Journal of Modern Physics, 2011, 2, 158-161
doi:10.4236/jmp.2011.23024 Published Online March 2011 (http://www.SciRP.org/journal/jmp)
Copyright © 2011 SciRes. JMP
Structural and Spin Polarization Effects of Cr, Fe and Ti
Elements on Electronical Properties of α–Al2O3 by First
Principle Calculations
Hossein Asghar Rahnamaye Alibad, Shaban Reza Ghorbani
Department of physics, Sabzevar Ta rbiat Moallem University, Sabzevar, Iran
E-mail: h.rahnama@sttu.ac.ir, h_rahnamay@yahoo.com
Received October 3, 2010; revised December 7, 2010; accepted December 11, 2010
Abstract
Structural and spin polarization effects of Cr, Fe and Ti elements on electronical properties of alumina have
been studied by using of Local spin density approximation within density functional theory. The calculated
results indicated that substituting aluminium atoms by these dopants have a significant influence on the
structural and electronic properties of α–Al2O3 crystals. Band gap of alumina decreases with the substitution
of these impurities. Results show that band gap is different for spin-up and down (spin splitting effect).
Among these impurities the effect of Ti on size of the energy gap is small in comparison with Cr and Fe. It is
suggested that the origin of electrons spin splitting is appeared from exchange energy of d-states. These
results may be useful to obtain a physical beheviour of transition metals for electrons spin polarization in
d-states.
Keywords: LSDA, Spin Polarization, Alumina, Transition Metals
1. Introduction
A unique combination of alumina with transition metals is
very important due to their possible industrial applications.
The corundum or sapphire phase of alumina (α–Al2O3)
has widespread applications in ceramic and semiconduc-
tor industry [1]. In order to improve electrical and optical
properties of alumina, it can be doped by other metals; this
requires the variation of electrical properties by theoreti-
cal calculations. Good substitutions on the Al atom sites
are transition metals (TM), because the d-bands in these
metals are partially filled and extended over the band gap.
The substitutions of these metals change the band gap size
and improve alumina properties.
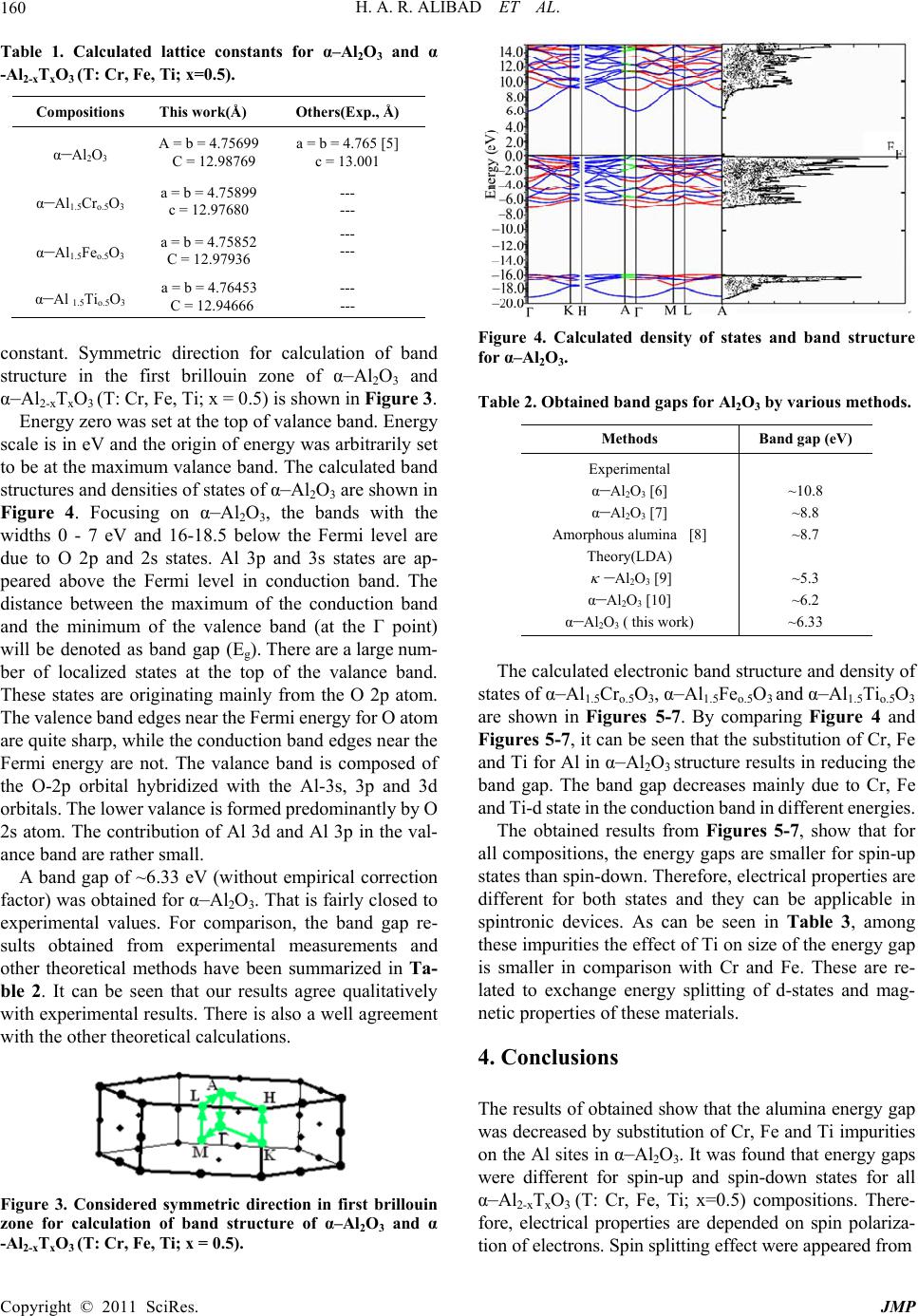
Nearly all atoms have multiple electrons but most of
them are paired up with another opposite spin electrons in
the orbital. Solid magnetic properties are derived from the
ground state properties of incompletely filled electron
shells. Observed magnetic response, in a particular system
largely depends on how the spin and orbital properties of
these electrons end up in consideration of Pauli’s exclu-
sion principle and minimizing Coulomb repulsion [2].
In this work, the influence of spin polarization Cr, Fe
and Ti elements on electrical properties of alumina have
been studied. Spin polarization is the first rule of Hound
for determining the ground state (lowest energy) of elec-
tronic configuration in an atom. According to these rules
and Pauli’s exclusion principle, electrons have been ar-
ranged in a way to have maximum total spin, S. In fact,
these minimize Coulomb energy so that two parallel spins
can not be in the same state. Energy decreasing due to the
preference of being parallel spins is called exchange en-
ergy. In solids, depending on whether the crystal is insu-
lating or conducting, magnetism has historically been
approached from two different schools of thinking: either
a localized or itinerant point of view. In the localized
concept of magnetism, the electrons and their magnetic
properties remain associated with their respective para-
magnetic ion in an insulating crystal. Conversely, in the
itinerant picture, the conduction electrons are responsible
for magnetism. The magnetic of ordering may arise based
on the specific alignment of the atomic magnetic mo-
ments, favoured by atomic exchange interactions and
itinerant magnetism which is associated with metallic
behaviour.
Normally, in a metal, there is an equal number of
spin-up and spin-down electrons which fill up states to the
Fermi energy. In the absence of an external field, a stable