Journal of Crystallization Process and Technology
Vol.3 No.1(2013), Article ID:27288,4 pages DOI:10.4236/jcpt.2013.31005
Synthesis, Spectral Characterization and Ligation of N-[2-(Phenylseleno)ethyl]phthalimide
![]()
1School of Studies in Chemistry, Jiwaji University, Gwalior, India; 2Department of Chemistry, Howard University, Washington DC, USA.
Email: *rastogirupali@ymail.com
Received September 6th, 2012; revised October 16th, 2012; accepted November 7th, 2012
Keywords: Phthalimide; Selenium; Monoclinic; Phenylseleno; Sodium Borohydride
ABSTRACT
PhSe−Na+ generated in situ by reduction of PhSeSePh, with sodium borohydride on reaction with N-(2-bromoethyl) phthalimide in N2 atmosphere results in the formation of N-[2-(phenylseleno)ethyl]phthalimide (L1). The title compound has been characterized by elemental analysis, FT-IR, 1H and 13C NMR techniques. The crystal structure of L1 has been solved by direct methods and refined by full-matrix least squares. The ligand L1 crystallize in the monoclinic space group. Selenium forms two Se-C linkages, one is due to Se-Calkyl and the other one to Se-Caryl. Further, the ligation reaction of L1 with complex 1 is also explored whose identities are characterized by spectroscopic techniques.
1. Introduction
There is current interest in the chemistry of a variety of polydentate organochalcogen ligands due to their hemilabile nature, and the possiblilty of using their metal complexes as precursor for II-VI semiconductors [1-4]. The synthesis and characterization of N-{2-(4methoxyphenyl telluro) ethylphthalimide} [5], potentially [Te, N, O2] type donor along with its palladium [5] and ruthenium complexes [6] were studied. Recently various bidentate heteroleptic ligands [7] containing donor groups (N, S and N, Se), have been prepared which offer opportunities in catalytic cycle of aryl bromides. In our previous study, we reported the synthesis, characterization, and crystal structure of the first example of selenium containing acetate salt (3-Phenylseleno) propylammonium acetate salt [8]. We have also reported the cleavage of L1 in two different ways upon reaction with selected metal salts [8]. In continuation of our studies on (Te, N, O) and (N, S and N, Se) donors, we have synthesized a novel (Se, N, O) donors. L1 was characterized by spectral and X-ray analysis.
2. Experimental
2.1. Materials
Diphenyl diselenide [9], was prepared according to the published method, N-(2-bromoethyl) phthalimide was used as received from Aldrich. Selenium powder and sodium borohydride were purchased from CDH (India). All glassware were cleaned with acetone, followed by copious rinsing with distilled water before rinsing with distilled water before drying at 150˚C in oven for a few hours.
2.2. Equipment
All reactions involving air sensitive compounds were performed under nitrogen atmosphere. The C, H and N analysis were carried out with an Elementar Vario EL III analyser. 1H and 13C NMR spectra were recorded with a JEOL AL300 FT NMR spectrometer at 300 MHz and 75.45 MHz, respectively, in CDCl3 and DMSO(d6) as solvents (tetra methyl silane (TMS) as internal standard). IR spectra in the range of 4000 - 400 cm−1 were recorded on a Shimadzu Prestige 21 FT-IR spectrometer as KBr pellets. The melting point was determined in open capillary are reported as such.
