 Journal of Geoscience and Environment Protection, 2014, 2, 37-43 Published Online July 2014 in SciRes. http://www.scirp.org/journal/gep http://dx.doi.org/10.4236/gep.2014.24006 How to cite this paper: Kollias, K., Mylona, E., Adam, K., Papassiopi, N., & Xenidis, A. (2014). Suppression of Pyrite Oxida- tion by Surface Silica Coating. Journal of Geoscience and Environment Protection, 2, 37-43. http://dx.doi.org/10.4236/gep.2014.24006 Suppression of Pyrite Oxidation by Surface Silica Coating K. Kollias, E. Mylona, K. Adam, N. Papassiopi, A. Xenidis School of Mining and Metallurgical Engineering, National Technical University of Athens, Athens, Greece Email: papasiop@metal.ntua.gr Received June 2014 Abstract The exposure of pyrite (FeS2) to atmospheric conditions during mining activity causes a series of complex oxidation reactions, resulting to acid generation and the subsequent release of toxic heavy metals in the surrounding aquatic and terrestrial ecosystems. The produced acidic mine waters, known as acid mine drainage (AMD), constitute one the major environmental problems of both operating and abandoned mixed sulphide, coal and other mine sites where sulphidic miner- als are encountered. A sustainable approach to the environmentally safe pyrite-bearing extractive waste management is related to the prevention of oxidation by developing artificial coatings on the pyrite surfaces. In this study, experiments performed to study the conditions of the silica coating formation on the FeS2 particles contained in a pyrite concentrate are presented. Batch tests involving the treatment of pyrite samples with a coating solution, consisting of Na2SiO3, H2O2 and buffered pH, were performed under a liquid to solid ratio (L/S) 100 l/kg. The effect of para- meters including SiO2 concentration (5 - 50 mM), pH values (5.0 - 8.0) and contact time up to 24 hours, was investigated. Parameters examined to monitor the silica coating formation process in- clude analysis of Fe, Si, and H2O2 in the aqueous phase. Scanning electron microscopy with energy dispersive spectrometry (SEM/EDS) was used for the examination of the chemically mod- ified surfaces of silica-treated pyrite samples. Keywords Acid Mine Drainage, Mine Wastes, Pyrite, Silica Coating 1. Introduction Acid mine drainage (AMD) constitutes one of the most severe environmental problems of the mining industry. AMD which is a low-pH aqueous phase enriched primarily in iron (Fe) and sulfate anions, but also containing many metals and metalloids, such as lead (Pb), copper (Cu), zinc (Zn), manganese (Mn), nickel (Ni), cadmium (Cd), mercury (Hg) etc., depending on the mineralogy of the ore/waste. These acidic waters are produced by the oxidation of sulphide minerals, contained in the mine wastes and rocks, under the action of atmospheric air and water and catalyzed by the presence of bacteria. Singer & Stumm (1970) described the pyrite oxidation, which may occur via direct and indirect mechanisms, according to the following