Y. Z. Fu et al. / J. Biomedical Science and Engineering 4 (2011) 403-409
408
Table 2. The results of the positive control experiments.
Coincidence
Positive
Control Theoretic
Sequence Experimental
Data Method 1 Method 2 Method 3
E.coli E.coli (1) 0.074 07 0.069 63 2.62E-04
E.coli E.coli (2) 0.074 074 074 074 074 100.066 666 666 666 666 708.20E-04
E.coli E.coli (3) 0.074 074 074 074 074 100.066 666 666 666 666 701.32E-04
E.coli E.coli (4) 0.074 074 074 074 074 100.066 666 666 666 666 708.76E-04
Part I
E.coli E.coli (5) 0.074 074 074 074 074 100.066 666 666 666 666 700.004 419 889 502 762 430
Salmonella Salmonella (1) 0.035 714 285 714 285 700.035 000 000 000 000 0004.28E-05
Salmonella Salmonella (2) 0.071 428 571 428 571 400.064 285 714 285 7143 0.015 463 917 525 773 200
Salmonella Salmonella (3) 0.035 714 285 714 285 700.035 000 000 000 000 0001.87E-04
Salmonella Salmonella (4) 0.035 714 285 714 285 700.035 000 000 000 000 0001.82E-05
Part II
Salmonella Salmonella (5) 0.035 714 285 714 285 700.035 000 000 000 000 0000.010 344 827 586 206 900
experiments.
Given the allowed tolerance during experiment and