2.3. Ligand Synthesis
Synthesis N-[2-(phenylseleno)ethyl]phthalimide (L1). To an aqueous THF solution (20 mL THF + 0.2 mL H2O) of diphenyl diselenide (3.14 g, 10 mmol) and NaOH (0.80 g, 20 mmol) under N2 atm, NaBH4 (0.75 g, 20 mmol) was added in pinches. The yellow color of the diselenide darkened and then gradually faded to colorless. To this colorless solution, N-(2-bromoethyl)phthalimide [5.08 gm, 20 mmol] in THF (20 mL) was added dropwise and the solution was stirred for 24 h at room temperature. It was hydrolyzed with 100 mL water and extracted into 200 mL chloroform, washed with 100 mL water and dried over anhydrous magnesium sulphate. The extract was concentrated to 5 mL under reduced pressure, then washed with diethyl ether to remove the precursor diphenyl diselenide, and kept overnight at room temperature. The resulting white precipitate was recrystallized with chloroform-diethyl ether mixture (1:2) and dried in vacuo. It was followed by crystallization with slow evaporation of a CHCl3/MeOH solution to yield a transparent crystals. Yield: 70% Mp: 120˚C Found: C, 58.04; H, 3.99; N, 4.17; Se, 23.82; Calcd for C16H13NO2Se (MW: 331 g/mol); C, 58.19; H, 3.96; N, 4.24; Se, 23.90%. IR (KBr, cm−1); 512 m, ν (Se-Calkyl); 1170 s, ν (C-N); 1708 s, ν (C=O); 1H-NMR (300 MHz, CDCl3, TMS): δH 7.81 - 7.21 (m, 9H, H-Ar), 3.19 (t, 2H, 3JH-H = 6.3 Hz, Se-CH2), 4.00 (t, 2H, 3JH-H = 7.2 Hz, N-CH2) ppm. 13C NMR (75 MHz, CDCl3, TMS) δC, 24.82 (C10, Se-CH2); 38.29 (C9, N-CH2); 129.03 (C11, Seipso); 168.04 (C8, C=O), 127.11 (C13), 123.27 (C14), 129.13 (C6), 132.00 (C7), 132.72 (C5), 133.94 (C12).
2.4. Complex Synthesis
Synthesis of [(HgCl2 (L1)] (1). A mixture of L1 (0.331 gm, 1 mmol) and mercuric chloride (0.271 g, 1 mmol) in methanol 5 mL was stirred at room temperature for 5 hr. The resultant white precipitate was filtered, washed with petroleum ether and dried in vacuo. It was recrystallized with a methanol:chloroform (9:1) mixture. Yield: 82% Mp: 90˚C Found: C, 31.84; H, 2.14; N, 2.36; Se, 13.04; Calcd for C16H13NO2SeHgCl2: C, 32.05; H, 2.17; N, 2.33; Se, 13.02%. IR (KBr, cm−1); 501 m, ν (Se-Calkyl); 1153 s, ν (C-N); 1708 s, ν (C=O); 1H NMR (300 MHz, DMSO (d6), TMS): δH, 7.74 - 7.82 (m, 9H), 3.81 (t, 2H, 3JH-H = 4.5 Hz, Se-CH2), 4.22 (t, 2H, 3JH-H = 6 Hz, N-CH2) ppm. 13C NMR (75 MHz, DMSO (d6), TMS): δC, 25.24 (C10, Se-CH2); 39.45(C9, N-CH2); 129.17 (C11, Seipso); 167.46 (C8, C=O), 126.63 (C13), 123.27 (C14), 129.17 (C6), 131.39 (C7), 132.10 (C5), 134.27 (C12) ppm.
3. Crystal Structure Analysis
X-ray data for N-[2-(phenylseleno)ethyl]phthalimide were collected with an Oxford Diffraction Gemini R CCD area detector using CrysAlisPro software and graphite-monochromated MoKα (l = 0.71073 Å) at 200 (2) K. The size of crystal used for data collection was 0.55 × 0.35 × 0.12 mm3. The structure was solved by direct methods using SHELXS97 [10] and all of the non-hydrogen atoms were refined anisotropically by full matrix least-squares on F2 using SHELXL97 [10]. The hydrogen atoms were placed in their calculated positions and included in the refinement using the riding model. An absorption correction was performed using CrysAlis RED and all calculations were performed using SHELXTL [11].