reactions:  K. Kollias et al. 2 22 234 FeS3.75O3.5H OFe(OH)2SO4H −+ + +→++ (1) 3 22 22 4 FeS14Fe8H O15Fe2SO16H ++ −+ ++→+ + (2) Once generation of acidic waters commences, the sources of acidity may remain active for a long time after the mine closure. So, the effects of AMD can be both pernicious and far-reaching since contamination of surface and/or underground water can carry the pollution considerable distances from the mining area. The acidic drainages are treated by conventional reclamation techniques, such as addition of alkaline mate- rials (mainly, limestone and lime), covering with water to prevent oxygen access or formation of low permeabil- ity soil covers to minimize oxidizing agents transfer to the sulphides. Apart from the conventional prevention and remediation methods, an emerging and appealing technique for the inhibition of acid generation from sul- phidic wastes involves the development of artificial coatings on sulphide grain surfaces (particularly on pyrite grains due to the abundance to all geological environments). This process is known as microencapsulation and was first described by Evangelou (1995a, 1995b), investigating the formation of phosphate coating on pyrite surfaces. Other published studies refer to the establishment of coatings using inorganic materials, FeOOH (Huminicki & Rimstidt, 2009), silicates (Zhang & Evangelou, 1998; Evangelou, 2001) and magnesium and manganese compounds (Glover, 2007) and organic coatings such as humic acid and other organic ligands (Lalvani et al., 1996; Elsetinow et al., 2003; Kargbo et al., 2004 ). The methodology, which was developed for the formation of silicic coating includes partial leaching/oxide- tion of the sulphidic waste using a solution containing hydrogen peroxide (H2O2), a silica source and adjusted to pH 4.0 - 6.0 (Evangelou, 1996). According to the study of Evangelou (2001), pyrite treatment with a solution consisting of H2O2 0.145 M, Si 0.0018 M, CH3COONa 0.01 M, NaCl 0.1 M and adjusted to pH 6 at room temperature results in the formation of a silica coating composed of an inner layer of Fe3+ oxy-hydroxides and an outer layer of Si hydroxides. The coating process reduced by 85% the amount of oxidized pyrite compared to the control test. Bessho et al. (2011) performed batch-type tests and an effective silica coating was established on the pyrite surfaces, after treatment with a solution of Si concentration higher than 2500 mg/L and pH: 7.0. However, coating technology needs further investigation. In this study, batch-type tests were performed in order to investigate the effect of pH and silica concentration on the formation of silica coating on the grains of a pyrite concentrate material. 