentration of 150 µM. Following this exposure period adherent cells were fixed, washed, and stained. The bound stain was solubilized and the absorbance was measured at 492 nm
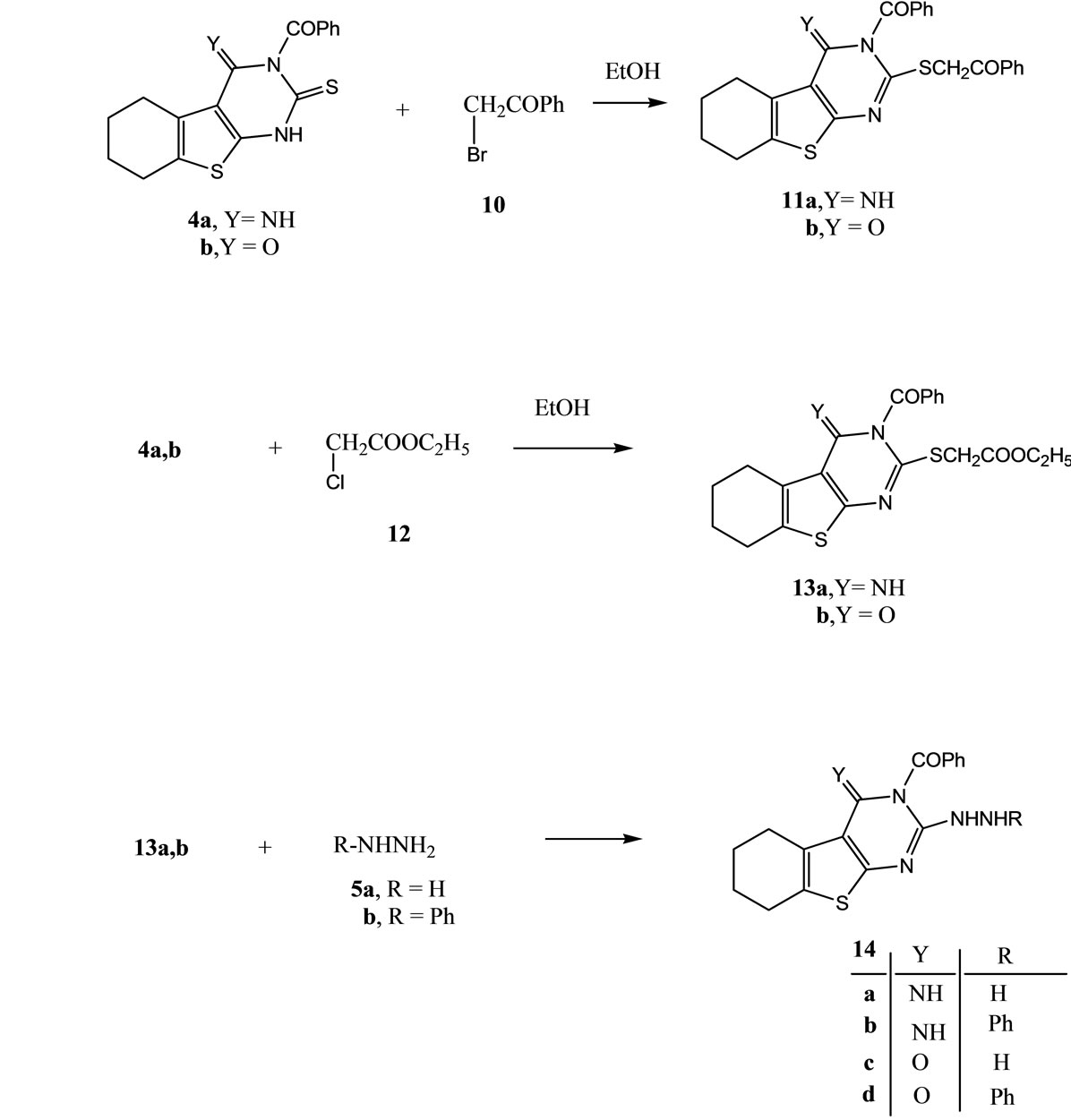
Scheme 3. Synthesis of compounds 11a,b-14a-d.
in a plate reader (Bio-Tek Instruments Inc., Powerwave XS, Wincoski, USA). For each test compound and cell line, a dose-response curve was obtained and the growth inhibition of 50% (GI50), corresponding to the concentration of the compounds that inhibited 50% of the net cell growth, was calculated as described elsewhere [19]. Doxorubicin was used as a positive control and tested in the same manner.
2.2. Effect on the Growth of Human Tumor Cell Lines
The effect of compounds 3a-14d was evaluated on the in vitro growth of three human tumor cell lines representing different tumor types, namely, breast adenocarcinoma (MCF-7), non-small cell lung cancer (NCI-H460) and CNS cancer (SF-268) after a continuous exposure for 48 h. The results are summarized in Table 1. All of the tested compounds were able to inhibit the growth of the tested human tumor cell lines in a dose-dependent manner (data not shown). The results indicated through Table 1 revealed that “compounds 11a and 14c showed the highest inhibitory effect against all the three tumor cell lines corresponding to reference standard material (Doxorubicin) for compounds 6a, 7d, and 14d showed the highest inhibitory effect against all the three tumor cell lines with respect to the remaining synthesized compounds”. While compounds 6d, 7b, 7c and 9 showed moderate inhibitory effects against the three cancer cell lines. The rest of the compounds showed a low growth inhibitory effect. Comparing compound 6a, 6b, 6c and 6d it is obvious that the presence of the NH group and R =H in compound 6a increased the inhibitory effect higher than that of the corresponding compounds 6b, 6c and 6d where as compound 6d with Y=O and R=Ph showed the lowest inhibitory effect. Comparing the tetrahydrobenzo [b]thiophene derivatives 7a, 7b, 7c and 7d it is obvious that compound 7d with the X=COOEt and the R=Ph showed the highest inhibitory effect among the fours compounds but compound 7c with X=COOEt, R=H showed moderate inhibitory effect among the four compounds.