the previous experience, we find the results of the coin-
cidence are all within acceptance, which reflects high
probability of the similarity between two species in the
comparison, and which also demonstrates that our soft-
ware is robust and accurate.
5. RELATED WORK
The software in this paper completes the comparison
between the known species in the databases and the un-
known species which has mass-intensity data generated
by MALDI-TOFMS. In the next phrase, we will do sta-
tistical analysis to amount of spectra from one species
and expect to compare affinity among unknown species.
Furthermore, we will try to model for species and search
the possible species range for the unknown species based
on its MALDI-TOFMS data.
6. CONCLUSIONS
In order to allow flexibly microbial matching in a user-
friendly way, we design the software “micromatcher”.
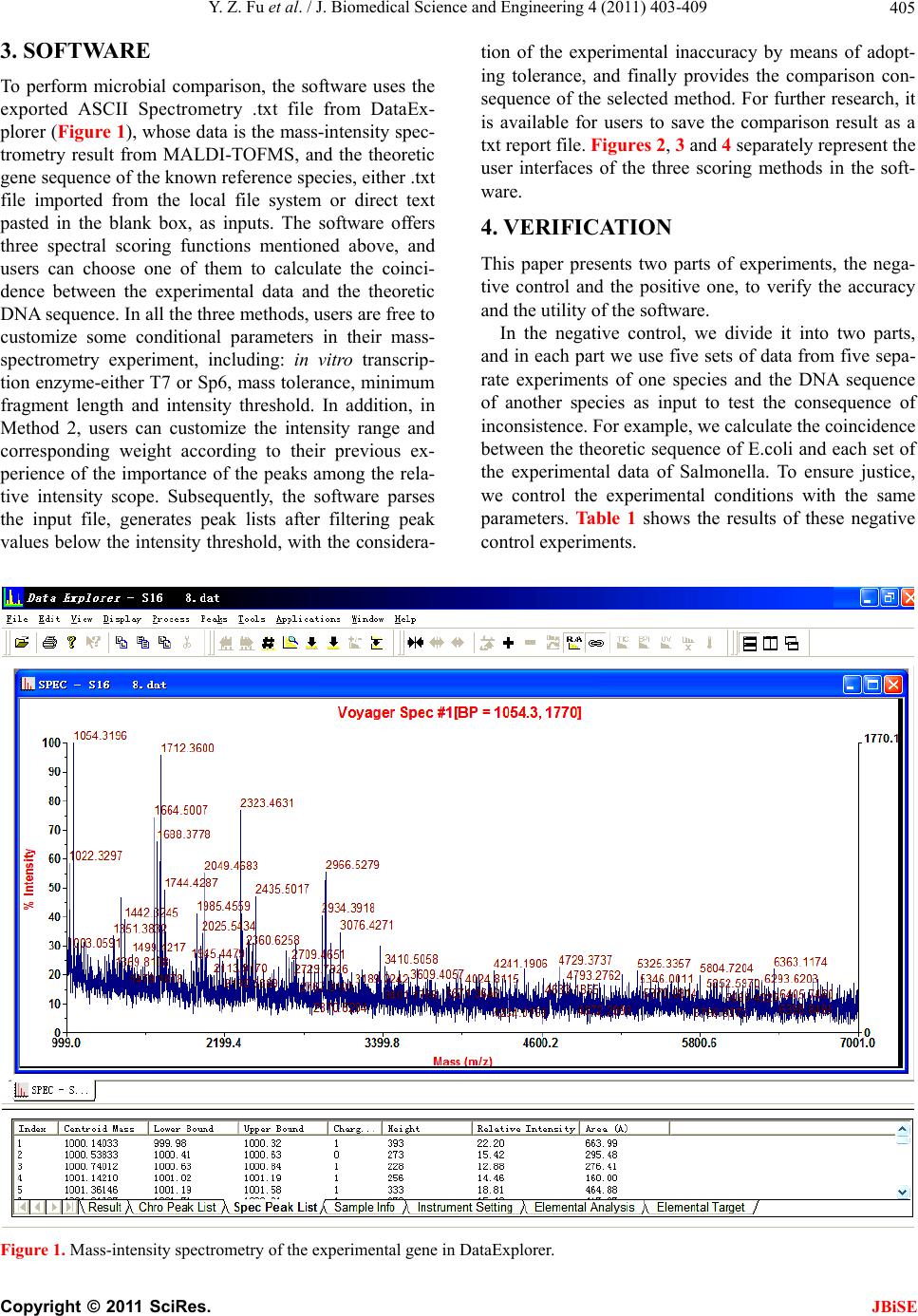
To perform microbial comparison, the software uses
exported ASCII Spectrometry .txt file from DataExplorer,
whose data is the mass-intensity spectrometry result
from MALDI-TOFMS and the theoretical gene sequence