4. Results and Discussion
The reaction given in Scheme 1 produce N-[2-(phenylseleno)ethyl]phthalimide (L1), which remain stable under ambient conditions for 2 - 3 months. It is soluble in ethanol, methanol, chloroform and dichloromethane. It was characterized by elemental analysis, IR, 1H, 13C NMR and single crystal structure. In the IR spectra of L1 due to presence of electronegative selenium, band of amide group is shifted to higher wavenumber 1708 cm−1 in comparison to the precursor N-[2-(bromo) ethyl] phthalimide at 1681 cm−1. The bands at 1170 and 512 cm−1 are attributed to ν (C-N) and ν (Se-CH2 ). The 1H and 13CNMR spectra of L1 display the expected resonances and peak multiplicities. 1:1 heterobimetallic adducts of L1 with HgCl2 was obtained by stirring a mixture of the two components in methanol: chloroform mixture (1:1) at r.t. (Scheme 2). Complex 1 remains stable under ambient conditions for 2 - 3 months. It is soluble in ethanol, methanol, chloroform and dichloromethane. It was characterized by elemental analysis, FAB mass IR, 1HNMR and 13CNMR. FAB mass spectrum of complex 1 confirms its monomeric character and the presence of a 1:1 metal:ligand stoichiometry in the compound. The molecular ion peak [M + H] is observed at m/z 604 with a very low intensity. The base peak at m/z 174 corresponds to [L1-SePh]. The peaks at m/z 533 [MH-2Cl] and m/z 254 [L1-Ph] are observed with low intensity whereas the peaks at m/z 568 [MH-Cl] and 331 [L1] observed with abundance. In the IR spectra of complex 1, the absorption due to ν (C-N) and ν (Se-CH2) are shifted to lower frequency and appear at 1153 and 501 cm−1 re-
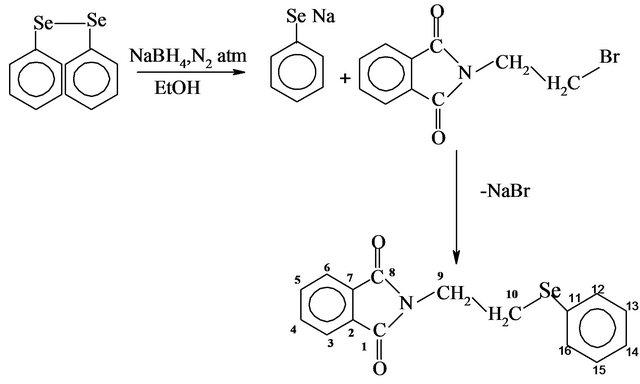
Scheme 1. Preparation of N-[2-(phenylseleno)ethyl]phthalimide (L1).
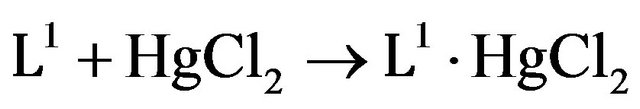 (1)
(1)
Scheme 2. Preparation of complexes 1.
spectively. The absorption due to ν (C=O) remain almost unaffected, indicating that L1 in complex 1 is coordinated through N and Se. The 1H and 13C NMR spectra of complex 1 are normal. In d10 system, coordination shifts in the NMR spectra is found to be insignificant [8]. In 1H NMR spectrum of complex 1 the downfield shift of SeCH2 and N-CH2 signals clearly establishes the coordination through Se and N to the metal atom. In the 13C {1H} NMR spectra of complex 1, the deshielding of Se-CH2 and N-CH2 signals by 0.42 and 1.16 ppm respectively with respect to those of free ligand L1, also argues for the coordination of the selenium and the nitrogen atom to the metal ion. Thus, the IR and NMR data shows that the ligand L1 ligates through (Se, N) in a bidentate mode [12-14] to the metal salt and support from previous work, the following most possible structure has been proposed on the presumption that geometry of complex 1 is tetrahedral (Figure 1)
The molecular and that of the unit cell, as determined by X-ray diffraction on a single crystal of N-[2-(phenylseleno)ethyl]phthalimide, are shown in Figures 2 and 3. It may act as a (Se, N) donor easily and perhaps (Se, N, O) donor also in bimetallic complexes. L1 crystallizes in the
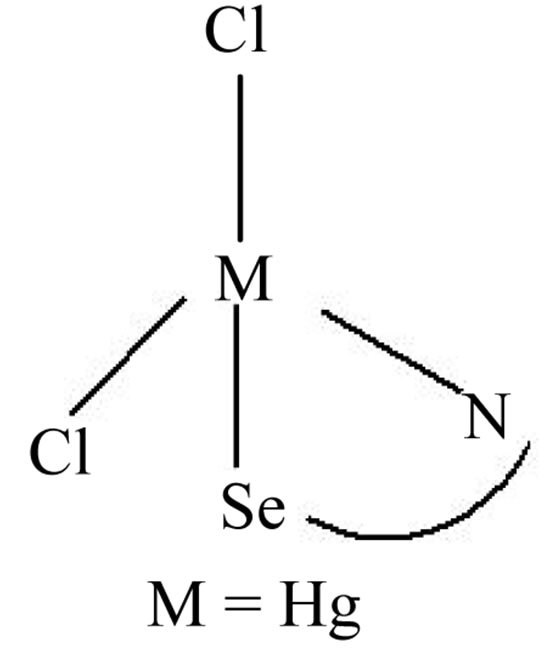
Figure 1. Proposed structure of complex 1.