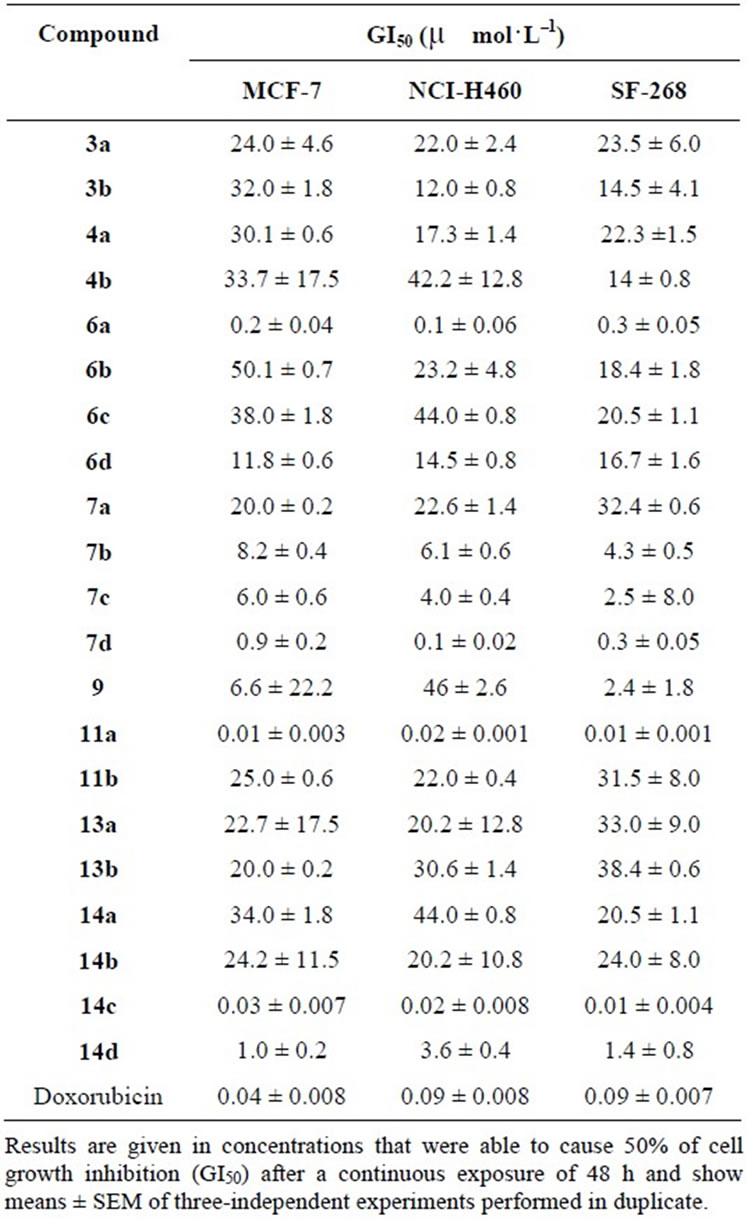
Table 1. Effect of compounds 3a-14d on the growth of three human tumor cell lines.
Finally for Hydrazopyrimidine derivatives 14a, 14b, 14c, 14d it is found that compounds 14c and 14d with the Y =O and the R=H or Ph respectively showed the highest inhibitory effect among the fours compounds but compounds 14a and 14b with the Y=NH and the R=H or Ph respectively showed the lowest inhibitory effect.
3. Experimental Section
3.1. General
All melting points were uncorrected; the IR spectra expressed in cm–1 and recorded using KBr pellets and a Pa-9721 IR spectrometer. 1H NMR spectra were obtained on a Varian EM-390 90 Hz spectrometer in DMSO-d6 as solvent and TMS as internal reference. Chemical shifts (d) are expressed in ppm. Elemental analyses were carried out by the Microanalytical Data Unit at the National Research Center, Giza, Egypt and the Microanalytical data Unit at Cairo University.
3.2. General Procedure for the Synthesis of: 3-Cyano-2-(N-benzoylthiouryl)-4,5,6,7-tetrahydro-benzo[b]-thiophene (3a) and Ethyl 2-(N-benzoylthiouryl)-4,5,6,7-tetrahydro-benzo[b]thiophen-3-carboxy-late (3b)
General procedure: Equimolar amounts of either 1a (5.34 g, 0.03 mol) or 1b (6.75 g, 0.03 mol) in 1,4-dioxane (50 mL), benzoylisothiocyanate (3.93 g, 0.03 mol) [prepared by adding ammonium isothiocyanate (0.03 mol) to a solution of benzoyl chloride (0.03 mol) in 1,4-dioxan (50 mL) and heat for 1/2 h followed by isolation of the byproduct, ammonium chloride] was added. The whole reaction mixture, in each case, was stirred at room temperature overnight and the formed solid product upon pouring onto ice/water was collected by filtration.