of the known species in the database as inputs. The soft-
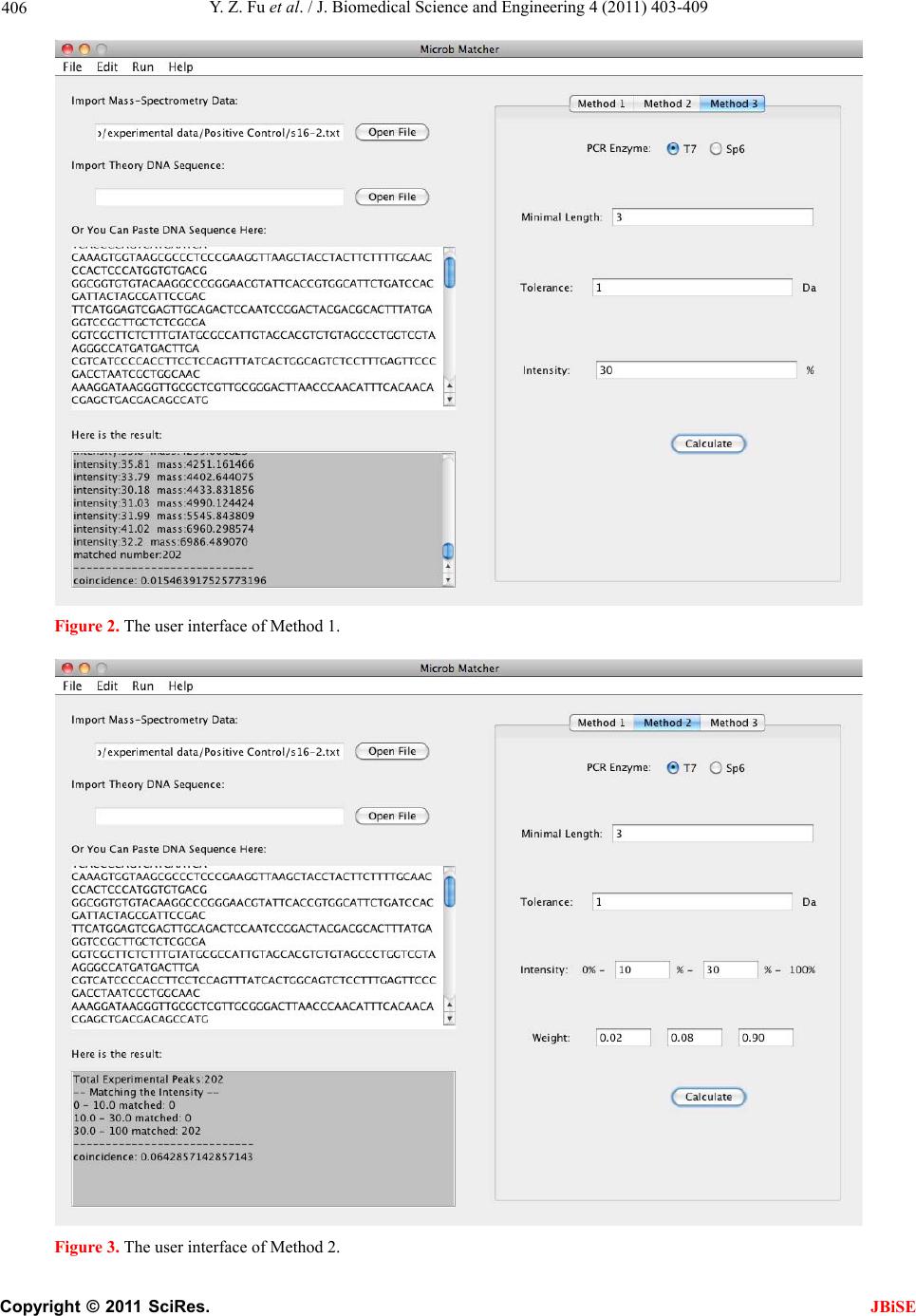
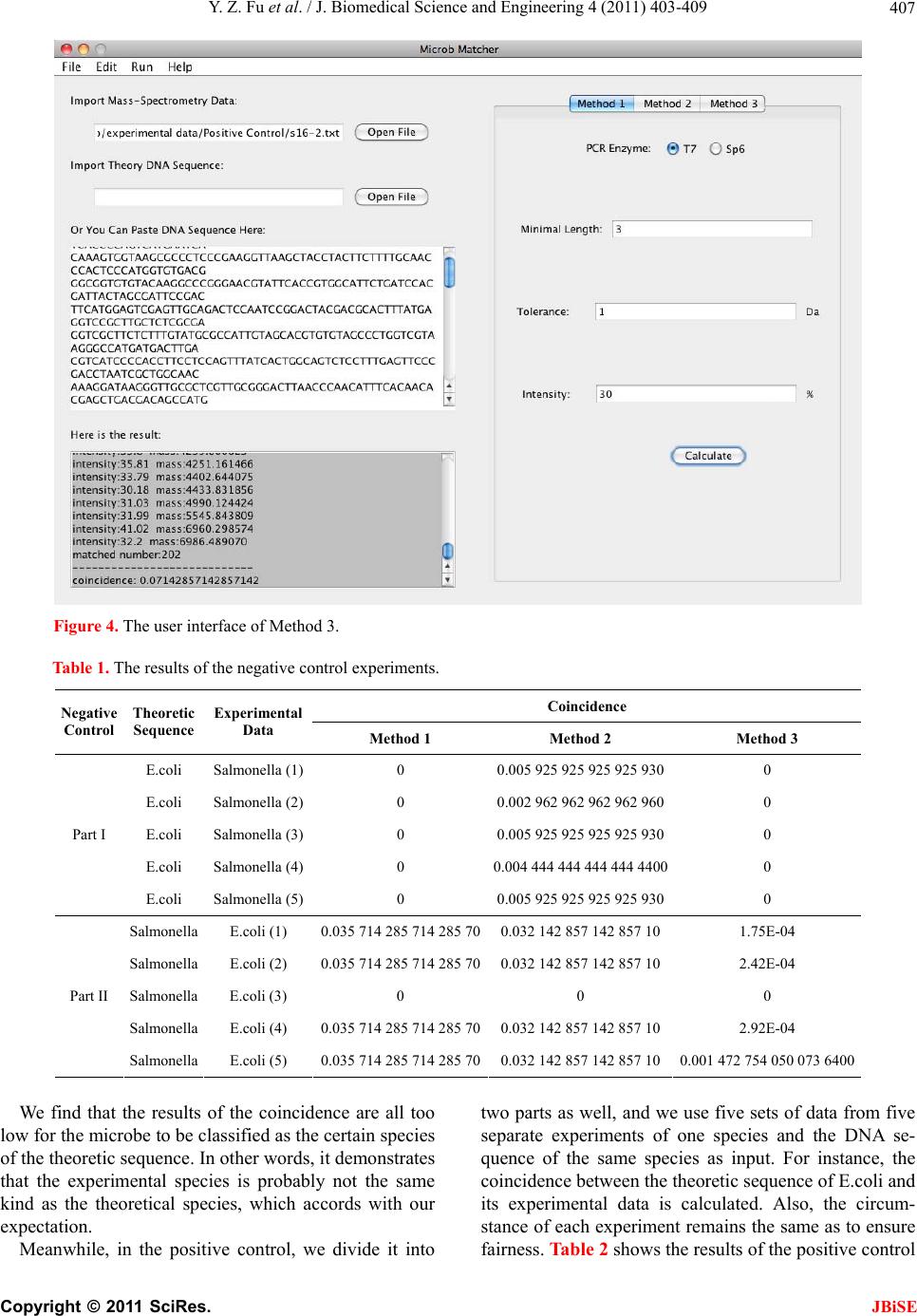
ware offers three spectral scoring functions and users
can choose one of them. Then users are free to customize
some comparison parameters, including: in vitro tran-
scription enzyme, mass tolerance, minimum fragment
length, intensity threshold and corresponding weight.
The software parses the input file, generates peek lists
after filtering peak values below the intensity threshold,
taking into account the experimental inaccuracy by
means of adopting tolerance and finally provides the
comparison consequences.
The software computes the genetic affinity between
the sample gene and genes of known species in the da-
tabase quickly, semi-automatically and accurately.
7. ACKNOWLEDGEMENTS
This paper is sponsored by National Science and Technology Major
Project 2009ZX10004-107 and The Natural Science Founds of Wuhan
University F020504.
REFERENCES
[1] Jarman, K.H., Cebula, S.T., Saenz, A.J., Petersen, C.E.,
Valentine, N.B., Kingsley, M.T. and Wahl, K.L. (2000)