2. Materials and Methods A pyrite concentrate material assaying 50% S was used. The pyrite sample was screened and the selected frac- tion for the experiments was (−125 + 75) μm. The secondary oxidation products formed on the pyrite were re- moved by a pre-treatment procedure in which the selected fraction was washed with HCl 1 M for 24 hours (liq- uid to solid ratio, L/S = 20 l/kg), thereafter rinsed repeatedly with deionised water. The samples were then dried at 40˚C for 24 hours and finally rinsed once with acetone. The purity of pyrite was confirmed by SEM/EDS and powder-XRD. A series of batch-type tests were executed under atmospheric conditions (T = 23˚C ± 1˚C) and the experi- mental conditions are presented in Table 1. Silica solution was prepared by dissolving pentahydrate sodium si- licate (Na2SiO3*5H2O) in deionized water and hydrochloric acid (HCl 3 N) was used to adjust pH to the selected Table 1. Experimental conditions with the individual variables studied. Parameter Value Size fraction (μm) −125 + 75 H2O2 (mM) 100 SiO2 (mM) 5, 10, 25, 50 pH 5.0, 6.0, 7.0, 8.0 L/S (l/kg) 100 Duration (h) 0.5, 1, 2, 4, 6, 24 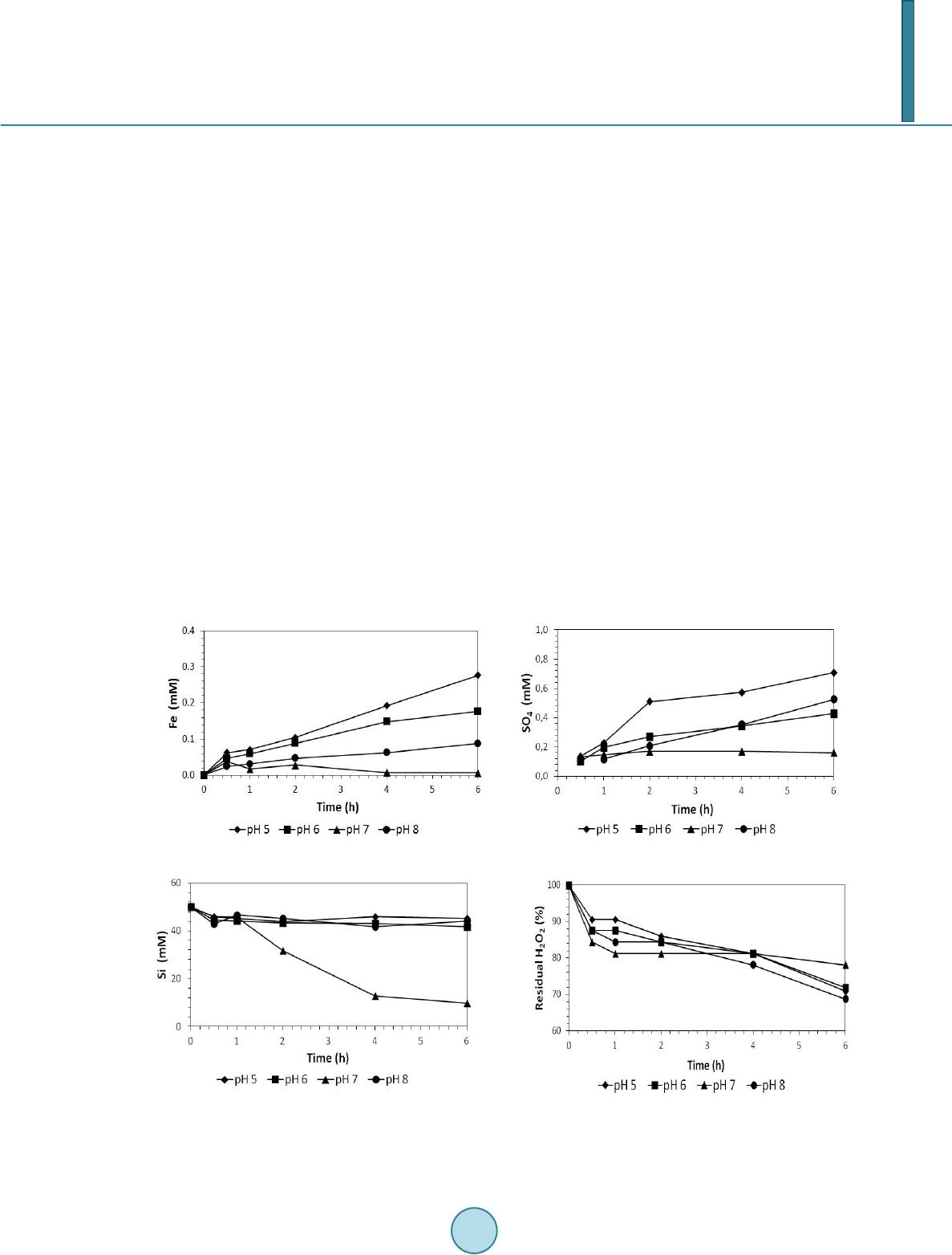 K. Kollias et al. pH values. Sodium acetate (CH3COONa) 0.2 M was used to buffer the coating solution to pH values 5.0 and 6.0, while THAM (tris-hydroxymethylaminomethane) 0.1 M was used to adjust to pH 7.0 and 8.0. The experimental procedure involved the preparation of suspensions containing 0.9 g of pyrite and 90 mL of solution. The suspensions were placed for agitation on a rotary shaker (10 rpm) and removed for analyses after 0.5, 1, 2, 4, 6 and 24 hours. Each suspension was filtered through a 0.45 μm pore size filter and the filtrate was analyzed for Fe, Si, SO4 and H2O2. It is mentioned that the filtration of the suspensions buffered at pH 7.0 was performed using a filter paper with pore size 4 - 12 μm due to their colloidal nature. Iron and silicon concentrations in the aqueous phase was determined by means of Atomic Absorption Spec- trophotometry (2100 Perkin Elmer). Sulfate concentrations were measured gravimetrically. The concentration of residual hydrogen peroxide in the aqueous phase was determined by volumetric titration with KMnO4 0.02 M. The surfaces of solid residues were examined and characterized by scanning electron microscopy with energy dispersive spectrometry (SEM/EDS). 3. Results and Discussion 3.1. Effect of pH and Silica Concentration The concentration of dissolved Fe and sulphates in the solutions after treatment of the pyrite sample with the coating solution consisting of H2O2 0.1 M, SiO2 50 mM and adjusted to pH: 5.0 - 8.0 vs. time is presented in Figure 1(a) and Figure 1(b). As seen in Figure 1(a), operation pH has a major effect on the dissolution of iron. The highest dissolution rate is observed at pH 5.0 with an almost linear kinetics, corresponding to the release of 0.28 mM of iron after 6 hours of treatment. Continuously increasing concentrations of iron in solution are also observed at pH 6.0 and 8.0, but the dissolution rates are lower and the final concentrations are 0.18 and 0.09 mM after 6 h of treatment. The best performance of the coating solution is observed at pH 7.0. At this pH, there is a slight mobilization of iron at the initial stages of treatment, not exceeding the level of 0.04 mM, but afterwards (a) (b) (c) (d) Figure 1. Concentration of dissolved (a) iron; (b) sulphates; (c) silicon and (d) % residual H2O2 in the aqueous phase vs. time at four operating pH values, 5.0, 6.0, 7.0, and 8.0. Initial Si 50 mM.  