Compound 3a: Yellow crystals from acetic acid, yield 70% (7.17 g), m.p. 200˚C - 203˚C. Anal. Calculated for C17H15N3OS2 (341.45): C, 59.80; H, 4.43; N, 12.31; S, 18.78. Found: C, 60.07; H, 4.42; N, 12.58; S, 19.01. IR (u/cm–1): 3456 - 3334 (2NH), 3030 (CH aromatic), 2888 (CH2), 2227 (CN), 1690 (CO), 1660 - 1649 (C=S), 1639 (C=C). 1H NMR (d ppm): 2.14 - 2.16 (m, 4H, 2CH2); 2.23 - 2.26 (m, 4H, 2CH2), 7.30 - 7.41 (m, 5H, C6H5), 8.26, 8.30 (2s, 2H, 2NH).
Compound 3b: Pale yellow crystals from acetic acid, yield 60% (6.99 g), m.p. 160˚C. Anal. Calculated for C19H20N2O3S2 (388.50): C, 58.74; H, 5.19; N, 7.21; S, 16.51. Found: C, 58.89; H, 5.24; N, 7.31; S, 16.44. IR (u/cm–1): 3466 - 3339 (2NH), 3042 (CH aromatic), 2986, 2893 (CH3, CH2), 1692, 1685 (2 CO), 1665 - 1652 (C=S), 1636 (C=C). 1H NMR (d ppm): 1.32 (t, 3H, CH3), 2.18 - 2.19 (m, 4H, 2CH2); 2.27 - 2.29 (m, 4H, 2CH2), 4.23 (q, 2H, CH2), 7.18 - 7.35 (m, 5H, C6H5) 8.27, 8.30 (2s, 2H, 2NH).
3-Benzoyl-4-imino-2-thioxo-4,5,6,7-tetrahydro-benzo[b]thieno[2,3-d]pyrimidin (4a), 3-Benzoyl- 4-oxo-2-thioxo-4,5,6,7-tetrahydro-benzo[b]thieno[2,3-d]-pyrimidin (4b)
General procedure: A suspension of either 3a (3.41 g, 0.01 mol), 3b (3.88 g, 0.01 mol), in sodium ethoxide (0.01 mol) [prepared by dissolving sodium metal (0.23 g, 0.01 mol) in absolute ethanol (40 mL)] was heated in a boiling water bath for 6 hr then left to cool. The solid product was formed upon pouring onto ice/water containing few drops from hydrochloric acid (till pH = 6) was collected by filtration.
Compound 4a: Colorless crystals from 1,4-dioxan, yield 70% (2.39 g), m.p. 233˚C - 235˚C. Anal. Calculated for C17H15N3OS2 (341.45): C, 59.80; H, 4.43; N, 12.31; S, 18.78. Found: C, 59.57; H, 4.35; N, 12.48; S, 18.99. IR (u/cm–1): 3446 - 3336 (2NH), 3038 (CH aromatic), 2887 (CH2), 1687 (CO), 1661 - 1652 (C=S), 1639 (C=C). 1H NMR (d ppm): 1.64 - 1.72 (m, 4H, 2CH2); 2.21 - 2.23 (m, 4H, 2CH2), 7.32 - 7.44 (m, 5H, C6H5), 8.26, 8.29 (2s, 2H, 2NH).
Compound 4b: Pale yelow crystals from ethanol, yield 88% (3.01 g), m.p. 185˚C - 187˚C. Anal. Calculated for C17H14N2O2S2 (342.44): C, 59.63; H, 4.12; N, 8.18; S, 18.73. Found: C, 59.87; H, 4.23; N, 8.38; S, 18.58. IR (u/cm–1): 3456 - 3336 (NH), 3042 (CH aromatic), 2890 (CH2), 1692, 1687 (2 CO), 1638 (C=C), 1668 - 1651 (C=S). 1H NMR (d ppm): 1.63 - 1.70 (m, 4H, 2CH2); 2.20 - 2.26 (m, 4H, 2CH2), 7.32 - 7.39 (m, 5H, C6H5), 8.22 (s, 1H, NH).
3-Benzoyl-2-hydrazono-4-imino-4,5,6,7-tetrahydro benzo[b]thieno[2,3-d]pyrimidine (6a), 3-benzoyl-2-phenylhydrazono-4-imino-4,5,6,7-tetrahy-drobenzo[b]thieno[2,3-d]pyrimidine (6b), 3-benzoyl-2-hydrazono-4-oxo-4,5,6,7-tetrahydrobenzo[b]thieno[2,3-d]pyramidine (6c) and 3-benzoyl-2-phenyl hydrazono-4-oxo-4,5,6,7-tetrahydrobenzo[b]thieno-[2,3-d]pyrimidine (6d)
Method (A): To a solution of either 4a (0.68 g, 0.002 mol) or 4b (0.68 g, 0.002 mol) in DMF (40 mL) either hydrazine hydrate (0.1 g, 0.002 mol) or phenylhydrazine (0.22 g, 0.002 mol) was added. The reaction mixture, in each case was heated under reflux for 2 hr till evolution of hydrogen sulphide ceased. The reaction mixture, in each case, was left to cool then poured onto ice/water containing few drops of hydrochloric acid (till pH 6) and the formed solid product was collected by filtration.