An algorithm for automated bacterial identification using
matrix-assisted laser desorption/ionization mass spec-
trometry. Analytical Chemistry, 72, 1217-1223.
doi:10.1021/ac990832j
[2] Jackson, G.W., McNichols, R.J., Fox, G.E. and Willson,
R.C. (2007) Universal bacterial identification by mass
spectrometry of 16S ribosomal RNA cleavage products.
International Journal of Mass Spectrometry, 261, 218-
226. doi:10.1016/j.ijms.2006.09.021
[3] Jackson, G
.W., McNichols, R.J., Fox, G.E. and Willson,
R.C. (2008) Toward universal flavivirus identification by
mass cataloging. Journal of Molecular Diagnostics, 10,
pp. 135-141. doi:10.2353/jmoldx.2008.070130
[4] Zhou, X.H., Gonnet, G., Hallett, M., Münchbach, M.,
Folkers, G. and James, P. (2001) Cell fingerprinting: An
approach to classifying cells according to mass profiles
of digests of protein extracts. Proteomics, 1, 683-690.
doi:10.1002/1615-9861(200104)1:5<683::AID-PROT68
3>3.0.CO;2-3
[5] Ressom, H.W., Varghese, R.S., Drake, S.K., Hortin, G.L.,
Abdel-Hamid, M., Loffredo, C.A. and Goldman, R.
C
opyright © 2011 SciRes. JBiSE