K. Kollias et al. reprecipitation of iron occurred resulting in a final concentration lower than 0.004 mM. The effect of pH on the oxidation of sulphates can be seen at Figure 1(b). The highest oxidation is observed at the acidic pH 5.0, corresponding to 0.7 mM after 6 h. At pHs 6.0 and 8.0 the concentration of SO4 in the solu- tion after 6 h of treatment is 0.4 and 0.52 mM, respectively. At pH 7.0, the release of SO4 in the aqueous phase is almost constant, varying between 0.15 - 0.17 mM, regardless of the treatment duration. The evolution of Si is shown in Figure 1(c). At the end of treatment tests, the concentration of Si was 45.5, 41.7 and 44.2 mM for the experiments executed under pH values 5.0, 6.0 and 8.0, indicating a very slight degree of precipitation compared to the initial Si concentration of 50 mM. On the contrary, the evolution of Si concen- tration at pH 7.0 suggests that a precipitation phenomenon occurs, resulting in a final concentration of Si after 6 h of treatment equal to 9.8 mM. According to several studies, Si concentration is strongly pH-depended and controlled by processes like polymerization, dissolution, absorption and precipitation (Alexander, 1954; Iler, 1979; Sjorberg, 1996). Our experimental data demonstrate that the higher removal of Si, i.e. 89% occur at pH 7.0 in accordance with the above studies. This may be attributed to the extensive silica polymerization, leading to the homogeneous nucleation of distinct SiO2 phases on the oxidized pyrite surface. Given that the higher removal of Si was associated with the lower level of dissolved iron, it may be deduced that silicic acid in the solution was associated with the iron hydroxides, leading to the formation of silica-iron(III) complexes on pyrite surfaces (Schenk & Weber, 1968; Herbillon & Vinh, 1969). The silicic acid association with hydrous ferric oxyhydroxides involves both absorption and polymerization processes and depends on the amount of H4SiO4 present at their surfaces, i.e. absorption as monomeric silicate on the surface, or via siloxane linkages (Si-O-Si) essentially polymerizing to form a separate silica phase (Swedlund & Webster, 1999; Swed- lund et al., 2011). According to Schwertmann & Cornell (1991), the Fe3+ hydroxides formed through co-preci- pitation with silicates are amorphous and/or poorly crystalline and the transformation of amorphous iron preci- pitates to more crystalline phases is blocked by the absorption of silicate species. The % residual H2O2 in the solution during treatment tests vs. time and pH is shown in Figure 1(d). The re- sidual hydrogen peroxide in the solutions decreased with time and after 6 h of treatment was corresponded to 69% - 78% of the initial