Method (B): 3-Benzoyl-2-hydrazono-4-oxo-4,5,6,7-tetrahydro-benzo[b]-thieno-[2,3-d]pyrimidine (6c)
General procedure: To a solution of 4b (1.03 g, 0.003 mol) in 1,4-dioxane (40 mL) hydrazine hydrate (0.15 g, 0.003 mol) was added. The reaction mixture was stirred at room temperature for 24 hr and the formed crystals was collected by filtration.
Compound 6a: Yellow crystals from 1,4-dioxane, yield 75% (0.51 g), m.p. 188˚C - 191˚C. Anal. Calculated for C17H17N5OS (339.41): C, 60.16; H, 5.05; N, 20.63; S, 9.45. Found: C, 60.23; H, 5.11; N, 20.88; S, 9.77. IR (u/cm–1): 3504 - 3332 (NH2, 2NH), 3043 (CH aromatic), 1690 (CO), 1678, 1665 (exocyclic 2C=N), 1643 (C=C). 1H NMR (d ppm): 1.62 - 1.70 (m, 4H, 2CH2); 2.21 - 2.27 (m, 4H, 2CH2), 4.22 (s, 2H, NH2), 7.29 - 7.38 (m, 5H, C6H5), 8.12 - 8.28 (2s, 2H, 2NH).
Compound 6b: Yellowish white crystals from 1,4-dioxan, yield 62% (0.52 g), m.p. 233˚C - 236˚C. Anal. Calculated for C23H21N5OS (415.51): C, 66.48; H, 5.09; N, 16.85; S, 7.72. Found: C, 66.53; H, 4.93; N, 16.90; S, 8.02. IR (u/cm–1): 3449 - 3324 (3NH), 3052 (CH aromatic), 1692 (CO), 1669, 1660 (exocyclic 2C=N), 1642 (C=C). 1H NMR (d ppm): 1.63 - 1.66 (m, 4H, 2CH2); 2.22 - 2.28 (m, 4H, 2CH2), 7.26 - 7.53 (m, 10H, 2C6H5), 7.88, 8.03, 8.25 (3s, 3H, 3NH).
Compound 6c: Yellow crystals from DMF, yield 86% (0.59 g), m.p. 180˚C - 183˚C. Anal. Calculated for C17H16N4O2S (340.4): C, 59.98; H, 4.74; N, 16.46; S, 9.42. Found: C, 60.21; H, 4.82; N, 16.68; S, 9.61. IR (u/cm–1): 3555 - 3312 (NH2, NH), 3050 (CH aromatic), 1698, 1690 (2CO), 1670 (exocyclic C=N), 1643 (C=C). 1H NMR (d ppm): 1.64 - 1.72 (m, 4H, 2CH2); 2.25 - 2.29 (m, 4H, 2CH2), 4.48 (s, 2H, NH2), 7.32 - 7.43 (m, 5H, C6H5), 8.22 (s, 1H, NH).
Compound 6d: Yellow crystals from DMF, yield 70 % (0.58 g), m.p. 266˚C - 269˚C. Anal. Calculated for C23H20N4O2S (416.50): C, 66.33; H, 4.84; N, 13.45; S, 7.70. Found: C, 66.06; H, 4.76; N, 13.74; S, 7.98. IR (u/cm–1): 3555 - 3312 (2NH), 3058 (CH aromatic), 1692, 1680 (2CO), 1662 (C=N), 1643 (C=C). 1H NMR (d ppm): 1.68 - 1.77 (m, 4H, 2CH2); 2.23 - 2.28 (m, 4H, 2CH2), 7.29 - 7.35 (m, 10H, 2C6H5), 8.24 - 8.26 (2s, 2H, 2NH).
1-(3-Cyano-4,5,6,7-tatrayhdrobenzo[b]thiophene-2- yl)-2-hydrazono-3-benzoyl urea (7a), 1-(3-Cyano-4,5, 6,7-tatrayhdrobenzo[b]thiophene-2-yl)-2-phenyl hydrazono-3-benzoyl urea (7b), 1-(Ethyl-4,5,6,7-tatrayhdrobenzo[b]thiophene-3-carboxylate-2-yl)-2-hydrazono-3-benzoyl urea (7c), 1-(Ethyl-4,5,6,7-tatrayhdrobenzo[b]thio-phene-3-carboxylate-2-yl)-2- phenyl hydra-zono-3-benzoyl urea (7d)
To a solution of either 3a (1.7 g, 0.005 mol), 3b (1.94 g, 0.005 mol) in ethanol (50 mL), either hydrazine hydrate (0.25 g, 0.005 mol) or phenyl hydrazine (0.59 g, 0.005 mol) was added. The reaction mixture, in each case, was heated under reflux for 6 hr then poured onto ice/water containing few drops of hydrochloric acid (till pH 6) and the formed solid product was collected by filtration.
Compound 7a: Yellowish white crystals from acetic acid, yield 78% (1.32 g), m.p. 167˚C - 170˚C. Anal. Calculated for C17H17N5OS (339.41): C, 60.16; H, 5.05; N, 20.63; S, 9.45. Found: C, 59.93; H, 4.86; N, 20.41; S, 9.62. IR (u/cm–1): 3578 - 3332 (NH2, NH), 3045(CH aromatic), 2227 (CN), 1688 (CO), 1660 (C=N), 1636 (C=C). 1H NMR (d ppm): 1.68 - 1.74 (m, 4H, 2CH2); 2.22 - 2.28 (m, 4H, 2CH2), 4.56 (s, 2H, NH2), 7.31 - 7.45 (m, 5H, C6H5), 8.20, 8.34 (2s, 2H, 2NH).