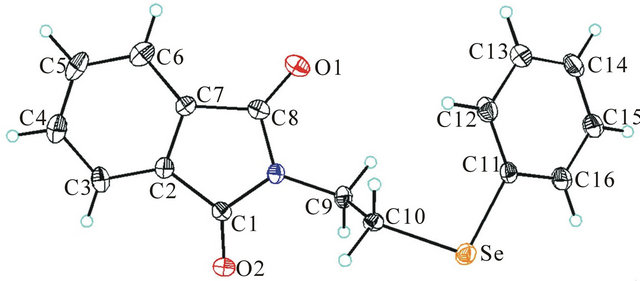
Figure 2. The molecular structure of the complex, C16H13- NO2Se, showing the atom numbering scheme and 30% probability displacement ellipsoids. Symmetry transformations used to generate equivalent atoms.
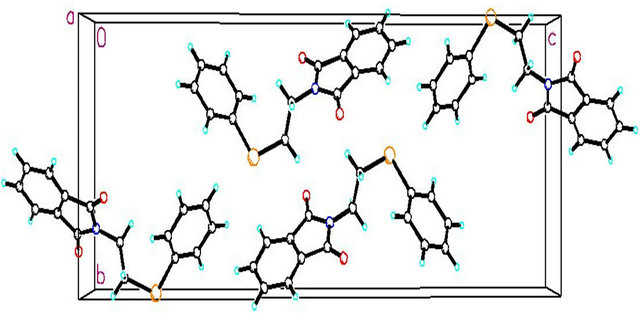
Figure 3. The molecular packing for C16H13NO2Se, viewed down the axis.
monoclinic space group P 21/n with unit cell parameters a = 5.0586 (5), b = 10.1316 (8), c = 27.050 (5), Å = 93.250 (11)˚, Z = 4. Crystal and experimental data for L1 are listed in Table 1. Bond lengths and bond angles are all within expected ranges, (see Table 2). In the present compound selenium is regarded as in oxidation state +2. The length of the C-Se bonds, 1.919 (3) and 1.952 (3)
Table 1. Crystal data and structure refinement for N-[2- (phenylseleno)ethyl]phthalimide.

Table 2. Bond lengths [Å] and angles [˚] for N-[2-(phenylseleno)ethyl]phthalimide.
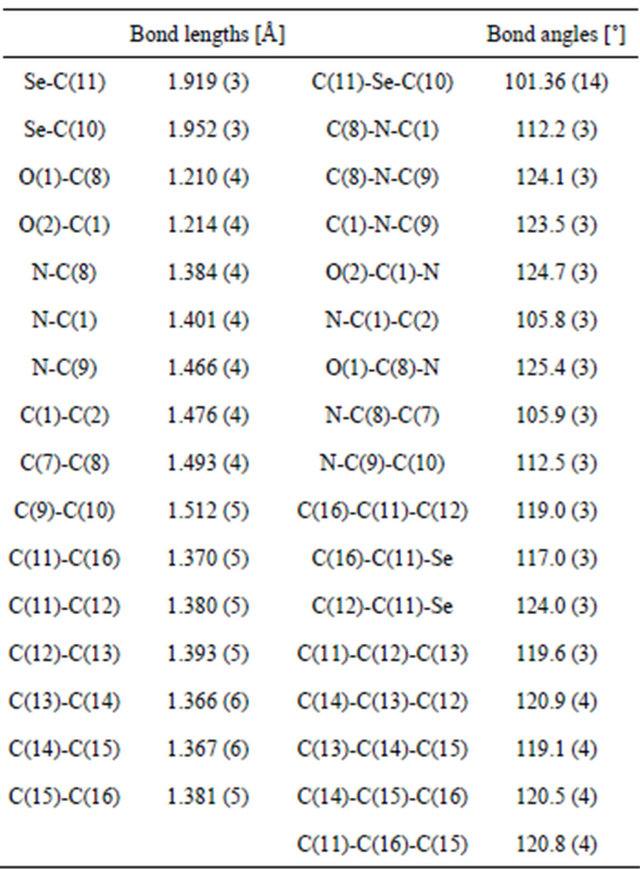
Ǻ and the angle between these two bonds, C-Se-C are consistent [15]. The Se-C(Ar) bond distance is somewhat shorter than the Se-C(alkyl) distance as expected [16].