amount. The consumption of the oxidizing agent does not correspond stoichiometrically to the amount of pyrite oxidation. It decomposes gradually and this reaction is mainly catalyzed by pyrite surfaces and dissolved iron ions in the solution, but also is catalyzed heterogeneously due to the presence of precipitated amorphous and/or poorly crystalline iron hydroxides (McKibben & Barnes, 1985; Lin & Gurol, 1998). The experimental results obtained with the silica solutions of variable concentrations are shown in Figure 2. It is noted that all these experiments were carried out at pH 7.0. As seen in Figure 2(a), when the treatment is conducted using the highest concentration of SiO2 (50 mM), the concentration of dissolved Fe increases during the first 2 h and then decreases at the final value of 0.004 mM. During the treatment tests with SiO2 25 mM, the decrease of Fe concentration down to the value of 0.004 mM occurs only after 24 h of treatment, indicating a slower kinetics of precipitation. Iron precipitation was not observed during the two other experiments that were carried out with lower SiO2 concentrations, i.e. 5 and 10 mM. Presumably, a longer treatment time is required for the appearance of precipitation process using these low SiO2 levels. The evolution of SO4 concentrations during the treatment with variable SiO2 concentration is shown in Figure 2(b). It is evident that the oxidation of sulphur is higher as the SiO2 concentration decreases. This is obviously related with the slower kinetics of formation of the silica coating at the lower SiO2 concentrations. The variation of SiO2 concentration during this experimental set is shown in Figure 2(c). It is evident that the kinetics of SiO2 removal from the aqueous phase depends greatly on the initial concentration of silica. 3.2. Characterization of Treated Pyrite Surfaces by SEM/EDS SEM microphotographs of pyrite grains treated with 50 mM SiO2 at pH 5.0, 7.0 and 8.0 are shown in Figure 3. Pyrite treated at pH 7.0 has a completely different surface morphology, which is due to the development of the Fe(OH)3-Si(OH)4 coating. EDS analyses of this grain indicated up to 16 wt% of Si and 12 wt% of O. Iron (52%) and sulphur (20%) measurements cannot be directly attributed to surface species due to the pyrite background. The presence of SiO2 aggregates of approximately 5 μm is also clear on the pyrite surface. Characteristic etch pits can be ob- served on the surface of pyrite grains treated at pH 5.0 and to a lesser extent at pH 8.0. According to the EDS analysis, grains at pH 5.0 have a very low content of Si, i.e. no more than 0.30 wt%. At pH 6.0 and 8.0, the con- centration of Si was up to 5 wt%. 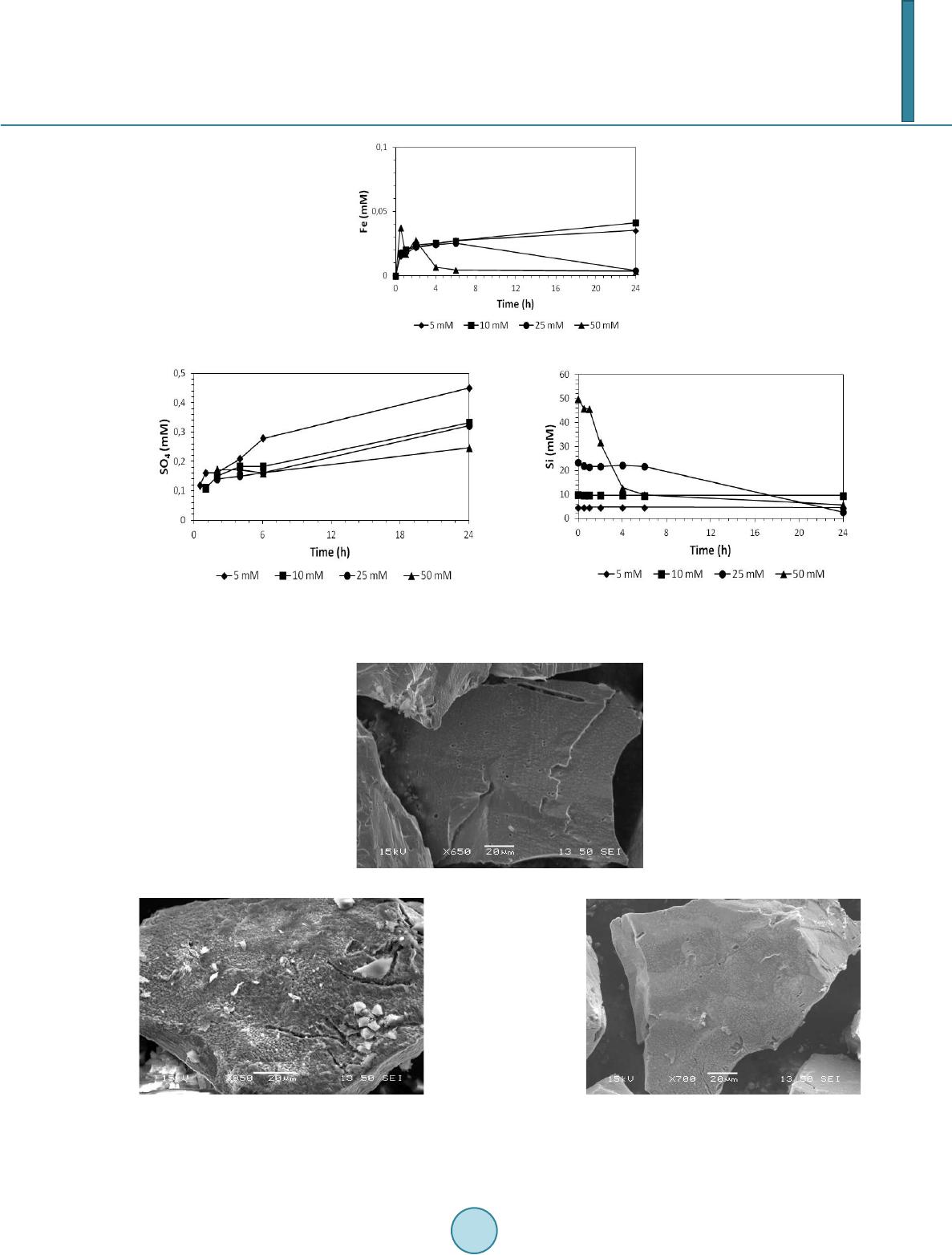 K. Kollias et al. (a) (b) (c) Figure 2. Concentration of dissolved (a) iron; (b) sulphates and (c) silicon vs. time for the pyrite treated with variable con- centrations of Si, 5, 10, 25 and 50 mM at pH 7.0. (a) (b) (c) Figure 3. SEM microphotographs of the (−125 + 75) μm pyrite size fraction treated with a solution containing SiO2 50 mM buffered at pH (a) 5.0, (b) 7.0 and (c) 8.0.  