Compound 7b: Yellow crystals from acetic acid, yield 66 % (1.37 g), m.p. 268˚C - 271˚C. Anal. Calculated for C23H21N5OS (415.51): C, 66.48; H, 5.09; N, 16.85; S, 7.72. Found: C, 66.32; H, 4.89; N, 16.95; S, 7.69. IR (u/cm–1): 3566 - 3332 (3 NH), 3055 (CH aromatic), 2223 (CN), 1688 (CO), 1662 (C=N), 1633 (C=C). 1H NMR (d ppm): 1.64 - 1.70 (m, 4H, 2CH2), 2.24 - 2.37 (m, 4H, 2CH2), 7.08 - 7.38 (m, 10H, 2C6H5), 8.02, 8.25 - 8.28 (3s, 3H, 3NH).
Compound 7c: Orange crystals from acetic acid, yield 69% (1.33 g), m.p. 190˚C - 192˚C. Anal. Calculated for C19H22N4O3S (386.47): C, 59.05; H, 5.74; N, 14.50; S, 8.30. Found: C, 59.28; H, 5.88; N, 14.79; S, 8.48. IR (u/cm–1): 3569 - 3322 (NH2, 2NH), 3057 (CH aromatic), 2983, 2856 (CH3, CH2), 1692, 1987 (2CO), 1670 (C=N), 1636 (C=C). 1H NMR (d ppm): 1.33 (t, 3H, J = 7.43 Hz, CH3), 1.64 - 1.72 (m, 4H, 2CH2); 2.26 - 2.29 (m, 4H, 2CH2), 4.23 (q, 2H, J = 7.43 Hz, CH2), 4.62(s, 2H, NH2), 7.30 - 7.38 (m, 5H, C6H5), 8.02, 8.24 (2s, 2H, 2NH).
Compound 7d: Buff crystals from DMF, yield 60% (1.39 g), m.p. 173˚C - 175˚C. Anal. Calculated for C25H26N4O3S (462.56): C, 64.91; H, 5.67; N, 12.11; S, 6.93. Found: C, 65.12; H, 5.92; N, 12.19; S, 7.02. IR (u/cm–1): 3484 - 3329 (3NH), 3051 (CH aromatic), 2992, 2890 (CH3, CH2), 1692, 1687 (2CO), 1663 (C=N), 1639 (C=C). 1H NMR (d ppm): 1.36 (t, 3H, J = 7.03 Hz, CH3), 1.68 - 1.73 (m, 4H, 2CH2), 2.24 - 2.28 (m, 4H, 2CH2), 4.26 (q, 2H, J = 7.03 Hz, CH2), 7.28 - 7.39 (m, 10H, 2C6H5), 8.02, 8.28, 8.34 (3s, 3H, 3NH).
3-Phenyl-5,6,7,8-tetrahydro-1H-9-thia-1,2,3a,10-tetraaza cyclopenta[b]fluoren-4-one (9):
Method (A): A solution of either 7a (0.68 g, 0.002 mol), 7c (0.77 g, 0.002 mol), in dimethylformamide (40 mL) containing piperidine (0.5 mL) was heated under reflux for 6 hr then left to cool. The solid product, formed upon pouring onto ice/water containing hydrochloric acid (till pH = 6) was collected by filtration.
Method (B): A solution of 6c (0.34 g, 0.001 mol) in dimethylformamide (30 mL) was heated under reflux for 4 hr. The reaction mixture was poured onto ice/water and the formed solid product was filtered off, crystallized from 1,4-dioxane and identified as compound 9 (m.p., mixed m.p.), yield 70% (0.23 g).
Compound 9: Pale yellow crystals from 1,4 dioxan, yield 70% (0.45 g), m.p. 210˚C - 212˚C. Anal. Calculated for C17H14N4OS (322.38): C, 63.33; H, 4.38; N, 17.38; S, 9.95. Found: C, 63.52; H, 4.55; N, 17.60; S, 10.05, IR (u/cm–1): 3380 - 3335 (NH), 3052 (CH aromatic), 1691 (CO), 1657 (C=N), 1644 (C=C). 1H NMR (d ppm): 1.60 - 1.66 (m, 4H, 2CH2); 2.20 - 2.32 (m, 4H, 2CH2), 7.32 - 7.37 (m, 5H, C6H5), 8.20 (s, 1H, NH).
3-Benzoyl-4-imino-2-phenylthioacetyl-4,5,6,7-tetrahydrobenzo[b]-thieno[2,3-d]pyrim-idine (11a) and 3-Benzoyl-2-phenylthioacetyl-4-oxo-4,5,6,7-tetrahydro benzo[b]-thieno[2,3-d]pyrimidine (11b)
To dry solid of either 4a (1.02 g, 0.003 mol) or 4b (1.03 g, 0.003 mol) in ethanol (40 mL), phenacylbromide (0.6 g, 0.003 mol) was added. The reaction mixture was heated under reflux for 2 hr. then left to cool and the remaining product was triturated with diethyl ether and the formed solid product was collected by filtration.