5. Conclusion
We have shown that NaBH4 can be used as reduction system to generate an organochalcogen anion. This anion reacts in a SN2 reaction on N-[2-bromoethyl]phthalimide to produce N-[2-(phenylseleno)ethyl]phthalimide (L1) under mild conditions. These organochalcogen derivatives, L1, was used to synthesize a metal complex 1, from HgCl2. L1 act as a polydentate ligand containing oxygen or nitrogen as a hard donor. And chelate 1 was characterized as 1:1 complex by various spectroscopic techniques (IR, FAB mass, 1H and 13C NMR).
6. Supplementary Material
For L1, crystal data, structural information (geometric bond distances, bond and torsion angles; hydrogen bonding) & refinement data are provided. Crystallographic data (CIF) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre and allocated the deposition numbers CCDC 766004 for L1. These data can be obtained, free of charge via www.ccdc.cam.ac.uk/data_request/cif, by sending an e-mail to data_request@ccdc.cam.ac.uk., from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax; +44 1223 336033
7. Acknowledgements
One of the authors (Rupali Rastogi) is grateful to UGC, New Delhi, India for financial assistance. The authors are thankful to the Director, Central Drug Research Institute, Lucknow, India and Chairman CISC, Banaras Hindu University, Varanasi, India for C, H, N analysis and 1H, 13C NMR spectra, respectively. Further, we are also thankful to the Head, School of Studies in Chemistry, Jiwaji University, Gwalior, India for FTIR. Crystallographic studies were supported in part by the Office of Naval Research (ONR) and the Naval Research Laboratory (NRL). RJB would like to acknowledge the NSFMRI program (CHE-0619278) for funds to purchase the X-ray diffractometer.
REFERENCES
- A. Panda, S. C. Menon, H. B. Singh, C. P. Morley, R. Bachman, T. M. Cocker and R. J. Butcher, “Synthesis, Characterization and Coordination Chemistry of Some Selenium-Containing Macrocyclic Schiff Bases,” European Journal of Inorganic Chemistry, Vol. 2005, No. 6, 2005, pp. 1114-1126. doi:10.1002/ejic.200400757
- P. R. Kumar, A. K. Singh, J. E. Drake, M. B. Hursthouse and M. E. Light, “First Structurally Characterized Complex of an Acyclic Tellurated Schiff Base [4-MeOC6H4- TeCH2CH2NiC(CH3)C6H4-2-OH (L1H)] Having MetalTellurium Bond; Synthesis and Crystal Structure of [PdCl (L1)],” Inorganic Chemistry Communications, Vol. 7, No. 4, 2004, p. 502.
- M. D. Milton, S. Khan, J. D. Singh, V. Mishra and B. L. Khandelwal, “Afacile Access to Chalcogen and Dichalcogen Bearing Dialkylamines and Diols,” Tetrahedron Letters, Vol. 46, No. 5, 2005, p. 755.
- K. A. Bala and B. L. Khandelwal, “Synthesis and Multinuclear NMR Studies of 3-Aminopropyl(aryl)chalcogenides, NH2CH2CH2CH2EAr (E = Se, Te), and Their Complexes with Pt(II) and Pd(II),” Journal of Organometallic Chemistry, Vol. 494, 1995, p. 199.