K. Kollias et al. Silicon concentration on the surfaces of pyrite grains treated with variable concentrations of SiO2 at pH 7.0 was low, as indicated by EDS, i.e. up to 0.40 and 0.46 wt% for experiments carried out with solution of SiO2 5 and 10 mM, respectively, after 24 hours of treatment. A higher content of Si was measured, i.e. up to 2.0 wt%, when the treatment was conducted with SiO2 25 mM. SiO2 aggregates of approximately 2 - 5 μm are also ob- vious on pyrite surface. 4. Conclusion Based on the results of this study, the following conclusions can be drawn: The treatment of the pyrite grains (−125 +75) μm with a solution consisting of H2O2 0.1 M, SiO2 50 mM and buffered at pH 7.0 for 6 hours re- sulted in the formation of an extensive coating on the pyrite surface, containing up to 16 wt% of Si. The forma- tion of this coating prevented pyrite oxidation, as demonstrated by the curves of iron and sulphates in the aqueous phase. The optimum pH for the development of surface coating is close to pH value 7.0. The kinetics of the process is greatly influence by the initial concentration of SiO2. For instance, precipitation of SiO2 is com- pleted after 6 hours starting with 50 mM of SiO2 and 24 hours with 25 mM SiO2. Experiments are ongoing to examine the stability of silica coatings under different conditions and evaluate their overall effectiveness in inhibiting pyrite oxidation. Acknowledgements This research has been co-financed by the European Union (European Social Fund—ESF) and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF)-Research Funding Program: THALES. Investing in knowledge society through the Euro- pean Social Fund. References Alexander, G. B. (1954). The Polymerization of Monosilicic Acid. Journal of the American Chemical Society, 76, 2094- 2096. http://dx.doi.org/10.1021/ja01637a017 Bessho, M., Wajima, T., Ida, T., & Nishiyama, T. (2011). Experimental Study on Prevention of Acid Mine Drainage by Si- lica Coating of Pyrite Waste Rocks with Amorphous Silica Solution. Environmental Earth Sciences, 64, 311-318. http://dx.doi.org/10.1007/s12665-010-0848-0 Elsetinow, A., Borda, M., Schoonen, M. A., & Strongin, D. R. (2003). Suppression of Pyrite Oxidation in Acidic Aqueous Environments Using Lipids Having Two Hydrophobic Tails. Advances Environmental Research, 7, 969-974. http://dx.doi.org/10.1016/S1093-0191(02)00101-6 Evangelou, V. P. (1996). Oxidation Proof Silicate Surface Coating on Iron Sulfides. U.S. Patent, Patent Number 5,494,703. Evangelou, V. P. (1995a). Potential Microencapsulation of Pyrite by Artificial Inducement of Ferric Phosphate Coatings. Journal of Environmental Quality, 24, 535-542. http://dx.doi.org/10.2134/jeq1995.00472425002400030021x Evangelou, V. P. (1995b). Pyrite Oxidation and Its Control. Boca Raton, FL: CPC Press. Evangelou, V. P. (2001). Pyrite Microencapsulation Technologies: Principles and