Compound 11a: Yellowish white crystals from acetic acid, yield 84% (1.16 g), m.p. 220˚C - 223˚C. Anal. Calculated for C25H21N3O2S2 (459.58): C, 65.33; H, 4.61; N, 9.14; S, 13.95. Found: C, 65.52; H, 4.43; N, 8.92; S, 14.01. IR (u/cm–1): 3489(NH), 3053 (CH aromatic), 2880 (CH2), 1710, 1682 (2CO), 1669 (C=N), 1637 (C=C), 1202 - 1191 (C-S). 1H NMR (d ppm): 1.64 - 1.68 (m, 4H, 2CH2); 2.25 - 2.28 (m, 4H, 2CH2), 2.86 (s, 2H, CH2), 7.03 - 7.38 (m, 10H, 2C6H5), 8.21 (s, 1H, NH)Compound 11b: Yellow crystals from acetic acid, yield 73% (1.0 g), m.p. 229˚C - 231˚C. Anal. Calculated for C25H20N2O3S2 (460.57): C, 65.20; H, 4.38; N, 6.08; S, 13.92. Found: C, 65.41; H, 4.41; N, 5.99; S, 13.89. IR (u/cm–1): 3056 (CH aromatic), 2890 (CH2), 1715 - 1688 (3CO), 1664 (C=N), 1637 (C=C), 1198 - 1190 (C-S). 1H NMR (d ppm): 1.68 - 1.72 (m, 4H, 2CH2); 2.23 - 2.24 (m, 4H, 2CH2), 2.74 (s, 2H, CH2), 7.33 - 7.42 (m, 10H, 2C6H5).
3-Benzoyl-2-ethyl-thioglycolato-4-imino-4,5,6,7-tetrahydrobenzo[b]-thieno[2,3-d]pyrimidine(13a) and 3-Benzoyl-2-ethyl-thioglycolato-4-oxo-4,5,6,7-tetra-hydrobenzo[b]-thieno[2,3-d]pyrimidine (13b)
A suspension of either 4a (1.02 g, 0.003 mol) 4b (1.03 g, 0.003 mol) in ethanol (50 mL), ethyl chloroacetate (0.37 g, 0.003 mol) was added. The reaction mixture was heated under reflux for 4 hr. then left to cool and the remaining product was triturated with diethyl ether and the formed solid product was collected by filtration.
Compo und 13a: Orange crystals from 1,4-dioxan, yield 61% (0.78 g), m.p. > 300˚C. Anal. Calculated for C21H21N3O3S2 (427.54): C, 58.99; H, 4.95; N, 9.83; S, 15.00. Found: C, 58.79; H, 4.72; N, 10.02; S, 14.83. IR (u/cm–1): 3378 (NH), 3059 (CH aromatic), 2971, 2885 (CH3, CH2), 1699, 1689 (2CO), 1670 (exocyclic C=N), 1644 (C=C), 1205 - 1192 (C-S). 1H NMR (d ppm): 1.36 (t, 3H, J = 7.13 Hz, CH3), 1.68 - 1.70 (m, 4H, 2CH2); 2.25 - 2.29 (m, 4H, 2CH2), 2.88(s, 2H, CH2), 4.23 (q, 2H, J = 7.13 Hz, CH2), 7.22 - 7.36 (m, 5H, C6H5), 8.26(s, 1H, NH).
Compound 13b: Orange crystals from DMF, yield 62% (0.8 g), m.p. 246˚C - 248˚C. Anal. Calculated for C21H20N2O4S2 (428.52): C, 58.86; H, 4.70; N, 6.54; S, 14.97. Found: C, 59.12; H, 4.53; N, 6.76; S, 14.94. IR (u/cm–1): 3049 (CH aromatic), 2986, 2876(CH3, CH2), 1698, 1693, 1685 (3CO), 1637 (C=C), 1203 - 1196 (C-S). 1H NMR (d ppm): 1.16 (t, 3H, J = 7.22 Hz, CH3), 1.65 - 1.74 (m, 4H, 2CH2); 2.22 - 2.27 (m, 4H, 2CH2), 2.75(s, 2H, CH2), 4.24 (q, 2H, J = 7.22 Hz, CH2), 7.28 - 7.38 (m, 5H, C6H5).
3-Benzoyl-2-hydrazino-4-imino-4,5,6,7 tetrahydrobenzo[b]-thieno[2,3-d]pyrimidine (14a), 3-Benzoy-4-imino l-2-phenylhydrazino-4,5,6,7-tetrahydrobenzo[b] thieno[2,3-d]pyrimidine (14b), 3-Benzoyl-2-hydrazino-4-oxo-4,5,6,7-tetrahydrobenzo[b]-thieno-[2,3-d] pyrimidine (14c) and 3-Benzoyl-2-phenylhydrazino-4-oxo-4,5,6,7-tetrahydro benzo[b]-thieno[2,3-d]pyrimidine (14d)
To a solution of either 13a (0.43 g, 0.001 mol) or 13b (0.43 g, 0.001 mol) in 1,4-dioxane (40 mL) either hydrazine hydrate (0.05 g, 0.001) or phenylhydrazine (0.11 g, 0.001) was added. The reaction mixture, in each case, was heated under reflux for 2 hr then poured onto ice/ water containing few drops of hydrochloric acid (till pH 6) and the formed solid product was collected by filtration.