- R. Batheja, S. K. Dhingra and A. K. Singh, “N-[2-(4-Methoxyphenyltelluro)ethyl]phthalimide: Synthesis and Complexation with Palladium(II),” Journal of Organometallic Chemistry, Vol. 487, No. 2, 1995, pp. 173-176. doi:10.1016/0022-328X(94)05105-K
- A. K. Singh, M. Kadarkaraisamy, G. S. Murthy, J. Srinivas, B. Varghese and R. J. Butcher, N-[2-(4-methoxyphenyltelluro)ethyl] Phthalimide (L1): Synthesis, Oxidation by Ruthenium(III) Chloride and Ligation with Ruthenium(II). Crystal Structures of L1, Its Oxidized Product and of [RuCl2 (p-Cymene)·L1],” Journal of Organometallic Chemistry, Vol. 605, No. 1, 2000, pp. 39-44. doi:10.1016/S0022-328X(00)00250-3
- C. Jones, A. J. Canty, M. G. Gardiner, B. W. Skelton, V. A. Tolhurst and A. H. White, “Synthesis and Structure of Dichloropalladium(II) Complexes of Heteroleptic N,Sand N,Se-Donor Ligands Based on the 2-Organochalcogenomethylpyridine Motif, and Mizoroki-Heck Catalysis Mediated by Complexes of N,S-Donor Ligands,” Inorganica Chimica Acta, Vol. 363, No. 1, 2010, pp. 77-87. doi:10.1016/j.ica.2009.10.002
- R. Rastogi, S. K. Srivastava, R. J. Butcher and J. P. Jansiki, “Synthesis, Structural Characterization and Crystal Structure of Selenium Bearing 3-(Phenylseleno) Propylammonium Acetate Salt and Its Cleavage on Reaction with Mercury(II) and Tin(IV) Salts,” Journal of Chemical Crystallography, Vol. 41, No. 3, 2011, pp. 391-400. doi:10.1007/s10870-010-9895-3
- K. B. Sharpless and M. W. Young, “Olefin Synthesis. Rate Enhancement of the Elimination of Alkyl Aryl Selenoxides by Electron-Withdrawing Substituents,” The Journal of Organic Chemistry, Vol. 40, No. 7, 1975, p. 947- 949. doi:10.1021/jo00895a030
- Bruker, AXS Inc., “SHELXTL. Version 6.10,” Bruker AXS Inc., Madison, 2000.
- G. M. Sheldrick, “SHELXS97, SHELXL97,” University of Gőttingen, Gőttingen, 1997.
- A. K. Singh and V. Srivastava, “Synthesis of Novel Bidentate (Te, N) Ligands-2-Aryltelluroethylamines and Their Complexation with Mercury (II),” Phosphorus Sulfur Silicon, Vol. 47, No. 4, 1990, pp. 471-475. doi:10.1080/10426509008038003
- S. K. Srivastava, Y. Pandey and S. B. Saxena, “Synthesis of Triphenyltin(IV) Compounds Containing Sn-S Bonds,” Journal of the Indian Chemical Society, Vol. 78, 2001, p. 254.
- T. N. Srivastava, M. A. Siddiqui and S. K. Srivastava, “Molecular ADDUCTS of triphenyltin (Ethyl Xanthates),” Indian Journal of Chemistry, Vol. 26A, 1987, p. 1162.
- B. Umezawa, O. Hoshino, S. Sawaki, H. Sashida, K. Mori, Y. Hamada, K. Kotera and Y. Iitaka, “A Synthesis of (±)-Δ2-α-Lycoren-7-One, the Key Intermediate for the Total Synthesis of (±)-Lycorine,” Tetrahedron, Vol. 40, No. 10, 1984, pp. 1783-1790.
- A. Kumar, M. Agarwal and A. K. Singh, “Selenated Schiff Bases of 2-Hydroxyacetophenone and Their Palladium(II) and Platinum(II) Complexes: Syntheses, Crystal Structures and Applications in the Heck Reaction,” Polyhedron, Vol. 27, No. 2, 2008, pp. 485-492. doi:10.1016/j.poly.2007.09.027
NOTES
*Corresponding author.