Potential Field Application. Ecological Engineering, 17, 165-178. http://dx.doi.org/10.1016/S0925-8574(00)00156-7 Glover, R. (2007). Permanganate Passivation of Pyrite Containing Ores: Scale up and Characterization. MS Thesis, Reno: University of Nevada. Herbillon, A. J., & Tran Vinh An, J. (1969). Heterogeneity in Silicon-Iron Mixed Hydroxides. Journal of Soil Science, 20, 223-235. http://dx.doi.org/10.1111/j.1365-2389.1969.tb01569.x Huminicki, D. M. C., & Rimstidt, J. D. (2009). Iron Oxyhydroxide Coating of Pyrite for Acid Drainage Control. Applied Geochemistry, 24, 1626-1634. http://dx.doi.org/10.1016/j.apgeochem.2009.04.032 Iler, R. K. (1979). The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties and Biochemistry of Silica. New York, NY: John Wiley and Sons. Kargbo, D. M., Atallah, G., & Chatterjee, S. (2004). Inhibition of Pyrite Oxidation by a Phospholipid in the Presence of Sili- cate. Environmental Science Technology, 38, 3432-3441. http://dx.doi.org/10.1021/es0352552 Lalvani, S. B., Zhang, G., & Lalvani, L. S. (1996). Coal Pyrite Passivation Due to Humic Acids and Lignin Treatment. Fuel Science and Technology International, 14, 1291-1313. http://dx.doi.org/10.1080/08843759608947640 Lin, S. S., & Gurol, M. D. (1998). Catalytic Decomposition of Hydrogen Peroxide on Iron Oxide: Kinetics, Mechanism, and  K. Kollias et al. Implications. Environmental Science and Technology, 32, 1417-1423. http://dx.doi.org/10.1021/es970648k McKibben, M. A., & Barnes, H. L. (1986). Oxidation of Pyrite in Low Temperature Acidic Solutions: Rate Laws and Sur- face Textures. Geochimica et Cosmochimica Acta, 50, 1509-1520. http://dx.doi.org/10.1016/0016-7037(86)90325-X Schenk, J. E., & Weber Jr., W. J. (1968). Chemical Interactions of Dissolved Silica with Iron(II) and (III). Journal of the American Water Works Association, 2, 199-212. Schwertmann, U., & Cornell, R. M. (1991). Iron Oxides in the Laboratory. Preparation and Characterization. VCH Verlags- gesellschaft, Weinheim. Singer, P. C., & Stumm, W. (1970). Acid Mine Drainage: The Rate-Determining Step. Science, 167, 1121-1123. http://dx.doi.org/10.1126/science.167.3921.1121 Sjoberg, S. (1996). Silica in Aqueous Environments. Journal of Non-Crystalline Solids, 196, 51-57. http://dx.doi.org/10.1016/0022-3093(95)00562-5 Swedlund, P. J., & Webster, J. G. (1999). Adsorption and Polymerization of Silicic Acid on Ferrihydrite, and Its Effect on Arsenic Adsorption. Water Research, 33, 3413-3422. http://dx.doi.org/10.1016/S0043-1354(99)00055-X Swedlund, P. J., Sivaloganathan, S., Miskelly, G. M., & Waterhouse, G. I. N. (2011). Assessing the Role of Silicate Polyme- rization on Metal Oxyhydroxide Surfaces Using X-Ray Photoelectron Spectroscopy. Chemical Geology, 285, 62-69. http://dx.doi.org/10.1016/j.chemgeo.2011.02.022 Zhang, Y. L., & Evangelou, V. P. (1998). Formation of Ferric Hydroxide-Silica Coatings on Pyrite and Its Oxidation Beha- vior. Soil Science, 163, 53-62. http://dx.doi.org/10.1097/00010694-199801000-00008
|