Compound 14a: Pale brown crystals from 1,4-dioxan, yield 74% (0.25 g), m.p. 192˚C - 195˚C. Anal. Calculated for C17H17N5OS (339.41): C, 60.16; H, 5.05; N, 20.63; S, 9.45. Found: C, 59.93; H, 4.82; N, 20.67; S, 9.72. IR (u/cm–1): 3455 - 3330 (NH2, 2NH), 3064 (CH aromatic), 1690 (CO), 1677 (exocyclic C=N), 1639 (C= C). 1H NMR (d ppm): 1.65 - 1.70 (m, 4H, 2CH2); 2.22 - 2.31 (m, 4H, 2CH2), 4.21 (s, 2H, NH2), 7.33 - 7.39 (m, 5H, C6H5), 8.38, 8.94 (2s, 2H, 2NH).
Compound 14b: Brown crystals from 1,4-dioxan, yield 62% (0.27 g), m.p. 171˚C - 173˚C. Anal. Calculated for C23H21N5OS (415.51): C, 66.48; H, 5.09; N, 16.85; S, 7.72. Found: C, 66.50; H, 5.02; N, 16.72; S, 7.48 IR (u/cm–1): 3442 - 3321 (3NH), 3052 (CH aromatic), 1693 (CO), 1636 (C=C). 1H NMR (d ppm): 1.66 - 1.73 (m, 4H, 2CH2); 2.24 - 2.28 (m, 4H, 2CH2), 7.31 - 7.40 (m, 10H, 2 C6H5), 8.18, 8.30, 8.39 (3s, 3H, 3NH).
Compound 14c: Pale brown crystals from 1,4-dioxan, yield 78% (0.27 g), m.p. 215˚C - 217˚C. Anal. Calculated for C17H16N4O2S (340.40): C, 59.98; H, 4.74; N, 16.46; S, 9.42. Found: C, 59.81; H, 4.88; N, 16.67; S, 9.70. IR (u/cm–1): 3455 - 3330 (NH2, NH), 3064 (CH aromatic), 1690, 1687 (2 CO), 1639 (C=C). 1H NMR (d ppm): 1.65 - 1.77 (m, 4H, 2CH2); 2.20 - 2.31 (m, 4H, 2CH2), 4.46 (s, 2H, NH2), 7.31 - 7.36 (m, 5H, C6H5), 8.36 (s, 1H, NH).
Compound 14d: Yallow crystals from 1,4-dioxan, yield 65% (0.27 g), m.p. 103˚C - 105˚C. Anal. Calculated for C23H20N4O2S (416.50): C, 66.33; H, 4.84; N, 13.45; S, 7.70. Found: C, 66.29; H, 4.62; N, 13.62; S, 7.60. IR (u/cm–1): 3442 - 3321 (2NH), 3052 (CH aromatic), 1693, 1684 (2CO), 1636 (C=C). 1H NMR (d ppm): 1.62 - 1.75 (m, 4H, 2CH2); 2.24 - 2.34 (m, 4H, 2CH2), 7.31 - 7.44 (m, 10H, 2C6H5), 8.26 - 8.30 (2s, 2H, 2NH).
4. Conclusion
The work described in this article showed the synthesis of fused thieophene derivatives, most of the newly synthesized products showed high antitumor activities. The 4,5,6,7-tetrahydrobenzo[b]-thieno[2,3-d]pyrimidine derivatives 11a and 14c showed the maximum inhibitory effect among the newly synthesized products where such activiity is higher than that of the reference doxorubicin.
5. Acknowledgements
The authors would like the research group working at both of the National Research Center and the National Cancer Institute for recording the antitumor evaluation for the synthesized products.
REFERENCES
- W. W. Wardakhan, H. M. Gaber, S. A. Ouf and S. M. Sherif, “Studies on 2-Amino Thiophenes: Synthesis, Transformations, and Biological Evaluation of FunctionallySubstituted Thiophenes and Their Fused Derivatives,” Phosphorus, Sulfur and Silicon, Vol. 180, No. 2, 2005, pp. 601-618. doi:10.1080/104265090517406
- A. A. Fadda, E. A. bdel-Latif and R. E. El-Mekawy, “Synthesis and Molluscicidal Activity of Some New Thiophene, Thiadiazole and Pyrazole Derivatives,” European Journal of Medicinal Chemistry, Vol. 44, 2009, pp. 1250- 1256. doi:10.1016/j.ejmech.2008.09.006
- S. Bondock, A. El-Gaber Tarhoni and A. A. Fadda, “Synthesis Utility of Cyanoacetic Acid Hydrazide in Heterocyclic Synthesis,” Online Journal of Organic Chemistry, Vol. 9, 2006, pp. 113-156.
- R. M. Mohareb, S. M. Sherif, and H. El-Dean Moustafa, “The Reaction of Cyclohexan-1,3-dione with Cyanomethylenes: Synthesis of Thiophenes and Their Fused Derivatives with Antifungal Activities,” Phosphorus, Sulfur, and Silicon, Vol. 184, No. 8, 2009, pp. 2078-2096. doi:10.1080/10426500802421127
- I. M. I. Fakhr, M. A. A. Radwan, S. El-Batran, O. M. E. Abd El-Salam and S. M. El-Shenawy, “Synthesis and Pharmacological Evaluation of 2-Substituted Benzo[b] thiophenes as Anti-Inflammatory and Analgesic Agents,” European Journal of Medicinal Chemistry, Vol. 44, No. 4, 2009, pp. 1718-1725. doi:10.1016/j.ejmech.2008.02.034
- K. I. Molvi, K. K. Vasu, S. G. Yerande, V. Sudarsanam and N. Haque, “Syntheses of New Tetrasubstituted Thiophenes as Novel Anti-Inflammatory Agents,” European Journal of Medicinal Chemistry, Vol. 42, No. 8, 2007, pp. 1049-1058. doi:10.1016/j.ejmech.2007.01.007
- P. R. Kumar, S. Raju, P. S. Goud, M. Sailaja, M. R. Sarma, G. Om Reddy, M. P. Kumar, V. V. R. M. K. Reddy, T. Suresh and P. Hegde, “Synthesis and Biological Evaluation of Thiophene[3,2-b]pyrrole Derivatives as Potential Anti-Inflammatory Agents,” Bioorganic & Medicinal Chemistry, Vol. 12, No. 5, 2004, pp. 1221-1230. doi:10.1016/j.bmc.2003.11.003
- N. Bharti, K. Husain, M. T. G. Garza, D. E. Cruz-Vega, J. Castro-Garza, B. D. Mata-Cardenas, F. Naqvia and A. Azama, “Synthesis and in Vitro Antiprotozoal Activity of 5-Nitrothiophene-2-carboxaldehyde Thiosemicarbazone Derivatives,” Bioorganic & Medicinal Chemistry Letters, Vol. 12, No. 23, 2002, pp. 3475-3478. doi:10.1016/S0960-894X(02)00703-5
- J. Valderrama, A. Fournet, C. Valderrama, S. Bastias, C. Astudillo, A. R. de Arias, A. Inchausti and G. Yaluff, “Synthesis and in Vitro Antiprotozoal Activity of Thiophene Ring-Containing Quinones,” Chemical & Pharmaceutical Bulletin, Vol. 47, No. 9, 1999, pp. 1221-1226. doi:10.1248/cpb.47.1221
- P. Dallemagne, L. Khanh, A. E. Alsaı̈di, I. Varlet, V. Collot, M. Paillet, R. Bureau and S. Rault, “Synthesis and Biological Evaluation of Five-Membered Heterocycles Fused to Cyclopenta[c]thiophene as New Antitumor Agents,” Bioorganic & Medicinal Chemistry, Vol. 11, No. 7, 2003, pp. 1161-1167. doi:10.1016/S0968-0896(02)00654-5
- A. E. Shchekotikhin, V. A. Glazunova, L. G. Dezhenkova, Y. N. Luzikov, Y. B. Sinkevich, L. V. Kovalenko, V. N. Buyanov, J. Balzarini, F. C. Huang, J.-J. Lin, H.-S. Huang, A. A. Shtil and M. N. Preobrazhenskaya, “Synthesis and Cytotoxic Properties of 4,11-Bis[(aminoethyl)amino] Anthra[2,3-b]thiophene-5,10-diones, Novel Analogues of Antitumor Anthracene-9,10-diones,” Bioorganic & Medicinal Chemistry, Vol. 17, 2009, pp. 1861-1869. doi:10.1016/j.bmc.2009.01.047
- M. E. Migaud, R. C. Wilmouth, G. I. Mills, G. J. Wayne, C. Risleuy, C. Chanbers, S. J. Macdonald and C. J. Schofield, “5,5-Fused Thiophene γ-Lactams as Templates for Serine Protease Inhibition,” Chemical Communications, Vol. 12, 2002, pp. 1274-1275. doi:10.1039/b111627d
- M. Pietsch and M. Gutschow, “Alternate Substrate Inhibition of Cholesterol Esterase by Thieno[2,3-d][1,3]oxazin- 4-ones,” The Journal of Biological Chemistry, Vol. 277, No. 27, 2002, pp. 24006-24013. doi:10.1074/jbc.M112252200
- R. M. Mohareb and F. M. Manhi, “Reaction of Ethyl 2-Diazo-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate with 3-Iminobutyronitrile: Synthesis of Pyridazines, Thiophenes, and Their Fused Derivatives,” Heteroatom Chemistry, Vol. 11, No. 6, 2000, pp. 403-412. doi:10.1002/1098-1071(2000)
- S. Reyes and K. Burgess, “Formation of Thiohydantoins from Amino Acids under Acylation Conditions,” The Journal of Organic Chemistry, Vol. 71, 2006, pp. 2507-2509. doi:10.1021/jo052576i
- R. M. Mohareb, A. Habashi, H. Z. Shams and S. M. Fahmy, “Polyfunctionally Substituted Pyridines from Cyanoacetamide and Cyanoacetanilide,” Archiv der Pharmazie, Vol. 320, No. 7, 1987, pp. 599-604. doi:10.1002/ardp.19873200706
- A. Z. A.-A. Elassar, “Novel Synthesis of Dihydroquinoxalinyl-Pyridazine Derivatives,” Journal of the Chinese Chemical Society, Vol. 53, No. 3, 2006, pp. 901-907.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. J. Boyd, “New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening,” National Cancer Institute, Vol. 82, No. 13, 1990, pp. 1107-1112.
- A. Monks, D. Scudiero, P. Skehan, R. Shoemaker, K. Paull, D. Vistica, C. Hose, J. Langley, P. Cronise, A. Vaigro-Wolff, M. Gray-Goodrich, H. Campbell, J. Mayo and M. J. Boyd, “Feasibility of a High-Flux Anticancer Drug Screen Using a Diverse Panel of Cultured Human Tumor Cell Lines,” National Cancer Institute, Vol. 83, No. 11, 1991, pp. 757-766.
NOTES
*Corresponding author.